
ABSTRACT
A DNA vaccine, SCIB1, incorporating two CD8 and two CD4 epitopes from TRP-2/gp100 was evaluated in patients with metastatic melanoma. Each patient received SCIB1 via intramuscular injection with electroporation. The trial was designed to find the safest dose of SCIB1 which induced immune/clinical responses in patients with or without tumour. Fifteen patients with tumor received SCIB1 doses of 0.4-8 mg whilst 20 fully-resected patients received 2–8 mg doses. Twelve patients elected to continue immunization every 3 months for up to 39 months. SCIB1 induced dose-dependent T cell responses in 88% of patients with no serious adverse effects or dose limiting toxicities. The intensity of the T cell responses was significantly higher in patients receiving 4 mg doses without tumor when compared to those with tumor (p < 0.01). In contrast, patients with tumor showed a significantly higher response to the 8 mg dose than the 4 mg dose (p < 0.03) but there was no significant difference in the patients without tumor. One of 15 patients with measurable disease showed an objective tumor response and 7/15 showed stable disease. 5/20 fully-resected patients have experienced disease recurrence but all remained alive at the cut-off date with a median observation time of 37 months. A positive clinical outcome was associated with MHC-I and MHC-II expression on tumors prior to therapy (p = 0.027).
We conclude that SCIB1 is well tolerated and stimulates potent T cell responses in melanoma patients. It deserves further evaluation as a single agent adjuvant therapy or in combination with checkpoint inhibitors in advanced disease.
Keywords:
Introduction
Checkpoint blockade has demonstrated anti-tumor responses in approximately 10–55% of melanoma patients.Citation1–4 However, many patients do not respondCitation5,Citation6 and may benefit from an effective vaccine that stimulates high avidity T cell responses either as a single agent or in combination with checkpoint blockade.
TRP-2 and gp100 are both crucial to melanin production and are therefore expressed by all pigment producing melanomas. Cloning of T cells from patients, showing spontaneous rejection of their melanomas, identified their targets as TRP-2180-188 and gp100174-190 and indicated that they were CD8 rejection epitopes.Citation7,Citation8 This demonstrated that, at least in these patients, there was a T cell repertoire recognizing self-antigens with sufficient avidity to kill tumour cells. The question remains as to whether an effective vaccine could stimulate these T cells in a wider cohort of patients.
We have previously shown in preclinical models that a DNA plasmid encoding T cell epitopes within the complementarity-determining regions of a human IgG1 antibody (ImmunoBody®)Citation9 and injected with electroporation (EP) stimulates high avidity T cell responses.Citation10 T cell avidity is critically important in both viral infection and tumor models as only high avidity T cells mediate viral clearance and tumor eradication.Citation11-16 EP increases DNA uptake over 1000-fold in comparison to injection alone and has an adjuvant effect resulting from local tissue damage and stimulation of pro-inflammatory cytokines.Citation17,Citation18 ImmunoBody® acts by direct uptake of the DNA into antigen presenting cells which is transcribed, translated and processed, with epitopes being presented on the cell surface in combination with MHC. ImmunoBody® can also be taken up by both antigen presenting cells and non-antigen presenting cells and the transcribed antibody protein secreted. The secreted antibody is internalized via the high affinity FcγR1 receptor (CD64) on antigen presenting cells, and is then processed and epitopes cross presented on MHC-I. It is this combination of direct and cross presentation that elicits T cells with sufficiently high avidity to eradicate established tumors in preclinical models.Citation9 Furthermore, the frequency and avidity of T cell responses induced by ImmunoBody® are superior to those induced by immunization with DNA encoding full-length antigen, using naked peptides or peptides loaded onto dendritic cells.Citation10,Citation19-21
In this first-in-human study, SCIB1 ImmunoBody®, incorporating HLA-A#0201 restricted epitopes from gp100 and TRP-2 plus HLA-DR#0401 and HLA-DR7/ DR53/DQ6 restricted epitopes from gp100 was assessed in HLA-A#0201 melanoma patients with at least one of the relevant HLA class-II types. The trial was designed to find the optimal dose of vaccine to induce immunological responses and to allow evaluation of safety/tolerability and clinical responses.
Results
Patient treatment
The trial design is shown in and patient demographics are shown in . A total of 35 patients were recruited, all were evaluated for toxicity and 33/35 for immunological responses. Fifteen patients had tumor present at baseline and 20 patients had fully-resected disease. The original premise for the study was that patients without a high tumor load might respond better to a cancer vaccine; however, first-in-human clinical trials are usually restricted to patients with advanced malignant disease. Although both patients with and without tumor present could be recruited into the initial planned dose escalation phase, most clinicians enrolled only patients with tumor (Part 1, cohorts 1–3, doses 0.4-4 mg, ). In the absence of any obvious toxicity, the highest dose of SCIB1 (4 mg) was selected to dose an expanded group of patients with no tumor present (Part 2, cohort 1). During recruitment of this cohort advances in the manufacturing methodology enabled us to generate plasmid DNA at higher concentrations giving us the potential to administer a higher, 8 mg, dose. A further safety cohort of patients receiving 8 mg was therefore added to the dose escalation phase in patients with tumor (Part 1, cohort 4) and then an additional cohort, again at 8 mg, was added to the expansion phase (Part 2, cohort 2). Patients with or without tumor present were permitted for this final group as an objective tumor response had been observed in a patient in the 8 mg safety cohort.
Figure 1. Patient recruitment and analysis. (A) Participant flowchart. Patients were initially recruited sequentially into Part 1, cohorts 1, 2 and 3. Resected patients were then recruited into Part 2 cohort 1 at a dose of 4 mg. A further cohort of patients was recruited in parallel with Part 2 cohort 1 at a higher dose of 8 mg (Part 1 cohort 4) and then Part 2 was expanded to dose patients at this 8 mg dose (Part 2 cohort 2). (B) Patient data was analysed based on whether tumor was present or not at screening and then by dose level. #One patient in each of Part 1 cohort 1 and cohort 3 only received a single injection and were replaced. ##Intra-patient dose escalation to 4 mg permitted.
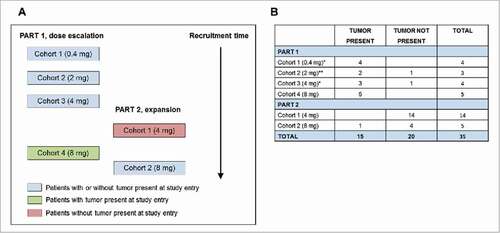
Table 1. Patient Demographics.
Dose escalation proceeded without dose limiting toxicity. Twenty-five patients received five doses and completed the study; 10 patients withdrew due to progressive disease, two after one dose (non-evaluable for immune response), two after three doses and six after four doses. In the 2 mg cohort all three patients had their dose escalated to 4 mg after receiving at least their first three immunizations (). Twelve patients received one or more doses of SCIB1 in the continuation phase of treatment at approximately 3-monthly intervals, including nine patients with resected disease. No patients received any concurrent melanoma therapy. The data presented was collected up to, and including, the point at which all patients had completed dosing in the main part of the study. As the hypothesis was that disease-free patients would elicit a stronger T cell response than patients with tumor, the data was analysed according to whether patients had tumor present or not at study entry and then by dose level, rather than by dose alone.
Table 2. Clinical findings and immune responses.
Immune responses
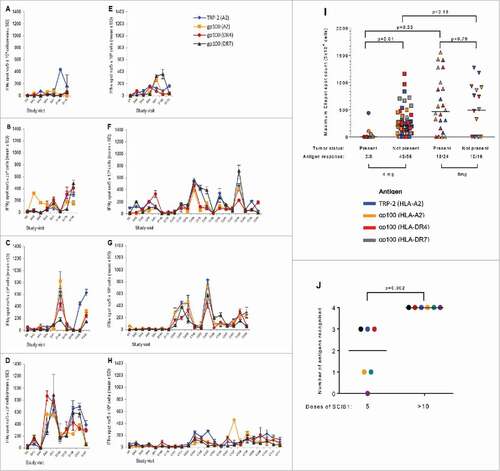
Thirty-three patients were assessed for T cell responses by cultured Elispot and/or proliferation assays (, ). The gp100 HLA-A#0201, CD8 epitope is nested within the HLA-DR7/DR53/DQ6 epitope so use of the long peptide does not discriminate between CD4 and CD8 responses The responses to the short peptide are likely to be CD8 responses as small hydrophobic peptides do not usually bind to HLA- DR7/DR53/DQ6. This is consistent with the observation that six patients responded only to the short CD8 peptide. We therefore counted these responses separately. Conversely, responses observed with the gp100 DR7/DR53/DQ6 long peptide could be against the nested HLA-A2-restricted short peptide. Indeed, all patients responding to the long peptide also react to the nested short peptide. Twenty-six patients were evaluated by Elispot and the results are summarized in . Six of eight tumor-bearing patients and 17/19 tumor-free patients made a detectable Elispot response. Two of the tumor-bearing patients received 4 mg doses; one responded to three peptides () whereas no responses were seen in samples from the other patient (data not shown). Six tumor-bearing patients received 8 mg doses and strong Elispot responses were detected in five patients as exemplified by . Sixteen tumor-free patients received 4 mg doses and 14 of them responded to at least one epitope with 10/14 responding to all four epitopes (exemplified by ). Four tumor-free patients received 8 mg doses and three of them responded to at least two epitopes. The intensity of the responses (maximal Elispot count) was significantly higher in patients without tumor in patients receiving 4 mg doses compared to those with tumor receiving 4 mg (p < 0.01; ). In contrast, patients with tumor showed a significantly higher response to the 8 mg dose than the 4 mg dose (p < 0.03) whereas there was no significant difference between doses in the patients without tumor (). This suggests that the lower dose of 4 mg was sufficient for the patients without tumor but a higher dose is required to overcome the immunosuppression associated with bulky tumors. None of the six fully-resected patients receiving the 4 mg dose, who continued therapy and eventually received at least 10 doses of SCIB1 responded to all four epitopes following the initial five doses; however, all six responded to all four epitopes following 10 SCIB1 administrations (). Overall, of the 26 patients evaluated by Elispot, three patients did not respond, three patients responded to one epitope, two patients responded to two, two patients responded to three and 16 patients responded to all four epitopes.
Table 3. HLA Typing and Immune Responses.
Figure 2. Generation of IFNγ (A-H) in response to immunization with SCIB1. PBMC were isolated at the indicated study visits and cultured for 10 – 17 days at 37°C with HLA-A2-restricted TRP-2 peptide or with HLA-A2-restricted, HLA-DR4-restricted or HLA-DR7/DR53/DQ6-restricted gp100 peptides. IFNγ was assayed on days 10 and 17 by Elispot following a 24 hour re-stimulation with the appropriate peptide. Data is shown from one timepoint for each patient. Representative Elispot data is shown for patient 04–16 (day 10), a patient with tumor present at screening and receiving 4 mg doses of SCIB1 (A); three patients with tumor present at screening and receiving 8 mg doses of SCIB1 (01-46 (day 10), 04–27 (day 17) and 04–28 (day 17) in panels B, C and D, respectively); and four fully-resected patients receiving 4 mg doses of SCIB1 (05-09 (day 10), 05–11 (day 10), 05–18 (day 17) and 05–21 (day 10) in panels E, F, G and H, respectively). The results shown are the mean number of IFNγ producing cells ± standard deviation (n = 4). Study visit days in the main study are denoted by the prefix “D” and in the continuation phase by “C” where Cx0y indicates x = weeks after dosing and y = continuation dose number. (I) Maximum mean of four wells IFNγ spot count for individual antigens determined for each responder. Non-parametric statistics (Mann Whitney test) were used for comparison of the results between cohorts. Antigen response defines the number of responses to any one epitope by any patient against the number of potential responses for the number of patients tested. (J) Administration of >10 doses of SCIB1 conveys a broader range of epitope recognition in fully-resected 4 mg patients compared to five administrations. The bar represents the median antigen recognition; the P value was calculated by the Mann Witney test. Individual colours represent individual patients i.e. 05–11 (black), 05–24 (red), 01–32 (blue), 05–19 (yellow), 05–21 (green) and 05–18 (purple).
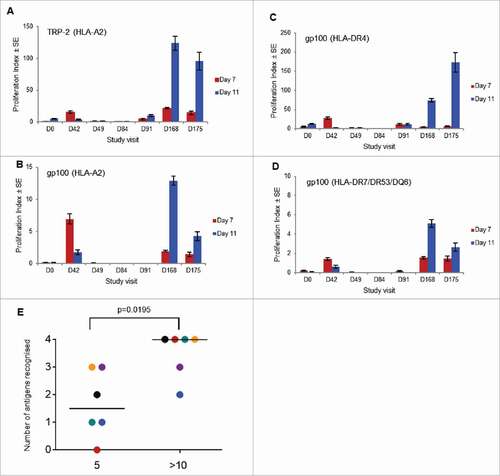
Thirty-three patients were evaluated by proliferation and the results are summarized in , and . Seven of 13 tumor-bearing patients and all 20 tumor-free patients made a detectable proliferation response. A typical proliferation response from a patient to all four epitopes is shown in . Only 1/3 tumor-bearing patients receiving 0.4 mg generated a proliferation response, and that was only to a single peptide; 3/3 patients receiving 2 mg/4 mg doses, 12/17 patients receiving 4 mg doses and 6/10 patients receiving 8 mg doses responded to at least two peptides. Proliferation responses to any peptide were observed after three or more doses, but they continued to increase with continued administration of SCIB1. Two of the six fully-resected patients receiving the 4 mg dose, who continued therapy and eventually received at least 10 doses of SCIB1, responded with proliferative responses to three epitopes following the initial five doses; however, five of six had proliferation responses to three or more epitopes following 10 SCIB1 administrations (). Overall, six patients did not respond, six patients responded to a single epitope, six patients responded to two, five patients responded to three and 10 patients responded to all four epitopes.
Figure 3. T cell proliferation in response to immunization with SCIB1. PBMCs were isolated from patient 05–13 (tumor not present at screening; 4 mg dose) at the indicated study visits (days) and cultured for up to 11 days at 37°C following stimulation with (A) TRP-2 (HLA-A2-restricted) peptide; (B) gp100 (HLA-A2-restricted) peptide; (C) gp100 (HLA-DR4-restricted) peptide; or (D) gp100 (HLA-DR7/DR53/DQ6-restricted) peptide. On days 7 and 11 cellular proliferation was assayed by the overnight incorporation of 3H-thymidine. The results shown are the mean of the calculated Proliferation Index (PI) ± standard error of the mean (n = 3). (E) Administration of >10 doses of SCIB1 conveys a broader range of epitope recognition in fully-resected 4 mg patients compared to five administrations. The bar represents the median antigen recognition; the P value was calculated by the Mann Witney test. Individual colours represent individual patients i.e. 05–11 (black), 05–24 (red), 01–32 (blue), 05–19 (yellow), 05–21 (green) and 05–18 (purple).
Patients showed similar responses in both assays used for immune monitoring with 27/33 (82%) evaluable patients responding in the proliferation assay, 23/26 (89%) in the Elispot assay, 21/26 (81%) in both assays and 29/33 (88%) in either assay. 67% (22/33) of patients responded to all four epitopes, 85% to TRP2, 76% to gp100A2, 76% to gp100DR4 and 67% to gp100DR7/DR53/DQ6 long peptide (shown in graphs as gp100DR7; , ). 100% of patients without detectable tumor responded in either assay with 80% responding to all four epitopes. In contrast, only 69% of patients with tumor present responded in either assay, with 46% responding to all four epitopes.
Correlation of HLA phenotype and T cell responses are shown in . Only one of the patients responding to the gp100 long peptide did not express any of the HLA-DR7/DR53/DQ6 alleles, but this could indicate a response to the nested CD8 epitope. Ten of 25 patients responding to the gp100 HLA-DR4 epitope did not express the HLA-DR4 allele suggesting that this epitope has a more promiscuous binding and is not just restricted to HLA-DR4. Indeed, the Immune Epitope Database and Analysis Resource (IEDB) (http://tools.iedb.org/mhcii/) predicts that this epitope will bind better to HLA-DQ4 than HLA-DR4 (supplementary Table 2).
Safety assessment
The vaccine was well tolerated (): 218 doses of SCIB1 were administered and there were no adverse events (AEs) leading to discontinuation of study treatment. AEs were reported as being related to either the study drug, SCIB1, or to the study electroporation device/procedure. Overall, 28 patients (80%) had AEs reported related to study drug; there was no obvious difference in the rate of AEs in patients with escalating doses or with tumor present at study entry and those with resected disease. The most common of these study drug-related events (>10%) were injection site hematoma (37%), injection site pain (20%), fatigue (14%), blurred vision, headache, and procedural pain (11%). In total, 32 patients (91%) experienced AEs related to the electroporation device and procedure, most commonly injection site hematoma (77%), injection site pain (37%), procedural pain (17%), and fatigue (11%). Grade ≥3 AEs were infrequent and were considered related to SCIB1/EP in only two patients (Grade 3 events of injection site hematoma, injection site pain, and anxiety). The discomfort associated with the EP device was generally described as tolerable. There were no clinically significant changes in laboratory values during the study that were associated with SCIB1 administration (data not shown).
Table 4. Common adverse events and relatedness to SCIB1 and electroporation procedure.
Clinical findings
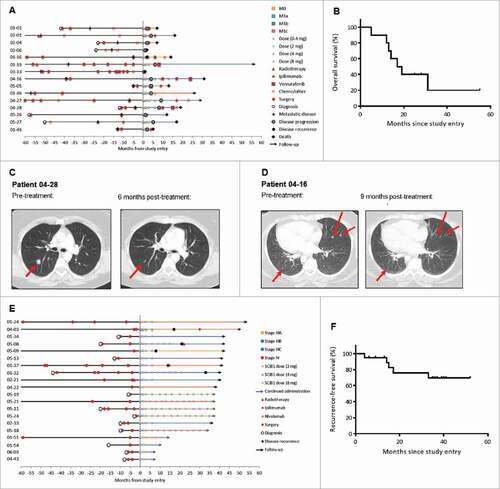
The disease stage, time on study, SCIB1 dosing and disease progression for the 15 patients with tumor present at study entry are shown in and in . Survival is shown in and in . Pre- and post-study treatments are shown in . Most patients had a number of prior lesions that were treated with surgery or decarbazine. No patients received checkpoint blockade prior to vaccination as these treatments had not been approved at the time of the SCIB1 trial. Four patients received ipilimumab post-vaccination and following disease progression. Three of these patients have died and one is still alive. One patient with stage IV (M1b) disease with lung metastases at study entry, treated at 8 mg, had a RECIST partial response of 29 weeks duration as determined during the main study period, per protocol (). After completing the main study period, new subcutaneous lesions identified progression of their disease prior to the patient's first continuation visit. The patient subsequently received vemurafenib and died 19 months after starting study participation. A second patient with stage IV (M1b) disease with metastases in the lung, lymph nodes, and subcutaneous space, treated at 4 mg, had a greater than 30% reduction in the size of their target lesions but progression of a non-target lesion (). The patient had a single continuation dose of SCIB1 before a new lesion was detected in the small bowel. Following further tumor excision the patient was treated with vemurafenib and died 31 months after starting the study. Both of these patients had shown strong Elispot and/or proliferative responses to all four SCIB1 epitopes. In addition to the patient with a partial response, seven patients had a period of stable disease on the study in excess of 16 weeks.
Figure 4. Clinical findings and tumor regression in patients immunized with SCIB1. (A) Swimmer plot for patients with tumor present at study entry (n = 15). Information about other treatments given post-SCIB1 was collected in follow-up; dates, duration and response data were not collected. (B) Kaplan-Meier analysis of the overall survival of patients who had tumor at study entry and received at least three doses (2-8 mg) of SCIB1 (n = 10). (C) CT scans of lung lesions of patient 04–28 before and 6 months after treatment with SCIB1. Lesion locations are indicated by arrows. (D) CT scans of lung lesions of patient 04–16 before and 9 months after treatment with SCIB1. (E) Swimmer plot for patients with fully-resected tumor at study entry (n = 20). (F) Kaplan-Meier analysis of the recurrence-free survival of patients with fully-resected tumor at study entry (n = 20).
The disease stage, time on study and SCIB1 dosing for the 20 patients who were disease-free at study entry are shown in and in . Disease recurrence is shown in and in . Pre- and post-study treatments are shown in . Most patients (17) had multiple lesions that were treated with surgery prior to trial entry. No patients received checkpoint blockade prior to vaccination as they had not been approved at the time of the trial. One patient received ipilimumab and one received nivolumab post-vaccination and both are still alive. The longest follow up is available for the 16 patients treated with 2 mg and 4 mg of SCIB1; at the cut-off date for reporting, all 16 patients were alive with a median observation time of 39 months (range 33 to 52 months). At 2 years, 12 (75%) of these patients remained disease-free without additional treatment beyond continued SCIB1. Disease-free survival was largely independent of stage at screening with 7/9 (78%) of stage III patients and 5/7 (71%) of stage IV patients alive and disease-free at 2 years. All patients with resected disease at study entry who received 8 mg doses of SCIB1 (who were recruited later than the 4 mg cohort) remained alive and disease-free at the data cut-off point.
Tumor analysis
Following an amendment to the original protocol, pre/post-treatment tumors were obtained from 21/35 patients and stained for expression of MHC-I/II, gp100/TRP-2 and PD-L1 plus infiltration of CD4, CD8 and Foxp3 positive cells (). There was great variability in expression of these markers between lesions from the same patient.
Table 5. Percentage Expression of Markers Present on Patient Tumor Biopsies.
Initially the biopsies taken prior to vaccination were analysed to see if any of the markers could predict response to SCIB1. Of interest was the finding that the pre-treatment tumors of 13 patients showed strong MHC-II expression (>15% of tumor cells expressing MHC-II) and this group of patients with MHC-II positive tumors included seven patients who remained alive and tumor-free post-resection and post-SCIB1 treatment plus both of the patients with CT scan findings of tumor regression. Of the eight patients whose pre-treatment tumors did not express high levels of MHC-II, only four were disease-free at the end of the main study period. Pre-treatment tumor samples from eight patients showed a loss of MHC-I (defined as ≤80% of tumor cells showing MHC-I expression in any pre-treatment biopsy sample). Five of these patients had disease recurrence and one had recurrence and then died (patient 04–28). In contrast, of the 13 patients whose tumors did not lose expression of MHC-I, only four were not disease-free (including patient 04–16 who also had evidence of tumor reduction). Fifteen patients expressed MHC-I (>80% of tumor cells), MHC-II (>15% of tumor cells) or both and only three of these patients had died/recurred or failed to show a reduction in tumor burden in response to the vaccine. Six patients had tumors that had lost expression of MHC-I (≤80% of tumor cells) and had no elevated expression of MHC-II (>15% of tumor cells). Only one of these patients remains disease free. Clinical benefit was superior in MHC-I/II positive patients (Fishers exact test p = 0.027).
All pre-treatment tumors tested showed some loss of TRP-2 expression (between 10–100% of cells showing no expression) and 14 showed some loss (10-100%) of gp100 expression. Expression of PD-L1, infiltration of CD4, CD8 and Foxp3 positive cells or CD4:Foxp3 or CD8:Foxp3 ratios did not predict disease recurrence or progression.
Tumors were obtained post-vaccination from six patients, three who had tumor present and three who were fully-resected at study entry. Tumors from one of the fully-resected patients (05-09) failed to express either target prior to vaccination and the patient did not benefit from the vaccine as they experienced tumor recurrence. One patient's recurrent tumor (04-16) had a reduction in expression of gp100 and TRP-2. One patient's post-vaccination tumor (01-19) showed a loss of MHC-I and TRP-2. Two patients' recurrent tumors excluded CD4T cells (04-03 and 04–28) and one patient's pre- and post-vaccination tumors showed no obvious changes (01-37).
Discussion
We conducted a first-in-human phase I/II trial to test the safety and efficacy of a gp100/TRP-2 antibody DNA vaccine, SCIB1, in melanoma patients. SCIB1 was safe and well tolerated. Use of the EP device to administer SCIB1 caused transient pain and, on occasion, injection site hematoma but was successfully given on 218 occasions, including administration to five patients who have now each received 15–17 immunizations over a period of up to 39 months. Discomfort from the EP procedure only limited treatment to three doses in a single patient.
The SCIB1 vaccine was developed to stimulate T cell responses to both MHC-I and MHC-II restricted epitopes from two different melanoma antigens. Eighty-eight percent of patients responded to one or more epitopes and 67% of patients responded to all four epitopes, with similar responses to both antigens. There were significantly stronger responses to the 8 mg dose than to the 2/4 mg doses in patients with tumor present, indicating that the former is the most appropriate dose for future studies in this population. The immune response rate compares favourably with other vaccines targeting gp100 (80% v 49%,Citation22,Citation23) but is a similar response rate to a DNA fusion vaccine targeting carcinoembryonic antigen (CEA,Citation24), although these comparisons are complicated by different assays being used to quantify the immune response in each study.
Also in line with the CEA study, we show that both the T cell Elispot responses were stronger in patients without tumor present at screening than in patients with detectable tumor, which suggests that tumor load may attenuate the response. It also suggests that previous vaccine studies in patients with tumor load may have underestimated the measurable effects due to systemic or local immune suppression. SCIB1 monotherapy may therefore be particularly effective in early stage patients with a low tumor burden. At present, interferon-α2a and ipilimumab are licensed for the adjuvant treatment of melanoma.Citation25,Citation26 Ipilimumab significantly improved median recurrence-free survival (RFS) from 17.1 to 26.1 months and the 3-year RFS of resected high-risk stage III patients from 35% to 47% when compared to placebo, but 52% of patients discontinued treatment due to toxicity.Citation25 Further follow-up has shown that this improvement in RFS led to a significant improvement in overall survival.Citation27 In the current study, all 20 of the fully-resected patients were alive at data cut-off with a median observation time of 37 months (range 6 – 52 months) from study entry; the median RFS has not been reached and there was minimal toxicity. The 2- and 3-year RFS for the stage III patients was 67% and 56%, respectively, and was 71% at both 2 and 3 years for the stage IV patients.
To prevent recurrence of melanoma it would be ideal to stimulate memory responses. In animal models, SCIB1 stimulated memory T cell responses that continued to increase in avidity as the T cells were selectively recruited into memory.Citation19 Similarly, in this current study, patients have shown stronger and broader responses following extended treatment with SCIB1. All patients on continuation responded to all four epitopes after five continuation doses at 21 months or after 10 immunizations; this suggests that vaccination for the prevention of recurrence should continue for at least 2 years post-surgery and that maintenance vaccination should be considered as it appears to improve the strength and breadth of the T cell response to vaccine-encoded epitopes. The former maybe related to memory but this is difficult to assess as we did not phenotype these responses. The latter may indicate de novo stimulation of new populations of T cells.
Thirty-eight percent of the tumors biopsied had reduced expression of MHC-I prior to vaccination, although none had total loss. One of six patients' tumors that were resected post-vaccination showed MHC-I loss (≤80% of cells expressing MHC-I) that was not apparent prior to treatment. Tumors lacking MHC-I expression can resist T cell attack and become the dominant cell type. Total loss of MHC-I is rare as it makes cells susceptible to attack by natural killer (NK) cells, but when it occurs it is usually due to loss or mutation of β2microglobulin.Citation28,Citation29 Loss of the specific allele recognized by the T cells is more frequent as this makes the tumor cells resistant to both T cell and NK attack. Unfortunately, allele specific monoclonal antibodies do not work in immunohistochemistry analyses on paraffin embedded tissue and therefore allele loss could not be assessed.
Most tumors do not express MHC-II; however, 62% of the tumors in this study showed strong MHC-II expression prior to vaccination. MHC-II expression on melanomas has previously been described as an indicator of poor prognosis.Citation30-32 However, in this study 9/13 of the patients whose tumors expressed MHC-II are either disease-free or their tumors regressed after SCIB1 treatment. This suggests that MHC-II expression could be a predictive biomarker of patients who are likely to respond to immunization. A recent study has shown that MHC-II positivity on tumors cells predicted response to anti-PD-1/PD-L1 therapy.Citation33 It also provides support for the growing evidence that tumor-specific CD4T cells play a vital role in anti-tumor immunity.Citation34-37
Some of the cells within all of the patients' pre-treatment tumors showed a loss of TRP-2 (with 10–100% of cells showing no expression) although, with the exception of one patient, expression did not decrease further on post-treatment, recurrent tumors. This suggests that there is a either a pre-existing T cell response to TRP-2 in patients which drives selection of antigen loss variants or it is lost due to genetic mutation or epigenetic dysregulation. The loss of TRP-2 prior to vaccination did not predict outcome; however, this could be explained by the expression of gp100 which is also a target for the vaccine and emphasizes the need for including more than one antigen in the vaccine design. Targeting these two antigens may also benefit from combination with other drugs which alleviate tumor immune suppression or epigenetic control. Indeed, preclinical studies have shown that in combination with checkpoint blockade, SCIB1 and SCIB2 (an ImmunoBody® targeting NY-ESO-1), induce increased infiltration and proliferation of T cells that result in significantly improved survival.Citation21,Citation39 In this trial, no patients received checkpoint inhibitors prior to vaccination but six patients (four with tumor and two with no tumor at study entry) received ipilimumab or nivolumab post-vaccination and three of these patients are still alive. Although these patient numbers are low, this suggests that SCIB1 vaccination may prime for responses to checkpoint inhibition.
In conclusion, SCIB1 is a novel class of anti-cancer immunotherapy that induces T cells which can cause tumor regression in patients with melanoma. The high frequency of responses, their breadth and durability suggest SCIB1 is worthy of further study in a larger cohort of patients. This is particularly the case in the adjuvant setting, where all of the patients responded immunologically and where absence of toxicity is an important clinical consideration. Furthermore, the stimulation of potent de novo immune responses by SCIB1 may provide an opportunity for synergistic combination therapy with checkpoint inhibitors in late stage disease.
Materials and Methods
Study design and treatment plan
This study is an open label, phase I/II dose escalation study in melanoma patients. The trial was designed to find the optimal dose of vaccine to induce immunological responses and to allow evaluation of safety/tolerability and clinical responses. Patients with stage III/IV melanoma (with or without tumor present at study entry (Part1)) were accrued in sequential cohorts to receive 0.4, 2 or 4 mg of SCIB1 via intramuscular injection with EP. Patients with fully-resected stage III/IV disease (Part 2) were treated at the maximum tolerated dose (MTD; or the highest dose administered in Part 1 if no MTD was determined). SCIB1 was administered at two injection sites every 3 weeks for three doses and then twice more at 12 and 24 weeks. Dose escalation was only permitted following a safety evaluation by the Cohort Review Committee of a minimum of three patients in each cohort followed up to the week 7 visit. Dose-limiting toxicities (DLTs) were defined as either Grade 3/4 neutropenia with fever and/or infection or any non-hematological toxicity or autoimmunity/allergy equal to or greater than Grade 3 (CTCAE, version 4.02). Patients receiving the 2 mg dose were permitted to escalate to the 4 mg dose in the absence of DLTs after their first three doses. Patients without any intercurrent toxicity by week 28 were permitted to continue treatment every 12–24 weeks for up to 5 years. All patients were followed up for a minimum of 2 years after study end or until death. The UK Gene Therapy Advisory Committee provided ethical approval for the study, which was conducted in accordance with the Declaration of Helsinki and with Good Clinical Practice as defined by the International Conference on Harmonization. As no MTD had been reached at 4 mg in the initial Part 1 dose escalation phase, a fourth cohort of five patients with tumors present was recruited and dosed with 8 mg SCIB1 (Part 1, cohort 4) in parallel with the recruitment of resected patients into the first Part 2 cohort. This was possible due to improvements in the manufacturing process for the DNA, which enabled a higher concentration of plasmid DNA solution to be prepared which, with the volume restrictions for intramuscular injection, enabled an 8 mg dose to be administered. Once safety at this higher dose had been demonstrated, an additional group of patients was then recruited into a second Part 2 cohort to receive 8 mg doses (either with or without tumor present at study entry).
Although, adjuvant interferon-α2a was a treatment option for fully-resected patients, the physicians involved in this trial did not routinely use it as standard of care. Ipilimumab was not licensed for adjuvant use when the patients were dosed. Ipilimumab and pembrolizumab became available for use as standard of care at a later date and, indeed, some of the patients received these treatments if they had progressive disease or a recurrence. The patients receiving SCIB1 therefore received no concurrent therapy for their melanoma.
Eligibility criteria
Patients were eligible for enrolment if they had histologically confirmed stage III or IV melanoma. Part 1 comprised of patients with either resected disease or patients with advanced disease who had measurable disease evaluable by the Response Evaluation Criteria in Solid Tumors (RECIST version 1.0). In Part 2, recruitment was limited to patients with resected disease (4 mg cohort) or patients with or without tumor present at screening (8 mg cohort). In both parts, patients with fully resected disease could not receive systemic therapy between resection and study registration. All patients needed to be positive for HLA-A2 and at least one of HLA-DR4, HLA-DR7, HLA-DR53 or HLA-DQ6. Other inclusion criteria included an Eastern Cooperative Oncology Group performance status of ≤2 and adequate liver function and lymphocyte counts. Exclusion criteria included the presence of brain metastases, a life-expectancy of less than 3 months, any prior systemic anti-cancer treatment or immunosuppressive therapy within 4 weeks of study entry, previous malignancy within 5 years of screening, the presence of any electronic stimulation device, cardiac abnormalities and women who were pregnant or lactating. All patients provided written informed consent prior to enrolment.
Evaluations at baseline and during treatment
Screening procedures and assessments consisted of a complete medical history, a full clinical examination, baseline electrocardiogram, ophthalmologic examination, assessment of vital signs, HLA tissue typing, pregnancy test (if appropriate) and standard biochemical, hematological and urine analysis. Tumor status at screening was determined by a CT scan of the head, thorax, pelvis and abdomen.
The safety and tolerability of SCIB1 and the EP device were monitored throughout by clinical examination, assessment of the injection site, evaluation of vital signs and laboratory parameters, recording of AEs and a patient tolerability questionnaire. Patients with tumor at screening were further assessed at weeks 9, 18 and 28 along with a CT scan prior to any additional SCIB1 dose given during continuation treatment. Patients were followed up in clinic for a minimum of 2 years with CT scans where appropriate for suspected disease progression.
Stimulation of naïve and memory immune responses and the response to multiple injections were assessed by standard proliferation and interferon-gamma (IFNγ) Elispot assays at baseline and before and after subsequent immunizations. Detailed methodology for these assays (in compliance with MIATA guidelines) and immunohistochemistry is reported in supplementary data. For the proliferation assays, a patient was designated as an immunological responder if on two or more time points post-dosing the proliferation index (PI) was double the pre-treatment PI; the PI was defined as the mean peptide-specific counts per minute (cpm) divided by the mean negative control cpm at a given time point. For the Elispot assays, a patient was designated as an immune responder if on two occasions or more post-dosing the mean peptide-specific Elispot response minus two Standard Deviations was greater than the mean pre-treatment peptide-specific response plus one Standard Deviation (of this mean) and the peptide-specific Elispot response was more than 50 spots per million peripheral blood mononuclear cells (PBMCs).
Clinical study statistics
The primary endpoint for Part 1 of the study was the safety and tolerability of SCIB1 administered by electroporation. No sample size was determined for this part as it was designed to seek an MTD that could not be predicted in advance. The sample size for Part 2 (4 mg dose) of the study was based on Fleming's single stage procedure.Citation40,Citation41 The highest immune response probability given the null hypothesis of no drug effect was set at 50% and the lowest immune response probability of the alternative hypothesis was set at 75%. This requires the study of at least 13 patients for a two-sided significance of 0.05 and 80% power. The sample size for the expansion of Part 2 (8 mg dose) was also based on Fleming's single stage procedure. As three patients (up to six) were to be enrolled in the Part 1 4 mg cohort, an additional 10 patients were planned to provide a total of 13 patients receiving the same dose.
Disclosure of interest
Lindy G. Durrant is a director and shareholder of Scancell Ltd. Sally E. Adams is a director and shareholder of Scancell Ltd. ImmunoBody® is a registered trademark of Scancell Ltd, Nottingham, UK. TriGrid™ is a registered trademark of Ichor Medical Systems, Inc., CA, USA. There are no further COI to declare.
Clinical trials repository link
Clinicaltrials.gov identifier NCT01138410.
supp_data.docx
Download MS Word (236.9 KB)Additional information
Funding
Related Research Data
References
- Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, Segal NH, Ariyan CE, Gordon RA, Reed K, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369:122–33. doi:10.1056/NEJMoa1302369. PMID:23724867.
- Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–23. doi:10.1056/NEJMoa1003466. PMID:20525992.
- Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, Daud A, Carlino MS, McNeil C, Lotem M, et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N Engl J Med. 2015;372:2521–32. doi:10.1056/NEJMoa1503093. PMID:25891173.
- Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–54. doi:10.1056/NEJMoa1200690. PMID:22658127.
- Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, Ivanova Y, Hundal J, Arthur CD, Krebber WJ, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature. 2014;515:577–81. doi:10.1038/nature13988. PMID:25428507.
- Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, Walsh LA, Postow MA, Wong P, Ho TS, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014;371:2189–99. doi:10.1056/NEJMoa1406498. PMID:25409260.
- Khong HT, Rosenberg SA. Pre-existing immunity to tyrosinase-related protein (TRP)-2, a new TRP-2 isoform, and the NY-ESO-1 melanoma antigen in a patient with a dramatic response to immunotherapy. J Immunol. 2002;168:951–6. doi:10.4049/jimmunol.168.2.951. PMID:11777994.
- Kawakami Y, Eliyahu S, Delgado CH, Robbins PF, Sakaguchi K, Appella E, Yannelli JR, Adema GJ, Miki T, Rosenberg SA. Identification of a human melanoma antigen recognized by tumor-infiltrating lymphocytes associated with in vivo tumor rejection. Proc Natl Acad Sci USA. 1994;91:6458–62. doi:10.1073/pnas.91.14.6458. PMID:8022805.
- Metheringham RL, Pudney VA, Gunn B, Towey M, Spendlove I, Durrant LG. Antibodies designed as effective cancer vaccines. MAbs. 2009;1:71–85. doi:10.4161/mabs.1.1.7492. PMID:20046577.
- Pudney VA, Metheringham RL, Gunn B, Spendlove I, Ramage JM, Durrant LG. DNA vaccination with T-cell epitopes encoded within Ab molecules induces high-avidity anti-tumor CD8+ T cells. Eur J Immunol. 2010;40:899–910. doi:10.1002/eji.200939857. PMID:20039301.
- Alexander-Miller MA, Leggatt GR, Berzofsky JA. Selective expansion of high- or low-avidity cytotoxic T lymphocytes and efficacy for adoptive immunotherapy. Proc Natl Acad Sci U S A. 1996;93:4102–7. doi:10.1073/pnas.93.9.4102. PMID:8633023.
- Gallimore A, Glithero A, Godkin A, Tissot AC, Pluckthun A, Elliott T, Hengartner H, Zinkernagel R. Induction and exhaustion of lymphocytic choriomeningitis virus-specific cytotoxic T lymphocytes visualized using soluble tetrameric major histocompatibility complex class I-peptide complexes. J Exp Med. 1998;187:1383–93. doi:10.1084/jem.187.9.1383. PMID:9565631.
- Sedlik C, Dadaglio G, Saron MF, Deriaud E, Rojas M, Casal SI, Leclerc C. In vivo induction of a high-avidity, high-frequency cytotoxic T-lymphocyte response is associated with antiviral protective immunity. J Virol. 2000;74:5769–75. doi:10.1128/JVI.74.13.5769-5775.2000. PMID:10846055.
- Yee C, Savage PA, Lee PP, Davis MM, Greenberg PD. Isolation of high avidity melanoma-reactive CTL from heterogeneous populations using peptide-MHC tetramers. J Immunol. 1999;162:2227–34. PMID:9973498.
- Zeh HJ, 3rd, Perry-Lalley D, Dudley ME, Rosenberg SA, Yang JC. High avidity CTLs for two self-antigens demonstrate superior in vitro and in vivo antitumor efficacy. J Immunol. 1999;162:989–94. PMID:9916724.
- Zhu Z, Singh V, Watkins SK, Bronte V, Shoe JL, Feigenbaum L, Hurwitz AA. High-avidity T cells are preferentially tolerized in the tumor microenvironment. Cancer Res. 2013;73:595–604. doi:10.1158/0008-5472.CAN-12-1123. PMID:23204239.
- Chiarella P, Massi E, De Robertis M, Sibilio A, Parrella P, Fazio VM, Signori E. Electroporation of skeletal muscle induces danger signal release and antigen-presenting cell recruitment independently of DNA vaccine administration. Expert Opin Biol Ther. 2008;8:1645–57. doi:10.1517/14712598.8.11.1645. PMID:18847301.
- van Drunen Littel-van den Hurk S, Hannaman D. Electroporation for DNA immunization: clinical application. Expert Rev Vaccines. 2010;9:503–17. doi:10.1586/erv.10.42. PMID:20450325.
- Brentville VA, Meteringham RL, Gunn B, Durrant LG. High Avidity cytotoxic T lymphocytes can be selected into the memory pool but they are exquisitely sensitive to functional impairment. PloS one. 2012;7:e41112. doi:10.1371/journal.pone.0041112. PMID:22829916.
- Saif JM, Vadakekolathu J, Rane SS, McDonald D, Ahmad M, Mathieu M, Pockley AG, Durrant L, Metheringham R, Rees RC, et al. Novel prostate acid phosphatase-based peptide vaccination strategy induces antigen-specific T-cell responses and limits tumour growth in mice. Eur J Immunol. 2014;44:994–1004. doi:10.1002/eji.201343863. PMID:24338683.
- Xue W, Metheringham RL, Brentville VA, Gunn B, Symonds P, Yagita H, Ramage JM, Durrant LG. SCIB2, an antibody DNA vaccine encoding NY-ESO-1 epitopes, induces potent antitumor immunity which is further enhanced by checkpoint blockade. Oncoimmunology. 2016;5:e1169353. doi:10.1080/2162402X.2016.1169353. PMID:27471648.
- Sosman JA, Carrillo C, Urba WJ, Flaherty L, Atkins MB, Clark JI, Dutcher J, Margolin KA, Mier J, Gollob J, et al. Three phase II cytokine working group trials of gp100 (210M) peptide plus high-dose interleukin-2 in patients with HLA-A2-positive advanced melanoma. J Clin Oncol. 2008;26:2292–8. doi:10.1200/JCO.2007.13.3165. PMID:18467720.
- Schwartzentruber DJ, Lawson DH, Richards JM, Conry RM, Miller DM, Treisman J, Gailani F, Riley L, Conlon K, Pockaj B, et al. gp100 peptide vaccine and interleukin-2 in patients with advanced melanoma. N Engl J Med. 2011;364:2119–27. doi:10.1056/NEJMoa1012863. PMID:21631324.
- McCann KJ, Mander A, Cazaly A, Chudley L, Stasakova J, Thirdborough S, King A, Lloyd-Evans P, Buxton E, Edwards C, et al. Targeting Carcinoembryonic Antigen with DNA Vaccination: On-Target Adverse Events Link with Immunologic and Clinical Outcomes. Clin Cancer Res. 2016;22:4827–36. doi:10.1158/1078-0432.CCR-15-2507. PMID:27091407.
- Eggermont AM, Chiarion-Sileni V, Grob JJ, Dummer R, Wolchok JD, Schmidt H, Hamid O, Robert C, Ascierto PA, Richards JM, et al. Adjuvant ipilimumab versus placebo after complete resection of high-risk stage III melanoma (EORTC 18071): a randomised, double-blind, phase 3 trial. Lancet Oncol. 2015;16:522–30. doi:10.1016/S1470-2045(15)70122-1. PMID:25840693.
- Grob JJ, Dreno B, de la Salmoniere P, Delaunay M, Cupissol D, Guillot B, Souteyrand P, Sassolas B, Cesarini JP, Lionnet S, et al. Randomised trial of interferon alpha-2a as adjuvant therapy in resected primary melanoma thicker than 1.5 mm without clinically detectable node metastases. French Cooperative Group on Melanoma. Lancet. 1998;351:1905–10.
- Eggermont AM, Chiarion-Sileni V, Grob JJ, Dummer R, Wolchok JD, Schmidt H, Hamid O, Robert C, Ascierto PA, Richards JM, et al. Prolonged Survival in Stage III Melanoma with Ipilimumab Adjuvant Therapy. N Engl J Med. 2016;375:1845–55. doi:10.1056/NEJMoa1611299. PMID:27717298.
- Bernal M, Ruiz-Cabello F, Concha A, Paschen A, Garrido F. Implication of the beta2-microglobulin gene in the generation of tumor escape phenotypes. Cancer Immunol Immunother. 2012;61:1359–71. doi:10.1007/s00262-012-1321-6. PMID:22833104.
- Zhao F, Sucker A, Horn S, Heeke C, Bielefeld N, Schrors B, Bicker A, Lindemann M, Roesch A, Gaudernack G, et al. Melanoma Lesions Independently Acquire T-cell Resistance during Metastatic Latency. Cancer Res. 2016;76:4347–58. doi:10.1158/0008-5472.CAN-16-0008. PMID:27261508.
- Hemon P, Jean-Louis F, Ramgolam K, Brignone C, Viguier M, Bachelez H, Triebel F, Charron D, Aoudjit F, Al-Daccak R, et al. MHC class II engagement by its ligand LAG-3 (CD223) contributes to melanoma resistance to apoptosis. J Immunol. 2011;186:5173–83. doi:10.4049/jimmunol.1002050. PMID:21441454.
- Martins I, Deshayes F, Baton F, Forget A, Ciechomska I, Sylla K, Aoudjit F, Charron D, Al-Daccak R, Alcaide-Loridan C. Pathologic expression of MHC class II is driven by mitogen-activated protein kinases. Eur J Immunol. 2007;37:788–97. doi:10.1002/eji.200636620. PMID:17304627.
- van der Stoep N, Quinten E, Alblas G, Plancke A, van Eggermond MC, Holling TM, van den Elsen PJ. Constitutive and IFNgamma-induced activation of MHC2TA promoter type III in human melanoma cell lines is governed by separate regulatory elements within the PIII upstream regulatory region. Mol Immunol. 2007;44:2036–46. doi:10.1016/j.molimm.2006.09.013. PMID:17067677.
- Johnson DB, Estrada MV, Salgado R, Sanchez V, Doxie DB, Opalenik SR, Vilgelm AE, Feld E, Johnson AS, Greenplate AR, et al. Melanoma-specific MHC-II expression represents a tumour-autonomous phenotype and predicts response to anti-PD-1/PD-L1 therapy. Nat Commun. 2016;7:10582. doi:10.1038/ncomms10582. PMID:26822383.
- Hung K, Hayashi R, Lafond-Walker A, Lowenstein C, Pardoll D, Levitsky H. The central role of CD4(+) T cells in the antitumor immune response. J Exp Med. 1998;188:2357–68. doi:10.1084/jem.188.12.2357. PMID:9858522.
- Xie Y, Akpinarli A, Maris C, Hipkiss EL, Lane M, Kwon EK, Muranski P, Restifo NP, Antony PA. Naive tumor-specific CD4(+) T cells differentiated in vivo eradicate established melanoma. J Exp Med. 2010;207:651–67. doi:10.1084/jem.20091921. PMID:20156973.
- Quezada SA, Simpson TR, Peggs KS, Merghoub T, Vider J, Fan X, Blasberg R, Yagita H, Muranski P, Antony PA, et al. Tumor-reactive CD4(+) T cells develop cytotoxic activity and eradicate large established melanoma after transfer into lymphopenic hosts. J Exp Med. 2010;207:637–50. doi:10.1084/jem.20091918. PMID:20156971.
- Zanetti M. Tapping CD4T cells for cancer immunotherapy: the choice of personalized genomics. J Immunol. 2015;194:2049–56. doi:10.4049/jimmunol.1402669. PMID:25710958.
- Tanaka A, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Cell Res. 2017;27:109–18. doi:10.1038/cr.2016.151. PMID:27995907.
- Xue W, Brentville VA, Symonds P, Cook KW, Yagita H, Metheringham RL, Durrant LG. SCIB1, a huIgG1 antibody DNA vaccination, combined with PD-1 blockade induced efficient therapy of poorly immunogenic tumors. Oncotarget. 2016;7:83088–100. doi:10.18632/oncotarget.13070. PMID:27825115.
- Fleming TR. One-sample multiple testing procedure for phase II clinical trials. Biometrics. 1982;38:143–51. doi:10.2307/2530297. PMID:7082756.
- Machin MC, M.J. Statistical tables for the design of clinical trails. Oxford, England: Blackwell Scientific Publications, 1987.