
ABSTRACT
Colorectal cancer (CRC) is one of the most commonly diagnosed cancers and a major cause of cancer mortality worldwide. At late stage of the disease CRC often shows (multiple) metastatic lesions in the peritoneal cavity which cannot be efficiently targeted by systemic chemotherapy. This is one major factor contributing to poor prognosis. Oxaliplatin is one of the most commonly used systemic treatment options for advanced CRC. However, drug resistance – often due to insufficient drug delivery – is still hampering successful treatment. The anticancer activity of oxaliplatin includes besides DNA damage also a strong immunogenic component. Consequently, the aim of this study was to investigate the effect of bacterial ghosts (BGs) as adjuvant immunostimulant on oxaliplatin efficacy. BGs are empty envelopes of gram-negative bacteria with a distinct immune-stimulatory potential. Indeed, we were able to show that the combination of BGs with oxaliplatin treatment had strong synergistic anticancer activity against the CT26 allograft, resulting in prolonged survival and even a complete remission in this murine model of CRC carcinomatosis. This synergistic effect was based on an enhanced induction of immunogenic cell death and activation of an efficient T-cell response leading to long-term anti-tumor memory effects. Taken together, co-application of BGs strengthens the immunogenic component of the oxaliplatin anticancer response and thus represents a promising natural immune-adjuvant to chemotherapy in advanced CRC.
Introduction
Colorectal cancer (CRC) is one of the most commonly diagnosed cancers and one of the leading causes of cancer mortality worldwide. In the past 20 years early stage disease recognition became much more frequent due to screening programs and techniques like colonoscopy. Consequently, therapeutic efficacy for CRC has distinctly improved, often based on curative surgery. Nonetheless, especially the so-called peritoneal carcinomatosis, which is frequently observed at the late stage of the disease, is still hampering successful curative CRC treatment.Citation1 Thus, in advanced disease and in 40–70% of recurrences dissemination occurs, often by transcoelomic spread to the peritoneal cavity, leading to multiple metastatic lesions.Citation2,Citation3 Since systemic chemotherapy and antibody therapies are not able to efficiently penetrate to these lesions confined to the inside of the peritoneum, this leads to very low overall survival (OS) rates of only a few months.Citation1,Citation4 Currently, the gold standard treatment strategy for peritoneal carcinomatosis –besides palliative treatment– is cytoreductive surgery, often as part of hyperthermic intraperitoneal chemotherapy (HIPEC).Citation5 In case of HIPEC treatment, after complete macroscopic removal of tumor lesions, a hyperthermic oxaliplatin solution (41– 43°C) is perfused through the abdominal cavity.Citation6 Although this local treatment strategy leads to an improved outcome and OS, complications during and after surgery as well as insufficient removal of tumor mass are the most critical obstacles for this treatment option. Consequently, there is intensive research for improvement of late stage CRC treatment by development of multimodal therapies containing immune-adjuvant components (like the cytokine IL27 or CAR-T cellsCitation8) to efficiently eliminate the remaining tumor residues.Citation1
Oxaliplatin is approved for treatment of stage III and advanced CRC (in combination with 5-fluorouracil and leucovorin in frame of the widely used FOLFOX regimen) and is considered to be the most effective therapeutic agent at this stage of the disease. This drug is a next generation platinum compound with a better safety profile than cisplatin and shows no cross-resistance to other platinum compounds.Citation9,Citation10 With regard to modes of action, the generation of platinum-DNA inter- and intrastrand crosslinks blocking DNA synthesis seems to be one main driver of its anticancer activity.Citation11-13 However, in contrast to cisplatin, oxaliplatin was found to additionally have a strong immunologic component and its anticancer activity strongly depends on the immune system.Citation14-18 Consequently, immunostimulating, anti-immunosuppressing as well as immunogenic effects were described.Citation18-21 The underlying mechanism probably involves the generation of tumor neo-antigens (platinum-DNA or -protein adducts), which subsequently arm the immune system against treated tumors. In more detail, oxaliplatin-treated cancer cells were found to undergo immunogenic cell death (ICD), a particular form of cell death, which is characterized by distinct hallmarks, including calreticulin (CRT) exposure as well as release of ATP and high mobility group box 1 (HMGB1).Citation22 Furthermore, the high efficacy of oxaliplatin against CRC was reported to rely on an intact colon microbiome.Citation23,Citation24 In line with these findings, patients under antibiotic treatment, characterized by a depleted or at least weakened intestinal flora, experienced a reduced responsiveness to oxaliplatin-containing treatment regimens.Citation23 Since the peritoneal cavity is known to harbor a high immune competenceCitation1,Citation25,Citation26 and oxaliplatin is strongly dependent on an active immune system, the combination of this drug with immunogenic adjuvants seems very promising. Therefore, we hypothesized that using intraperitoneal (i.p.) administration of oxaliplatin combined with potent immune-stimulating bacterial ghosts (BGs) could be an effective therapeutic approach to treat CRC peritoneal metastasis. BGs are empty (non-living) envelopes of gram-negative bacteria, which are produced by controlled expression of the lysis gene E, leading to the formation of a tunnel structure in the bacterial membranes, resulting in release of the cytoplasmic content.Citation27-29 Due to their preserved intact surface structures, BGs are still attaching to mammalian cells,Citation30 and thus capable of inducing an immune-stimulatory response. Numerous in vitro and in vivo studies have already proven that BGs are non-toxic and well tolerated.Citation29,Citation31
In this study, we show that the combination of immune-stimulating BGs with oxaliplatin is a promising new strategy to treat intraperitoneal carcinomatosis of CRC.
Results
Addition of BGs to oxaliplatin enhances ICD in vitro
As a first approach, the impact of BGs on the ICD induction potential of oxaliplatin was evaluated in vitro using the murine CRC line CT26. To this end, surface CRT levels were tested after incubation with either oxaliplatin and BGs alone or in combination. Immunofluorescence staining revealed a distinctly increased exposure of the so-called “eat me” signal protein CRT on the surface of cells after 4 h incubation with the oxaliplatin/BGs combination (). In more detail, the combination resulted in both a significantly higher percentage of CRT-positive cells as well as an increased amount of surface CRT spots per cell compared to BGs or oxaliplatin treatment alone ( and ). Additionally, experiments at different time points (3 h, 4 h, 5 h) indicated that the combination did not only increase the surface CRT levels but also resulted in an earlier onset of CRT presentation (Suppl. Fig. 1). Thus, in our hands, oxaliplatin mono-treatment exerted effects only after 5 h incubation, while in the combination-treated cells CRT exposure was detectable already after 3 h (Suppl. Fig. 1). Similar results were found in the murine ovarian carcinoma cell line ID8, a second model of intraperitoneal carcinomatosis (Suppl. Fig. 2).
Figure 1. Impact of BGs on the ICD hallmark induction of oxaliplatin in cell culture. (A) Immunofluorescent staining of CRT (green), membrane stain with wheat germ agglutinin (WGA, magenta) and DAPI nucleic stain of CT26 cells was performed after short term treatment (4 h) with oxaliplatin (oxali) and 500 BGs/cell. Representative confocal microphotographs at a magnification of 400x are shown. (B) Quantification of the cell number positive for CRT on the surface relative to the total number of cells, as well as (C) the amount of CRT spots per cell, was performed by counting. A minimum of 50 cells per treatment were counted. (D, E) CT26 cells were treated with the indicated agents and supernatants collected after the indicated incubation times. Subsequently, (D) ATP and (E) HMGB1 release was determined by a luminescent ATP detection assay or an ELISA kit, respectively. Data are represented as mean value ± S.D. of quadruplicates of one representative experiment. Statistical significance was calculated by one-way ANOVA and Bonferroni posttest * p < 0.05; **p < 0.01, significantly different from control.
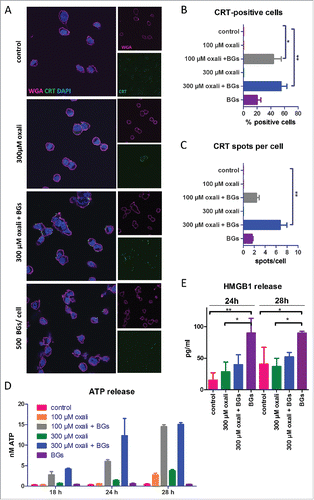
Figure 2. Synergistic antitumor activity of BGs with oxaliplatin in the murine carcinosis model CT26. (A) Male BALB/c mice were injected i.p. with 1 × 105 CT26 cells on day 0 (D0) and treated according to TS1 (n = 4 per group). (B) On day 10 mice were dissected, the tumors visually identified, collected and weighed. Statistical significance was calculated by nonparametric one-way ANOVA and Bonferroni posttest (* p < 0.05, significantly different from solvent group). (C) In a follow-up experiment, mice were treated with TS1 and dissection was performed on day 13. Median tumor and spleen weights were evaluated. Statistical significance was calculated by unpaired t test (** p < 0.01). (D) The second, another treatment group continued therapy as shown in TS2. (E) In these animals, treatment resulted in significantly prolonged survival (**p < 0.01 calculated by Log-rank test and Mantel-Cox posttest). One of the animals experienced complete remission. This cured mouse was subsequently rechallenged i.p. with CT26 cells (1 × 105) on day 80 (indicated by the arrow).
As a next step, ATP and HMGB1 release, two further hallmarks of ICD, were evaluated after several time points ( and ). These experiments revealed a distinctly enhanced ATP release, again together with an earlier onset (18 h vs. 28 h) upon combination treatment compared to oxaliplatin mono-treatment. In contrast to the CRT assay, treatment with BGs alone did not induce a significant ATP release (). Interestingly, when investigating HMGB1 release into the same supernatant samples, BGs mono-treatment was the strongest inducer of this hallmark, while the combination treatment only showed a slightly higher effect than the platinum compound alone (). This was also seen in the ID8 model (data not shown). Finally, to exclude the possibility that addition of BGs enhances oxaliplatin cytotoxicity in vitro, viability assays were performed. These experiments revealed that BGs did not alter tumor cell viability, neither alone nor in combination with oxaliplatin. This indicates the need of an active immune system to enhance cytotoxicity (Suppl. Fig. 3)
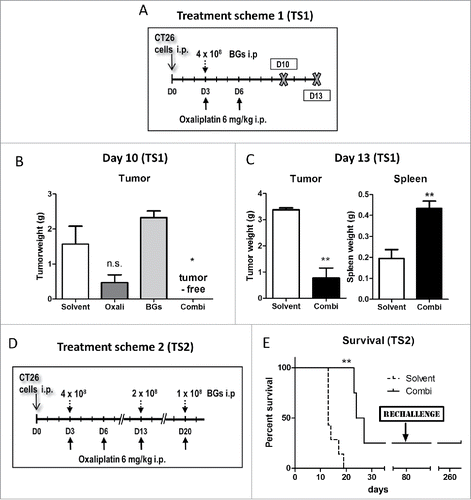
Figure 3. Enhanced efficacy of repeated combination treatment. (A) Female BALB/c mice were injected i.p. with 1 × 105 CT26 cells on day 0 (D0) and treated according to TS3 (n = 7 per group). After one or two cycles of therapy, mice were dissected on day 17 and tumor weight assessed. (B) Pictures of the collected tumor tissues are shown (one of the solvent-treated animals died on day 16, thus no tumors were available here; ✓ marks tumor-free mice). (C) Individual and median tumor weights per treatment group are plotted. Statistical significance was calculated by nonparametric one-way ANOVA and Bonferroni posttest (***p < 0.001, as compared to solvent group). (D) Female C57 BL/6 mice were injected i.p. with 1 × 106 ID8 cells and treated according to TS3 with two therapy cycles (n = 4 per group). The experiment was terminated on day 35 and the intraperitoneal fat tissue containing the small tumor lesions was collected, histologically reprocessed, and H&E-stained. Number of tumor nodules was evaluated from stained slides by counting. In addition, tumor area was digitally measured from scanned H&E-stained slides using Panoramic Viewer Software. Statistical significance was calculated by unpaired t test (* p < 0.05, as compared to solvent group).
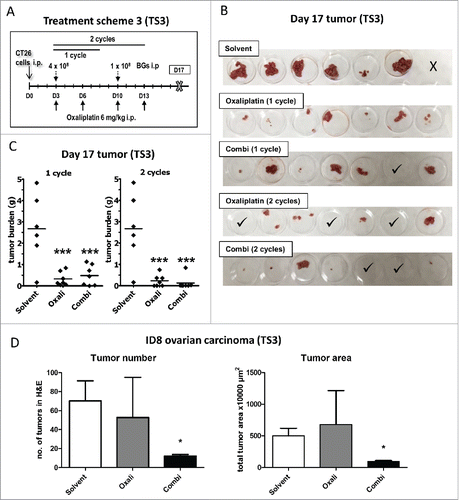
Combination of BGs with oxaliplatin shows significant antitumor activity in two peritoneal carcinomatosis models in vivo
After finding evidence for the induction of ICD in cell culture, the next step was to investigate this drug combination in vivo. Therefore, male BALB/c mice were injected i.p. with CT26 cells (day 0), which resulted in formation of multiple tumors, spread throughout the whole peritoneal cavity with a propensity to colonize the pancreatic niche (Suppl. Fig. 4A). Treatment started on day 3 with oxaliplatin, BGs, or their combination, followed by a second application of oxaliplatin on day 6 (, Treatment scheme 1, TS1). Dissection on day 10 revealed a high tumor burden (average total weight of ∼ 1.6 g) in the peritoneum of both control- and BG-treated mice, while oxaliplatin mono-treatment led to a clearly visible but non-significant reduction to ∼ 0.5 g of tumor burden. Remarkably and opposed to that, all combination-treated mice were tumor-free ( and Suppl. Fig. 4B, upper panel). In order to test whether treatment leads to a complete eradication of cancer cells, a follow-up experiment was performed, in which animals were sacrificed at a later time point (day 13). As shown in and Suppl. Fig. 4B, lower panel, all four combination-treated animals carried significantly (p < 0.01) smaller but clearly visible tumors, indicating that these complete responses after one treatment cycle were only of a transient nature. Noteworthy, in contrast to the multiple malignant lesions (n > 15) of the control animals, the combination treatment group had a distinctly reduced number of lesions (n = 1 to 3) (Suppl. Fig. 4B, lower panel). In addition, in both experiments, the immunogenic component of the BGs activity was evident, as all treated mice had a significantly increased spleen size and weight () indicative for activation of the immune system.Citation32,Citation33
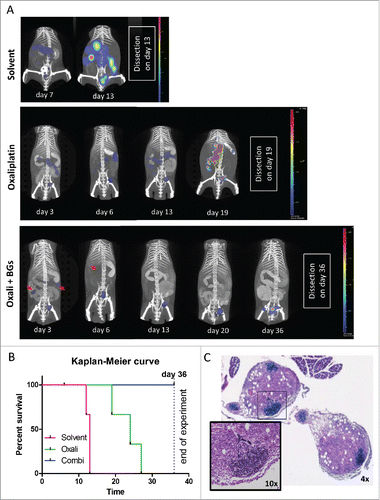
Figure 4. Longitudinal evaluation of treatment response in vivo using CT26F-luc cells. Male BALB/c mice were injected i.p. with 1 × 105 CT26F-luc cells on day 0 (D0) and treated according to TS2 (n = 3 per group). Following s.c. luciferin injection, DLIT/µCT was performed to evaluate tumor burden on the indicated days during treatment. (A) One representative animal is depicted for each treatment showing the overlay of the individual tumor nodules as 3D-luminescent signals (in photons per second) with the µCT scans (red arrows highlight tumor nodules responding to therapy). (B) OS of the treated mice is shown as a Kaplan Meier curve. Statistical significance was calculated by Log-rank test together with Cox Mantle posttest (* p < 0.05). Of note, the combination-treated animal #9 died during drug application on day 6. (C) Histological evaluation of tumor tissue collected from mouse #9 was done by H&E-stain. Infiltration of small, densely packed lymphocytes with a high nuclear/cytoplasmic ratio into two tumor nodules is shown (4x and 10x magnification).
Prolonged combination treatment results in long-lasting complete response and even antitumor vaccination
Based on the finding of clearly visible tumors on day 13, we decided to extend the combination BG/oxaliplatin drug treatment for two additional applications (, TS2). In this experiment, all animals experienced a distinctly and significantly prolonged OS (> 200 % increase in life span, as compared to controls) (). Noteworthy, one of these animals experienced a complete response without relapse for more than 80 days. To investigate whether the complete response was associated with the development of an immunologic memory effect, on day 80 this animal in remission was re-challenged (i.p.) with CT26 cells (arrow in ). In line with the strong immunogenic component of our treatment scheme, indeed the cells were rejected and not able to form tumors within 185 days after injection, thus proving an anti-CT26 vaccination (Suppl. Fig. 4C). Similar results were also gained in a comparable experiment using female mice, where complete responders were also able to reject s.c. injected CT26 cells (data not shown).
Based on these promising results, we evaluated the difference in effectivity between one and two cycles of treatment against peritoneal cancer in more detail (, TS3). In line with the first experiments, dissection on day 17 revealed visible tumor regrowth in both oxaliplatin- as well as combination-treated animals after application of a single treatment cycle. Only one of the combination-treated animals experienced a complete remission and was found to be tumor-free. In contrast, in animals treated for 2 cycles, the anticancer activity of our combination setting was superior to oxaliplatin. Here, 85% of the combination-treated mice had a tumor mass of less than 0.03 g (compared to a mean mass of ∼ 2.7 g in the solvent control group), while in the oxaliplatin group these were only 43% of animals ( and ).
Also in ID8-bearing mice, the combination-treatment resulted in a stronger reduction in tumor burden ( and Suppl. Fig. 5). Thus, both a significantly (p < 0.05) reduced number of tumor lesions, as well as reduced tumor size were found in the combination-treated group as compared to the oxaliplatin-only treated mice ( and Suppl. Fig. 5C).
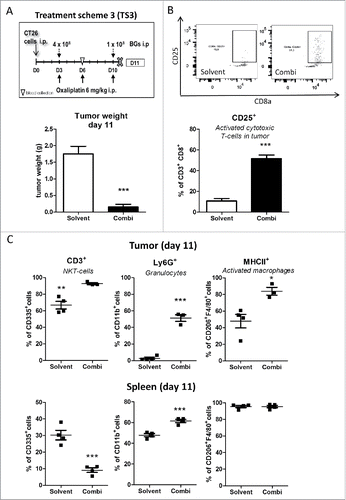
Figure 5. Impact of the combination treatment on the immune cells in tumor and spleen tissue. (A) Male BALB/c mice were injected i.p. with 1 × 105 CT26 cells on day 0 (D0) and treated according to TS3 (n = 4 per group). Animals were sacrificed on day 11 and tumor as well as spleen tissue collected. Statistical significance of the impact of combination treatment was calculated by unpaired t test (***p < 0.001). (B) Impact of treatment on tumor-associated activated cytotoxic T-cells (CD25+) was analysed by flow cytometry. In addition, representative dot plots from each group are shown. (C) Therapy-induced changes on NK-T cells (CD335+/CD3+), granulocytes (CD11b+/Ly6G+) and active macrophages (F4/80+/MHCII+) in tumor vs. spleen tissue were evaluated by multicolor flow cytometry. Each point represents one individual mouse. Statistical analysis was done using unpaired t test (*p < 0.05;**p < 0.01; ***p < 0.001).
Diffuse-light imaging tomography reveals in situ tumor response during therapy
To allow the longitudinal evaluation of the impact of our combination treatment, we stably transfected CT26 cells with firefly luciferase (CT26F-luc) and used diffuse-light imaging tomography (DLIT) combined with micro-computed tomography (µCT) to detect and quantify tumor signals in situ at several time points. In parallel, magnetic resonance imaging (MRI) measurements were used to check for formation of luminescence-silent tumor lesions (data not shown). In line with the experiments described above, the solvent-treated animals had multiple tumor lesions in the abdominal cavity on day 13 (indicated by the high intensity and spread of the photon signal, , upper row and Suppl. Fig. 6). On this day the experiment needed to be terminated due to severe ascites caused by exponentially growing CT26F-luc cells. In contrast, when treated according to TS2, tumor growth started at a later time point, with the effect in the combination group being again superior to oxaliplatin mono-treatment (, middle and lower row and Suppl. Fig. 7 and 8). The longitudinal DLIT measurements indicated that oxaliplatin treatment led to transient tumor stabilization followed by disease progression. In contrast, the addition of BGs to oxaliplatin resulted in complete response of several initially detectable tumor nodules (indicated by arrows). Luminescence quantification of the total tumor load of the individual mice is shown in Suppl. Fig. 9A. Accordingly, at the end of the experiment on day 36 (with the exception of one mouse, which died during drug application on day 6) all combination-treated mice were still alive, while the oxaliplatin-treated animals had to be sacrificed due to tumor-associated ascites on day 19, 24 and 27, respectively ( and Suppl. Fig. 7). In line with the strong antitumor effect of the combination treatment seen in our first experiments (please refer to ), in each of the two remaining combination-treated animals only one single tumor lesion was found. The tumor of mouse #7 had a superficial localization in the caudal abdomen, while the tumor of mouse #8 had infiltrated into the abdominal muscle layers, ventral to the urinary bladder and was vascularized solely via skin vessels (Suppl. Fig. 8, encircled). Consequently, both combination-treated mice had tumors (on the injection site), which probably were not accessible for the i.p. applied BGs. By use of a macromolecular contrast agent, also in this experiment distinct impact of the therapy on the spleen size was visible (Suppl. Fig. 9B and C). Thus, combination-treated mice showed a dramatic increase in spleen volume especially during the second week of treatment suggesting a strong immune cell proliferation response.
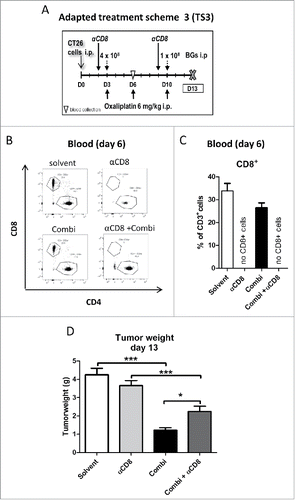
Figure 6. Effect of CD8+ cells depletion on activity of combination treatment. (A) Male BALB/c mice were injected i.p. with 1 × 105 CT26 cells on day 0 (D0) and treated according to the modified TS3 (n = 4 per group). Blood samples were taken on day 6 and mice were dissected on day 13. (B) Impact of treatment on cytotoxic T-cells (CD8+) was verified in blood samples, by flow cytometry. Representative dot plots and (C) quantification as fraction of CD8+ cells in CD3+ population are shown. (D) Tumor burden is plotted as mean ± SD. Statistical significance was calculated by unpaired t test (*p < 0.05;**p < 0.01;***p < 0.001)
As a next step, the tumor samples collected from the DLIT experiment at the respective dissection days were analyzed by H&E stainings. Noteworthy, especially the stains of the tumor samples from the combination-treated mouse #9 (which died during drug application on day 6) were characterized by the formation of areas with densely packed cells with small nuclei and little cytoplasmic content (). Subsequent immunohistochemical staining identified a fraction of these cells as CD3-positive T-cells (Suppl. Fig. 10A). An increased presence of CD3-positive cells was also found in oxaliplatin mono-treated tumors (Suppl. Fig. 10B) from the first in vivo experiment (compare ), correlating with a decreased tumor mass as compared to control. Combination-treated tumors were not available for staining, due to the strong synergistic antitumor effect.
Adjuvant combination of BGs to oxaliplatin leads to induction of a cellular immune response
To further investigate the nature of the invading immune cells after combination treatment, multicolor flow cytometry was performed. To this end, animals were treated according to TS3 and sacrificed on day 11, 12 h after the last combination treatment (, upper panel) and tumors as well as spleens, analyzed for 9 immune cell types and their activation status. In this experiment again a strong impact of the combination treatment on tumor load was seen (, lower panel). In line with the hypothesis that adjuvant BGs added to oxaliplatin treatment lead to an enhanced response and invasion of immune cells, the amount of CD45+ cells was increased in the tumor tissue. Thus, when analyzing T-cell subpopulations, a slight increase of CD8+ T-cells was detected in the combination-treated tumors (Suppl. Fig. 11). Noteworthy, these cells revealed a highly significantly increased activation (CD8+CD25+; p < 0.001; ) as compared to the solvent-treated group. In addition, also another immune cell type capable of direct tumor cell killing, namely NK-T-cells (CD335+ CD3+), was significantly (p < 0.01) upregulated upon combination treatment (). No change was seen with regard to NK cells (CD335+). In the spleen, the CD8+ T-cell subpopulation remained unchanged (Suppl. Fig. 11) and also no activation was detected (data not shown). In contrast, a remarkable and significant (p < 0.01) decrease in splenic NK-T (CD335+ CD3+) and NK (CD335+) cells was induced by drug treatment ( as well as Suppl. Fig. 11).
With regard to the myeloid compartment (CD11b+ cells), granulocytes (Ly6G+) were highly increased (p < 0.001) in the tumor and also, to a lesser extent, in the spleen (), indicating a typical systemic anti-bacterial response. Interestingly, in the tumor the percentage of F4/80+ cells was unchanged between the two groups. However, the fraction of activated (MHCII+) macrophages was significantly increased. This cell type was also evaluated by measurement of mean fluorescent intensity ( and Suppl. Fig. 12): in the spleen, although the fraction of F4/80+ MHCII+ cells was indistinguishably high in both groups (), mean fluorescence intensity quantification was increased in the combination-treated group (Suppl. Fig. 12). With regard to monocytic (Ly6 C+) cells, both tumor and spleen showed an increased percentage of MHCII+ cells, upon treatment, which may be precursors of tumor infiltrating dendritic cells or macrophages (Suppl. Fig. 12).
In addition, blood was collected from the submandibular vein on day 6. Interestingly, an increase of CD4+ T-helper cells upon treatment was observed, whereas NK (CD335+) cells were – comparable to the observations made in the spleens – reduced (Suppl. Fig. 13 A). Changes similar to the spleen were also detected for monocytes (Ly6 C+) and granulocytes (LY6G+) (Suppl. Fig. 13B). Overall, the decrease of CD8+ cells together with NK cells in the peripheral blood followed by influx of these cell populations into the malignant tissue could indicate stimulation of a specific anti-tumor immune response against CT26 cells treated with oxaliplatin and adjuvant BGs.
Application of αCD8 antibody confirms the tumor-specific activity of cytotoxic T-cells induced upon combination treatment
Based on the observed strong activation of CD8+ tumor-infiltrating cytotoxic T-lymphocytes (CTLs) after combination treatment, as a next step we investigated the impact of a CD8+-depleting antibody on the anticancer activity of our new drug combination. Therapy was started one day after the first αCD8 antibody application (). Depletion of cytotoxic CD8+ T-cells was verified by flow cytometry of blood samples collected on day 6 ( and ). Treatment with the depleting antibody resulted in significantly reduced anticancer activity of our drug combination, reflected by higher tumor weight in the combination group depleted of CD8+ cells (). This finding is in line with our hypothesis that the potent anticancer activity of our drug/adjuvant combination is mediated – at least in part – by activation of a specific CD8+ T-cells response against tumor cells.
Discussion
Until the year 2030, the burden of CRC is predicted to increase by 60% worldwide and one in two persons diagnosed with CRC will die as a consequence of this malignancy, if no new treatments will be available for the deadly late stage of the disease.Citation34 Anticancer immunotherapy with immune checkpoint inhibitors has recently revolutionized therapy of several tumor types with high mutational load including melanoma, lung and renal cancer.Citation35-37 Unfortunately, in case of CRC, responses to immune checkpoint inhibition were comparably rare and restricted to patients harboring mismatch repair deficiencies.Citation38 Peritoneal carcinomatosis, the dissemination of CRC to the peritoneum, is a major contributor to a high mortality rate in recurrent CRC.Citation2,Citation3 Since the peritoneal cavity is known to harbor a high immune competence and the activity of oxaliplatin (the most important drug against CRC) is strongly dependent on an active immune system, the combination of this drug with locally administered immunogenic adjuvants seems auspicious.Citation9,Citation26,Citation27 Here, we report that using i.p. administration of oxaliplatin combined with potent immune-stimulating BGs is an effective therapeutic approach to treat CRC peritoneal metastasis, resulting in strong responses including even cure and anti-tumor vaccination of some treated animals.
In general, the idea of exploiting the anti-bacterial immune response for anticancer therapy is a renaissance of an old concept which dates back to 1891, when William Coley, the father of immunotherapy already achieved first successes with bacteria against solid tumors of diverse origins.Citation39 Recently, several preparations of living, attenuated or genetically engineered bacteria (e.g. the streptococcal preparation OK-432 (Picibanil),Citation40 Listeria monocytogenesCitation41 or Salmonella typhimuriumCitation42) were tested for anticancer therapy in human, with Bacillus Calmette-Guérin even being used as first-line local immunotherapy of superficial bladder carcinomaCitation43,Citation44 and OK-432 being approved for lymphangioma in Japan since 1995.Citation40 The molecular basis of the interaction between bacterial and immune cells are so-called pathogen-associated molecular patterns (PAMPs), like e.g. lipopolysaccharide (LPS), lectin or triacyl lipopeptide residing on the bacterial surface.Citation45 These conserved bacterial components can be detected by the host immune cells by binding to the suited pattern recognition receptors (PRRs), of which the largest and most important is the toll-like receptor (TLR) family.Citation45 PRRs are predominantly found on macrophages and dendritic cells (DCs), but also on tumor cells, where they can activate pro-inflammatory signaling. Still, the main drawback in treatment with living bacteria is their intrinsic toxicity as well as the difficulty to control colonization intensity.Citation43 Hence, the strategy of using BGs, which are empty envelopes of non-living, Gram-negative bacteria, circumvents these drawbacks, as it exploits only the PAMPs potential and thus the external immunological properties of bacteria in a controllable fashion.Citation27-29 Examples of PAMPs found on BGs are LPS, flagellin or peptidoglycan.Citation46 On top of that, BGs safety and applicability have already been demonstrated in numerous studies.Citation28,Citation30,Citation31,Citation46 Moreover, given as an adjuvant in subcutaneous vaccination, using oncolysate as a source of tumor antigen, BGs have already proven to mobilize CTLs and prolong survival in a murine metastasizing lung carcinoma model.Citation47
When it comes to oxaliplatin, different groups have reported its outstanding capability of ICD induction.Citation18,Citation48 ICD is characterized by stressed cells releasing selected endogenous molecules as danger signals known as damage-associated molecular patterns (DAMPs).Citation49 Briefly, endoplasmatic reticulum (ER) stress is induced, which leads to relocation of the ER-resident chaperone protein CRT to the outer cell membrane (“eat me” signal). In turn, CRT binds to the low density lipoprotein receptor-related protein (LRP1; also known as CD91) on the surface of immature DCs and macrophages, essential phagocytic and antigen-presenting cells of the innate immunity.Citation50 In addition, release of ATP (“find me” signal) together with HMGB1 (“danger” signal) results in binding to their adequate PRRs (the purinergic P2RX7 and the TLR4 receptor, respectively) on the DC surfaces. This DAMP binding subsequently effects DC recruitment, maturation and activation, as well as improved (tumor) antigen presentation by DCs.Citation51-53 However, there are so far only limited reports on the anticancer combination of bacteria with systemic or local anticancer chemotherapy and their impact on the immunogenic activity of oxaliplatin is still unknown.Citation47
Interestingly, our study revealed that addition of BGs to oxaliplatin treatment elicited a distinct and synergistic increase of some of the major ICD hallmarks (CRT exposure and ATP but not HMGB1 release) already in cell culture experiments, in two different cancer models. Overall, this seems surprising as there are no DCs present to interact with BGs in this setting. Nevertheless, BGs also have the potential to provide stimulatory signals to other than antigen-presenting cells.Citation46 Therefore, one explanation for the shown effects might be the observation that colon cancer cells in general and also CT26 cells in particular themselves express TLR4.Citation54 Consequently, the LPS on the BG surface could be able to directly activate TLR4 signaling in vitro in these cells. This subsequently leads to activation of downstream-inflammatory NF-κB signaling, ER stress (CRT exposure is also an ER-stress marker) and ROS induction.Citation55,Citation56 Notably, Wang et al. described in CT26 cells, that TLR4-activation-alone would lead to tumor immune-escape in vivo.Citation54 This solitary effect seems to be diminished by our combination with oxaliplatin, as we have demonstrated in the presented animal experiments.
We hypothesize that a major advantage of our drug combination is the induction and enhanced release of tumor neoantigens by chemotherapeutic action next to the conjunction of bacterial PAMPs with DAMPs generated upon treatment with ICD-inducers like oxaliplatin.Citation48 In fact, PAMPs-associated receptors are often shared with those for DAMPs and thus these signals interact in concert with competent immune cells.Citation57 The sum of these signals is integrated by antigen-presenting cells like DCs and macrophages, resulting in their maturation, enhanced (tumor) antigen uptake, processing and finally presentation to naïve T-cells by using MHC class I and II molecules.Citation58 Remarkably, it recently has been shown, that oncolysate-loaded BGs are capable of enhancing the tumor-associated antigen delivery to DCs and induce DC maturation ex vivo. Consequently, these armed DCs were able to stimulate and activate T-cells –which are specific for the native cancer cells used for the oncolysate– in co-culture.Citation59 Similar properties were found for OK-432.Citation60 Accordingly, there is already clinical evidence that by stimulating dendritic cells (and also NK cells) via in vitro cytokine treatment, the anti-CRC activity of the FOLFOX scheme can be improved.Citation61 The mechanism of DC activation by BGs, via PAMPs, relies on direct induction of PRR signalling (e.g. TLR 4), which are generally found on antigen-presenting cells.Citation62 Noteworthy, TLR4 is also the main HMGB1-binding receptor and is known to be essential in the action of ICD-inducers and in particular of oxaliplatin, for anti-tumor T-cell response via DCs.Citation18,Citation58 Moreover, there are already claims of using TLR4-activation to enhance effectivity of ICD inducers in patients with insufficient immune cell induction after oxaliplatin monotreatment.Citation15 Our data on the adjuvant addition of BGs to oxaliplatin therapy strongly supports these hypotheses.
In accordance with the immune-stimulating activity of the various PAMPs on BG surface, we have found activation of a typical cellular response to bacteria, namely an increased number of (Ly6G+) neutrophilic granulocytes in both blood and tumors of treated animals. However, the function of this immune cell type regarding malignant growth is controversial. Liu et al. have found these cells to even act in a tumor-suppressive manner,Citation63 while Hu et al. observe them to be immunosuppressive in vitro and tumor-promoting in vivo.Citation64 Furthermore, we found that macrophages, a type of terminally differentiated myeloid cells, to carry significantly more MHCII molecules in the tumors after combination treatment. Still, this interesting finding cannot be interpreted as it stands, since this F4/80+ cell population found in all tumors was both CD206+ and MHCII+, thus giving no information about macrophage polarization. Therefore, we here are not able to clearly distinguish a tumoricidal from a tumor-promoting action and further investigations would be needed for clarification.Citation65 Albeit, the role of the macrophage population in tumor therapy is quite divisively discussed in the literature.Citation66
An increased influx of CTLs into the tumor has been repeatedly reported to correlate with longer patient survival in a broad range of tumor types.Citation67-69 Higher CTL invasion into the tumor microenvironment was found not only upon treatment with BGs (Kudela, Sramko, Lubitz, manuscript in preparation) but also with other bacterial preparations, such as OK-432 or Bacillus Calmette-Guérin.Citation70,Citation71 Also in our hands, the combination of oxaliplatin with BGs resulted in higher numbers of CTLs in the tumor microenvironment. But even more importantly, while Tel et al.Citation72 have found that oxaliplatin alone leads to a decreased proportion of activated T-cells in the tumor, we in fact observed a strong activation (CD8+CD25+) of CTLs. This is of high relevance as these CD25-carrying cells are proliferating, (tumor) specific effector CTLs, which besides direct tumor cell killing can also finally lead to generation of memory T cells and a long-lasting immunity against the specific antigens.Citation46 Indeed, we have observed that once an animal experienced a complete remission, it is also able to repel living CT26 tumor cells upon rechallenge. Moreover, depletion of the CTL population by an antibody directed against CD8+ cells was able to significantly diminish the anticancer activity of adjuvant BGs added to oxaliplatin, confirming the importance of CTLs for this synergistic action. Still, a significantly reduced tumor load was found after triple-combination treatment, as compared to αCD8 alone, while the latter did not have any impact on tumor growth. This indicates that activated CTLs are main players, but not the whole truth when it comes to the mode of action of our synergistic combination, leaving space for new hypotheses concerning other immune cell populations acting as part of the anticancer immunotherapeutic action of oxaliplatin with BGs. The only other immune cell type capable of direct tumor cell killing, namely NK cells, was decreased after combination treatment in the blood and, in the spleen. In contrast to the ability of OK-432 to induce NK cell influx in a hepatocellular carcinoma rat model on day 3,Citation73 we found this population to be unchanged in the tumor microenvironment on day 11. Thus, as part of the early, first-line defense innate immunity, NK cells may have already carried out their anti-tumor effect, but further studies at an earlier time-point would be necessary to clarify this speculation. Yet, in tumors of our combination-treated animals the NKT cell fraction –a type of lymphocytes thought to be important in bridging innate and adaptive immunity– was significantly increased. Some clinical studies correlated a higher NKT-cell frequency with improved cancer patient prognosis.Citation74,Citation75
Taken together, we demonstrate that the combination of adjuvant BGs with oxaliplatin is a promising new immunochemotherapeutic strategy against colorectal carcinomatosis in vivo, based on a potent activation of an antitumor T-cell response.
Materials and methods
Chemicals
Oxaliplatin was purchased from Selleck Chemicals (#S1224). All other reagents were purchased from Sigma-Aldrich.
Production of BGs
BGs from E. coli Nissle 1917 were produced by controlled expression of the phage-derived lysis protein E as described previously.Citation76 For the experiments, the lyophilized BGs were suspended in sterile 5% glucose at a concentration of 6 × 109 BGs/ml.
Cells and culturing conditions
CT26 murine colon carcinoma cells (CRL-2638, purchased from ATCC) were cultured in Dulbecco's modified eagle's medium (DMEM)/F12 medium (1:1 from Sigma; #D6421) supplemented with 10% heat-inactivated fetal calf serum (FCS). The ID8 murine ovarian carcinoma cells (a kind gift of Prof. Kathy Roby, University of Kansas)Citation77 were grown in DMEM supplemented with 4% FCS, insulin (5µg/ml), transferrin (5 µg/ml), and sodium selenite (5 ng/ml). Cells were maintained at 37.0°C in a 5 % carbon dioxide-humidified atmosphere.
Immunofluorescent CRT staining
CT26 or ID8 cells were seeded (4 × 103 cells per spot) on Spot slides (Thermo Fisher Scientific; #101432648EPOXY) and allowed to recover for 24 h. Then they were treated for the indicated times with oxaliplatin (100 or 300 µM) and/or BGs (500 BGs/cell) and, after a washing step in phosphate-buffered saline (PBS), fixed with 4% formaldehyde. Blocking was done using 20% FBS in PBS for 1 h at room temperature. The cells were incubated with the primary CRT antibody (1:200, Cell Signaling Technology; #12238) for 1 h at 4°C. After washing, the cells were incubated with a solution containing the secondary antibody (FITC-conjugated anti-rabbit, 1:200, Sigma; #F9887), DAPI (2.5 µg/ml; #D9542) and WGA (1:500, Vector Laboratories; #R1022) for 1 h at room temperature. Subsequently, confocal microscopy was performed on a Zeiss LSM 700 (Carl Zeiss AG). Quantification of the CRT exposure was done with Fiji/Image J software by evaluation of at least 50 cells per sample.
ATP and HMGB1 release assay
CT26 (5 × 104 cells per well) or ID8 (1 × 105 cells per well) were plated in 24-well plates and left to recover for at least 24 h. Treatment was performed as indicated and all supernatants collected. Dying tumor cells, BGs or cell debris were removed by centrifugation (8 min, 100 g). ATP levels in the supernatants were measured using the Cell Titer Glo kit (Promega; #G9681) and quantified using an ATP (Sigma Aldrich, #A3377) standard curve. Quantification of released HMGB1 was assessed by enzyme-linked immunosorbent assay according to the manufacturer's instructions. (Cloud-Clone Corp, #SEA399Mu)
Animals
Six- to eight-week-old BALB/c mice and C57BL/6J were purchased from Janvier (imaging), Envigo and Jackson Laboratories, respectively. The animals were kept in a pathogen-free environment and every procedure was done in a laminar airflow cabinet. Experiments were done according to the regulations of the Ethics Committee for the Care and Use of Laboratory Animals at the Medical University Vienna (proposal number BMWF-66.009/0081-WF/V/3b/2015), the U.S. Public Health Service Policy on Human Care and Use of Laboratory Animals as well as the United Kingdom Coordinating Committee on Cancer Prevention Research's Guidelines for the Welfare of Animals in Experimental Neoplasia. To ensure animal welfare throughout the experiment, the body weight of the mice was assessed once a day. At weight loss exceeding 10 % (in less than two days) or occurrence of ascites, animals were sacrificed by cervical dislocation.
Carcinomatosis allograft models
CT26 or ID8 cells were harvested and re-suspended in serum-free cell culture medium. 1 × 105 CT26 cells and 5 × 106 ID8 cells, were applied i.p. to BALB/c or C57 BL/6 J mice, respectively. Treatment started on day 3 with the indicated schemes. Oxaliplatin was applied i.p. at a dose of 6 mg/kg (dissolved in 5% glucose), while BGs were given in doses, depending on treatment scheme and day (4 × 108 – 1 × 108 per mouse, suspended in 5% glucose, i.p.). Animals were sacrificed on the indicated days per cervical dislocation and tumors as well as organs collected.
Histology
Tumors and organs were fixed in 4% formaldehyde for 24 h (Carl Roth, # P087.3) and paraffin-embedded using a KOS machine (Milestone). For histological evaluation, tumor tissues were sliced in 4 µm thick sections and hematoxylin/eosin stained by routine procedures. For the C57BL/6J experiment, tumor burden was evaluated by Panoramic Viewer Software (3D Histech). Lesions consisting of >20 cells were considered tumors, marked using free hand annotation and each area automatically measured using the annotation managing tool.
CT26 transfection with F-Luciferase
CT26F-luc cells were generated by retroviral transduction in CT26 cells. Briefly, firefly luciferase cDNA was cloned from plasmid pGL3 (pGL3 Luciferase Reporter Vectors, Promega; #E1751) into the retroviral expression vector pQCXIN (pQCXIN Retroviral Vector, Clontech; #631514). Production of viral particles was performed in HEK293 cells as described in ref.Citation78. CT26 cells were transduced with the viral particles in 6-well plates and stable integrants were selected with 500 µg/ml G418.
DLI/TµCT
To follow exact tumor growth, non-invasive DLIT combined with µCTCitation79 was performed using an IVIS Spectrum CT imaging system (IVIS Spectrum CT preclinical imaging system, PerkinElmer Inc.) as previously described.Citation80 Briefly, mice were anaesthetized with 5 % of isoflurane (Isoflurane CP 1 ml/ml, CP-Pharma; #1214). Subsequently, D-luciferin bioluminescent substrate (D-Luciferin potassium salt, 120 mg/kg in PBS, Intrace Medical SA) was injected s.c.. DLIT was performed within 30 min post injection to guarantee a steady light emission. For evaluation of the gained data Living Image software was used. In order to enhance spleen and liver contrast ExiTron nano 12000 (Miltenyi Biotec; #130-095-698) was applied i.v. as described.Citation81 In addition, an iodine-based contrast agent was used to visualize the gastro-intestinal tract (Gastrografin, Bayer Vital GmbH; #00268169). Here, a solution in sterile water was provided ad libitum overnight prior to each imaging time point.Citation82 Spleen volume was calculated with Amira Software (Mercury Computer Systems).
Flow cytometric analysis of tumor, spleen and blood cell populations
Dissected tumors and spleens were digested, strained to give a single cell suspension and auto-fluorescent red blood cells were removed. In more detail, digestion was effected using 1 mg/ml Collagenase VIII and DNAse I, at 37°C for 15 (spleen) and 25 (tumor) minutes, respectively. The cells were then first blocked using Fc-Antibody Solution 1 ng/µl (Biolegend, #101321) and then stained for 30 min at room temperature with two different antibody panels purchased from Biolegend. T- and B-cell marker-specific antibodies: CD45 #103116, CD3e #100306, CD4 #100422, CD8a #100738, CD19 #115528 and CD335 (NKp46) #137610). The gating strategy used for this panel was: live cells/single cells FSC/ single cells SSC/CD45+ overall for hematopoetic cells and from that population CD3e+ for T-cells, CD3e+/CD4+ for T-helper cells, CD3e+/CD8 a+ for cytotoxic T-cells, CD19+ for B-cells, CD335+ for NK cells and CD335+/CD3e+ for NK T-cells. CD25 was stained as an activation marker for T-cells. Macrophage and monocyte specific markers: CD45 #103116, CD11b #101216, CD11c #117312, Ly-6 C #128024, Ly-6G #127626, I-A/I-E #107620, F4/80 #123110 and CD206 (MMR) #141732). The gating strategy for the myeloid panel was: live cells/single cells FSC/ single cells SSC/CD45+/CD11b+ overall for hematopoetic cells and from that population CD11b+ for all myeloid cells, CD11b+/Ly6CloLy6G+ for neutrophilic granulocytes, CD11b+/Ly6G−LyC+ for monocytes, CD11c+/CD206+F4/80+ for macrophages and MHCII as a marker of activation for these populations. Blood samples were taken from the submandibular vein of mice during treatment on the indicated days (by using heparin-coated tubes to prevent coagulation), as an alternative to terminal bleeding. Red blood cells were lysed from the whole blood samples and subsequently stained with the same two marker panels as the tumor and spleen samples. All samples were analyzed on an LSR Fortessa (BD Biosciences) and analyzed using Flowjo (Treestar).
In vivo depletion of CD8+ cells
Cytotoxic T-cell depletion was achieved by weekly i.p. administration of 300 µl of a 2 mg/ml αCD8a antibody (clone 53.6.7, rat [Lou/Ws1/M] IgG2aK) solution, isolated and purified from hybridoma supernatants (as described in Drobits et al.Citation83). One day after the first αCD8a antibody application, therapy with the combination treatment scheme started as indicated. To confirm depletion of CD8a+ T-cells blood samples were taken from the submandibular vein on day 6 and 13 and analyzed by multicolor flow cytometry as described above. In addition, tumor samples were analyzed for immune cells on day 13.
Statistics
The imaging data was statistically analyzed by using SPSS Statistics software (IBM) and a paired Student's t-test (P-value = 0.05). All the other data were analyzed using GraphPad Prism software using the adequate statistical tests and p-values, as indicated in the figure legends.
Abbreviation
| µCT | = | micro-computed tomography |
| BG | = | bacterial ghosts |
| CRT | = | calreticulin |
| CRC | = | colorectal cancer |
| CTL | = | cytotoxic (CD8+) T-lymphocyte |
| DAMP | = | damage-associated molecular pattern |
| DC | = | dendritic cell |
| DLIT | = | diffuse light imaging tomography |
| ER | = | endoplasmatic reticulum |
| HIPEC | = | hyperthermic intraperitoneal chemotherapy |
| HMGB1 | = | high mobility group box 1 |
| ICD | = | immunogenic cell-death |
| LOD | = | limit of detection |
| MRI | = | magnetic resonance imaging |
| OS | = | overall survival |
| PAMP | = | pathogen-associated molecular pattern |
| PRR | = | pattern recognition receptor |
| TLR | = | toll-like receptor |
| TS | = | treatment scheme |
Groza_et_al._Supplementary_Figures_proof_read.pptx
Download MS Power Point (13.3 MB)2017ONCOIMM0988-file004.docx
Download MS Word (12.3 KB)Acknowledgments
BGs were kindly provided by BIRD-c. We thank Gerhard Zeitler for devoted animal care, Alexandra Bogusch for isolation and purification of the αCD8a antibody and Gerald Timelthaler for help with the digital tissue analysis.
Disclosure statement
Werner Lubitz is the founder and CEO of BIRD-C GmbH & CoKG Vienna, Austria, which has licensed the rights to the Bacterial Ghosts Technology, inventor and proprietor of Patent P2591798B1 (Vaccine for use in tumor immunotherapy). Pavol Kudela and Marek Sramko are employees of BIRD-C GmbH, Vienna, Austria. Pavol Kudela is also inventor of Patent EP2591798B1 (Vaccine for use in tumor immunotherapy). All other authors declare no conflict of interests.
Additional information
Funding
References
- Strohlein MA, Heiss MM, Jauch KW. The current status of immunotherapy in peritoneal carcinomatosis. Expert Rev Anticancer Ther. 2016;16:1019–27. doi:10.1080/14737140.2016.1224666. PMID:27530056.
- Lemoine L, Sugarbaker P, Van der Speeten K. Pathophysiology of colorectal peritoneal carcinomatosis: Role of the peritoneum. World J Gastroenterol. 2016;22:7692–707. doi:10.3748/wjg.v22.i34.7692. PMID:27678351.
- Sugarbaker PH. Improving oncologic outcomes for colorectal cancer at high risk for local-regional recurrence with novel surgical techniques. Expert Rev Gastroenterol Hepatol. 2016;10:205–13. doi:10.1586/17474124.2016.1110019. PMID:26643935.
- Cao C, Yan TD, Black D, Morris DL. A systematic review and meta-analysis of cytoreductive surgery with perioperative intraperitoneal chemotherapy for peritoneal carcinomatosis of colorectal origin. Annals Surg Oncol. 2009;16:2152–65. doi:10.1245/s10434-009-0487-4.
- Morano WF, Aggarwal A, Love P, Richard SD, Esquivel J, Bowne WB. Intraperitoneal immunotherapy: historical perspectives and modern therapy. Cancer Gene Therapy. 2016;23:373–81. doi:10.1038/cgt.2016.49. PMID:27834358.
- Gonzalez-Moreno S, Gonzalez-Bayon LA, Ortega-Perez G. Hyperthermic intraperitoneal chemotherapy: Rationale and technique. World J Gastrointestinal Oncol. 2010;2:68–75. doi:10.4251/wjgo.v2.i2.68.
- Perrin P, Cassagnau E, Burg C, Patry Y, Vavasseur F, Harb J, Le Pendu J, Douillard JY, Galmiche JP, Bornet F, et al. An interleukin 2/sodium butyrate combination as immunotherapy for rat colon cancer peritoneal carcinomatosis. Gastroenterology. 1994;107:1697–708. doi:10.1016/0016-5085(94)90810-9. PMID:7958681.
- Katz SC, Point GR, Cunetta M, Thorn M, Guha P, Espat NJ, Boutros C, Hanna N, Junghans RP. Regional CAR-T cell infusions for peritoneal carcinomatosis are superior to systemic delivery. Cancer Gene Therapy. 2016;23:142–8. doi:10.1038/cgt.2016.14. PMID:27080226.
- Graham J, Mushin M, Kirkpatrick P. Oxaliplatin. Nat Rev Drug Discov. 2004;3:11–2. doi:10.1038/nrd1287. PMID:14756144.
- Heffeter P, Jungwirth U, Jakupec M, Hartinger C, Galanski M, Elbling L, Micksche M, Keppler B, Berger W. Resistance against novel anticancer metal compounds: differences and similarities. Drug Resist Updat. 2008;11:1–16. doi:10.1016/j.drup.2008.02.002. PMID:18394950.
- Di Francesco AM, Ruggiero A, Riccardi R. Cellular and molecular aspects of drugs of the future: oxaliplatin. Cell Mol Life Sci. 2002;59:1914–27. doi:10.1007/PL00012514. PMID:12530522.
- Raymond E, Faivre S, Woynarowski JM, Chaney SG. Oxaliplatin: mechanism of action and antineoplastic activity. Seminars Oncol. 1998;25:4–12.
- Raymond E, Faivre S, Chaney S, Woynarowski J, Cvitkovic E. Cellular and molecular pharmacology of oxaliplatin. Mol Cancer Ther. 2002;1:227–35. PMID:12467217.
- Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, Mignot G, Maiuri MC, Ullrich E, Saulnier P, et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. 2007;13:1050–9. doi:10.1038/nm1622. PMID:17704786.
- Galluzzi L, Senovilla L, Zitvogel L, Kroemer G. The secret ally: immunostimulation by anticancer drugs. Nat Rev Drug Discov. 2012;11:215–33. doi:10.1038/nrd3626. PMID:22301798.
- Jungwirth U, Xanthos DN, Gojo J, Bytzek AK, Korner W, Heffeter P, Abramkin SA, Jakupec MA, Hartinger CG, Windberger U, et al. Anticancer activity of methyl-substituted oxaliplatin analogs. Mol Pharmacol. 2012;81:719–28. doi:10.1124/mol.111.077321. PMID:22331606.
- Terenzi A, Pirker C, Keppler BK, Berger W. Anticancer metal drugs and immunogenic cell death. J Inorg Biochem. 2016;165:71–9. doi:10.1016/j.jinorgbio.2016.06.021. PMID:27350082.
- Tesniere A, Schlemmer F, Boige V, Kepp O, Martins I, Ghiringhelli F, Aymeric L, Michaud M, Apetoh L, Barault L, et al. Immunogenic death of colon cancer cells treated with oxaliplatin. Oncogene. 2010;29:482–91. doi:10.1038/onc.2009.356. PMID:19881547.
- Liu WM, Fowler DW, Smith P, Dalgleish AG. Pre-treatment with chemotherapy can enhance the antigenicity and immunogenicity of tumours by promoting adaptive immune responses. Br J Cancer. 2010;102:115–23. doi:10.1038/sj.bjc.6605465. PMID:19997099.
- Martins I, Kepp O, Schlemmer F, Adjemian S, Tailler M, Shen S, Michaud M, Menger L, Gdoura A, Tajeddine N, et al. Restoration of the immunogenicity of cisplatin-induced cancer cell death by endoplasmic reticulum stress. Oncogene. 2011;30:1147–58. doi:10.1038/onc.2010.500. PMID:21151176.
- Lesterhuis WJ, Punt CJ, Hato SV, Eleveld-Trancikova D, Jansen BJ, Nierkens S, Schreibelt G, de Boer A, Van Herpen CM, Kaanders JH, et al. Platinum-based drugs disrupt STAT6-mediated suppression of immune responses against cancer in humans and mice. J Clin Invest. 2011;121:3100–8. doi:10.1172/JCI43656. PMID:21765211.
- Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Ann Rev Immunol. 2013;31:51–72. doi:10.1146/annurev-immunol-032712-100008.
- Iida N, Dzutsev A, Stewart CA, Smith L, Bouladoux N, Weingarten RA, Molina DA, Salcedo R, Back T, Cramer S, et al. Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science. 2013;342:967–70. doi:10.1126/science.1240527. PMID:24264989.
- Viaud S, Saccheri F, Mignot G, Yamazaki T, Daillere R, Hannani D, Enot DP, Pfirschke C, Engblom C, Pittet M, et al. The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science. 2013;342:971–6. doi:10.1126/science.1240537. PMID:24264990.
- McCully ML, Madrenas J. Dendritic cells as arbiters of peritoneal immune responses. Perit Dial Int. 2006;26:8–25. PMID:16538869.
- Melichar B, Freedman RS. Immunology of the peritoneal cavity: relevance for host-tumor relation. Int J Gynecol Cancer. 2002;12:3–17. doi:10.1046/j.1525-1438.2002.01093.x. PMID:11860531.
- Witte A, Wanner G, Sulzner M, Lubitz W. Dynamics of PhiX174 protein E-mediated lysis of Escherichia coli. Arch Microbiol. 1992;157:381–8. doi:10.1007/BF00248685. PMID:1534215.
- Paukner S, Stiedl T, Kudela P, Bizik J, Al Laham F, Lubitz W. Bacterial ghosts as a novel advanced targeting system for drug and DNA delivery. Expert Opinion Drug Delivery. 2006;3:11–22. doi:10.1517/17425247.3.1.11.
- Kudela P, Koller VJ, Lubitz W. Bacterial ghosts (BGs)-Advanced antigen and drug delivery system. Vaccine. 2010;28:5760–7. doi:10.1016/j.vaccine.2010.06.087. PMID:20619379.
- Paukner S, Kohl G, Lubitz W. Bacterial ghosts as novel advanced drug delivery systems: antiproliferative activity of loaded doxorubicin in human Caco-2 cells. J Controlled Release Official J Controlled Release Soc. 2004;94:63–74. doi:10.1016/j.jconrel.2003.09.010.
- Stein E, Inic-Kanada A, Belij S, Montanaro J, Bintner N, Schlacher S, et al. In vitro and in vivo uptake study of Escherichia coli Nissle 1917 bacterial ghosts: cell-based delivery system to target ocular surface diseases. Invest Ophthalmol Visual Sci. 2013;54:6326–33. doi:10.1167/iovs.13-12044.
- Bronte V, Pittet MJ. The spleen in local and systemic regulation of immunity. Immunity. 2013;39:806–18. doi:10.1016/j.immuni.2013.10.010. PMID:24238338.
- Cesta MF. Normal structure, function, and histology of the spleen. Toxicol Pathol. 2006;34:455–65. doi:10.1080/01926230600867743. PMID:17067939.
- Arnold M, Sierra MS, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global patterns and trends in colorectal cancer incidence and mortality. Gut. 2017;66:683–91. doi:10.1136/gutjnl-2015-310912. PMID:26818619.
- Axelrod ML, Johnson DB, Balko JM. Emerging biomarkers for cancer immunotherapy in melanoma. Semin Cancer Biol. 2017. doi:10.1016/j.semcancer.2017.09.004. PMID:28917578.
- Marrone KA, Brahmer JR. Using immune checkpoint inhibitors in lung cancer. Oncology (Williston Park). 2016;30:713–21. PMID:27528240.
- Tsiatas M, Grivas P. Immunobiology and immunotherapy in genitourinary malignancies. Ann Transl Med. 2016;4:270. doi:10.21037/atm.2016.06.29. PMID:27563657.
- Boland PM, Ma WW. Immunotherapy for Colorectal Cancer. Cancers (Basel). 2017;9:E50. doi:10.3390/cancers9050050. PMID:28492495.
- Coley WB. Late results of the treatment of inoperable sarcoma by the mixed toxins of erysipelas and Bacillus prodigiosus. Am J Med Sci. 1906;131:375–430.
- Oba MS, Teramukai S, Ohashi Y, Ogawa K, Maehara Y, Sakamoto J. The efficacy of adjuvant immunochemotherapy with OK-432 after curative resection of gastric cancer: an individual patient data meta-analysis of randomized controlled trials. Gastric Cancer. 2016;19:616–24. doi:10.1007/s10120-015-0489-9. PMID:25804300.
- Singh R, Wallecha A. Cancer immunotherapy using recombinant Listeria monocytogenes: transition from bench to clinic. Hum Vaccin. 2011;7:497–505. doi:10.4161/hv.7.5.15132. PMID:21422819.
- Toso JF, Gill VJ, Hwu P, Marincola FM, Restifo NP, Schwartzentruber DJ, Sherry RM, Topalian SL, Yang JC, Stock F, et al. Phase I study of the intravenous administration of attenuated Salmonella typhimurium to patients with metastatic melanoma. J Clin Oncol. 2002;20:142–52. doi:10.1200/JCO.2002.20.1.142. PMID:11773163.
- Dzutsev A, Badger JH, Perez-Chanona E, Roy S, Salcedo R, Smith CK, Trinchieri G. Microbes and cancer. Annu Rev Immunol. 2017;35:199–228. doi:10.1146/annurev-immunol-051116-052133. PMID:28142322.
- Fuge O, Vasdev N, Allchorne P, Green JS. Immunotherapy for bladder cancer. Res Rep Urol. 2015;7:65–79. PMID:26000263.
- Tang D, Kang R, Coyne CB, Zeh HJ, Lotze MT. PAMPs and DAMPs: signal 0 s that spur autophagy and immunity. Immunol Rev. 2012;249:158–75. doi:10.1111/j.1600-065X.2012.01146.x. PMID:22889221.
- Muhammad A, Champeimont J, Mayr UB, Lubitz W, Kudela P. Bacterial ghosts as carriers of protein subunit and DNA-encoded antigens for vaccine applications. Expert Rev Vaccines. 2012;11:97–116. doi:10.1586/erv.11.149. PMID:22149712.
- Krasko JA, Zilionyte K, Darinskas A, Strioga M, Rjabceva S, Zalutsky I, Derevyanko M, Kulchitsky V, Lubitz W, Kudela P, et al. Bacterial ghosts as adjuvants in syngeneic tumour cell lysate-based anticancer vaccination in a murine lung carcinoma model. Oncol Rep. 2017;37:171–8. doi:10.3892/or.2016.5252. PMID:27878261.
- Hato SV, Khong A, de Vries IJ, Lesterhuis WJ. Molecular pathways: the immunogenic effects of platinum-based chemotherapeutics. Clin Cancer Res. 2014;20:2831–7. doi:10.1158/1078-0432.CCR-13-3141. PMID:24879823.
- Garg AD, Nowis D, Golab J, Vandenabeele P, Krysko DV, Agostinis P. Immunogenic cell death, DAMPs and anticancer therapeutics: an emerging amalgamation. Biochim Biophys Acta. 2010;1805:53–71. PMID:19720113.
- Gardai SJ, McPhillips KA, Frasch SC, Janssen WJ, Starefeldt A, Murphy-Ullrich JE, Bratton DL, Oldenborg PA, Michalak M, Henson PM. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell. 2005;123:321–34. doi:10.1016/j.cell.2005.08.032. PMID:16239148.
- Michaud M, Martins I, Sukkurwala AQ, Adjemian S, Ma Y, Pellegatti P, Shen S, Kepp O, Scoazec M, Mignot G, et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science. 2011;334:1573–7. doi:10.1126/science.1208347. PMID:22174255.
- Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol. 2017;17:97–111. doi:10.1038/nri.2016.107. PMID:27748397.
- Sims GP, Rowe DC, Rietdijk ST, Herbst R, Coyle AJ. HMGB1 and RAGE in inflammation and cancer. Annual Rev Immunol. 2010;28:367–88. doi:10.1146/annurev.immunol.021908.132603.
- Wang L, Liu Q, Sun Q, Zhang C, Chen T, Cao X. TLR4 signaling in cancer cells promotes chemoattraction of immature dendritic cells via autocrine CCL20. Biochem Biophys Res Commun. 2008;366:852–6. doi:10.1016/j.bbrc.2007.12.030. PMID:18083111.
- O'Leary DP, Bhatt L, Woolley JF, Gough DR, Wang JH, Cotter TG, Redmond HP. TLR-4 signalling accelerates colon cancer cell adhesion via NF-kappaB mediated transcriptional up-regulation of Nox-1. PLoS One. 2012;7:e44176. doi:10.1371/journal.pone.0044176. PMID:23071493.
- Akira S, Hemmi H. Recognition of pathogen-associated molecular patterns by TLR family. Immunol Lett. 2003;85:85–95. doi:10.1016/S0165-2478(02)00228-6. PMID:12527213.
- Lotze MT, Zeh HJ, Rubartelli A, Sparvero LJ, Amoscato AA, Washburn NR, Devera ME, Liang X, Tör M, Billiar T. The grateful dead: damage-associated molecular pattern molecules and reduction/oxidation regulate immunity. Immunol Rev. 2007;220:60–81. doi:10.1111/j.1600-065X.2007.00579.x. PMID:17979840.
- Fang H, Ang B, Xu X, Huang X, Wu Y, Sun Y, Wang W, Li N, Cao X, Wan T. TLR4 is essential for dendritic cell activation and anti-tumor T-cell response enhancement by DAMPs released from chemically stressed cancer cells. Cell Mol Immunol. 2014;11:150–9. doi:10.1038/cmi.2013.59. PMID:24362470.
- Michalek J, Hezova R, Turanek-Knotigova P, Gabkova J, Strioga M, Lubitz W, Kudela P. Oncolysate-loaded Escherichia coli bacterial ghosts enhance the stimulatory capacity of human dendritic cells. Cancer Immunol Immunother. 2017;66:149–59. doi:10.1007/s00262-016-1932-4. PMID:27864613.
- Nakahara S, Tsunoda T, Baba T, Asabe S, Tahara H. Dendritic cells stimulated with a bacterial product, OK-432, efficiently induce cytotoxic T lymphocytes specific to tumor rejection peptide. Cancer Res. 2003;63:4112–8. PMID:12874015.
- Liu Y, Zheng Z, Zhang Q, Zhou X, Feng Y, Yan A. FOLFOX regimen plus dendritic cells-cytokine-induced killer cells immunotherapy for the treatment of colorectal cancer: a meta-analysis. Onco Targets Ther. 2017;10:2621–33. doi:10.2147/OTT.S138011. PMID:28572734.
- Schijns V, Tartour E, Michalek J, Stathopoulos A, Dobrovolskiene NT, Strioga MM. Immune adjuvants as critical guides directing immunity triggered by therapeutic cancer vaccines. Cytotherapy. 2014;16:427–39. doi:10.1016/j.jcyt.2013.09.008. PMID:24280238.
- Liu Y, O'Leary CE, Wang LS, Bhatti TR, Dai N, Kapoor V, Liu P, Mei J, Guo L, Oliver P, et al. CD11b+Ly6G+ cells inhibit tumor growth by suppressing IL-17 production at early stages of tumorigenesis. Oncoimmunology. 2016;5:e1061175. doi:10.1080/2162402X.2015.1061175. PMID:26942073.
- Hu X, Zhou Y, Dong K, Sun Z, Zhao D, Wang W, Yu G, Liu W, Xu G, Han Z, et al. Programming of the development of tumor-promoting neutrophils by mesenchymal stromal cells. Cell Physiol Biochem. 2014;33:1802–14. doi:10.1159/000362959. PMID:24923759.
- Mantovani A, Sica A, Allavena P, Garlanda C, Locati M. Tumor-associated macrophages and the related myeloid-derived suppressor cells as a paradigm of the diversity of macrophage activation. Hum Immunol. 2009;70:325–30. doi:10.1016/j.humimm.2009.02.008. PMID:19236898.
- Takeya M, Komohara Y. Role of tumor-associated macrophages in human malignancies: friend or foe? Pathol Int. 2016;66:491–505. doi:10.1111/pin.12440. PMID:27444136.
- Nguyen N, Bellile E, Thomas D, McHugh J, Rozek L, Virani S, Peterson L, Carey TE, Walline H, Moyer J, et al. Tumor infiltrating lymphocytes and survival in patients with head and neck squamous cell carcinoma. Head Neck. 2016;38:1074–84. doi:10.1002/hed.24406. PMID:26879675.
- Deschoolmeester V, Baay M, Van Marck E, Weyler J, Vermeulen P, Lardon F, Vermorken JB. Tumor infiltrating lymphocytes: an intriguing player in the survival of colorectal cancer patients. BMC Immunol. 2010;11:19. doi:10.1186/1471-2172-11-19. PMID:20385003.
- Barnes TA, Amir E. HYPE or HOPE: the prognostic value of infiltrating immune cells in cancer. Br J Cancer. 2017;117:451–60. doi:10.1038/bjc.2017.220. PMID:28704840.
- Li L, Wang W, Pan H, Ma G, Shi X, Xie H, Liu X, Ding Q, Zhou W, Wang S. Microwave ablation combined with OK-432 induces Th1-type response and specific antitumor immunity in a murine model of breast cancer. J Transl Med. 2017;15:23. doi:10.1186/s12967-017-1124-9. PMID:28137271.
- Ratliff TL, Gillen D, Catalona WJ. Requirement of a thymus dependent immune response for BCG-mediated antitumor activity. J Urol. 1987;137:155–8. doi:10.1016/S0022-5347(17)43909-7. PMID:3491909.
- Tel J, Hato SV, Torensma R, Buschow SI, Figdor CG, Lesterhuis WJ, de Vries IJ. The chemotherapeutic drug oxaliplatin differentially affects blood DC function dependent on environmental cues. Cancer Immunol Immunother. 2012;61:1101–11. doi:10.1007/s00262-011-1189-x. PMID:22193989.
- Uehara K, Ichida T, Sugahara S, Ishikawa T, Yamagiwa S, Yoshida Y, Nomoto M, Katoh M, Satoh H, Watanabe H, et al. Systemic administration of liposome-encapsulated OK-432 prolongs the survival of rats with hepatocellular carcinoma through the induction of IFN-gamma-producing hepatic lymphocytes. J Gastroenterol Hepatol. 2002;17:81–90. doi:10.1046/j.1440-1746.2002.02675.x. PMID:11895558.
- Berzins SP, Ritchie DS. Natural killer T cells: drivers or passengers in preventing human disease? Nat Rev Immunol. 2014;14:640–6. doi:10.1038/nri3725. PMID:25103356.
- Bricard G, Cesson V, Devevre E, Bouzourene H, Barbey C, Rufer N, Im JS, Alves PM, Martinet O, Halkic N, et al. Enrichment of human CD4+ V(alpha)24/Vbeta11 invariant NKT cells in intrahepatic malignant tumors. J Immunol. 2009;182:5140–51. doi:10.4049/jimmunol.0711086. PMID:19342695.
- Langemann T, Koller VJ, Muhammad A, Kudela P, Mayr UB, Lubitz W. The Bacterial Ghost platform system: production and applications. Bioengineered Bugs. 2010;1:326–36. doi:10.4161/bbug.1.5.12540. PMID:21326832.
- Roby KF, Taylor CC, Sweetwood JP, Cheng Y, Pace JL, Tawfik O, Persons DL, Smith PG, Terranova PF. Development of a syngeneic mouse model for events related to ovarian cancer. Carcinogenesis. 2000;21:585–91. doi:10.1093/carcin/21.4.585. PMID:10753190.
- Grusch M, Schelch K, Riedler R, Reichhart E, Differ C, Berger W, Inglés-Prieto Á, Janovjak H. Spatio-temporally precise activation of engineered receptor tyrosine kinases by light. EMBO J. 2014;33:1713–26. doi:10.15252/embj.201387695. PMID:24986882.
- Kuo C, Coquoz O, Troy TL, Xu H, Rice BW. Three-dimensional reconstruction of in vivo bioluminescent sources based on multispectral imaging. J Biomed Opt 2007; 12:024007. doi:10.1117/1.2717898. PMID:17477722.
- Geyer A, Taschauer A, Alioglu F, Anton M, Maier J, Drothler E, Simlinger M, Yavuz S, Sami H, Ogris M. Multimodal Fluorescence and Bioluminescence Imaging Reveals Transfection Potential of Intratracheally Administered Polyplexes for Breast Cancer Lung Metastases. Hum Gene Ther. 2017;28(12):1202–1213. doi:10.1089/hum.2017.137. PMID:28874076.
- Boll H, Figueiredo G, Fiebig T, Nittka S, Doyon F, Kerl HU, Nölte I, Förster A, Kramer M, Brockmann MA. Comparison of Fenestra LC, ExiTron nano 6000, and ExiTron nano 12000 for micro-CT imaging of liver and spleen in mice. Academic Radiol. 2013;20:1137–43. doi:10.1016/j.acra.2013.06.002.
- Rampurwala M, Ravoori MK, Wei W, Johnson VE, Vikram R, Kundra V. Visualization and quantification of intraperitoneal tumors by in vivo computed tomography using negative contrast enhancement strategy in a mouse model of ovarian cancer. Translational Oncol. 2009;2:96–106. doi:10.1593/tlo.08199.
- Drobits B, Holcmann M, Amberg N, Swiecki M, Grundtner R, Hammer M, Colonna M, Sibilia M. Imiquimod clears tumors in mice independent of adaptive immunity by converting pDCs into tumor-killing effector cells. J Clin Invest. 2012;122:575–85. doi:10.1172/JCI61034. PMID:22251703.