
ABSTRACT
Interleukin-12 (IL12) (p35/p40 complex) is a heterodimeric cytokine with potent anti-tumor activity. However, its short serum half-life and high dose-related toxicities limit its clinical efficacy. Here, we constructed heterodimeric immunoglobulin Fc-fused mouse IL12 (mIL12) in a monovalent binding format (mono-mIL12-Fc) to generate long-acting mIL12 in the naturally occurring heterodimeric form. Mono-mIL12-Fc exhibited a much longer plasma half-life than recombinant mIL12, enabling twice-weekly systemic injections to remove established tumors in syngeneic mouse models. Mono-mIL12-Fc was more potent than wild-type Fc-based bivalent-binding IL12-Fc (bi-mIL12-Fc) for eradicating large established immunogenic tumors without noticeable toxicities by enhancing interferon-γ production and the proliferation of immune effector cells in tumors. More importantly, mono-mIL12-Fc triggered weaker IL12 signaling than bi-mIL12-Fc, favoring the generation of functional and protective memory CD8+ T cells. Our results demonstrate that heterodimeric-Fc-fused IL12 is a suitable format for augmenting adaptive CD8+ T cell immune responses, providing a practical alternative to the systemic administration of IL12 for antitumor therapy.
Introduction
Interleukin-12 (IL12) is a pleiotropic cytokine linking the innate and adaptive immune responses, and shows potent immune responses against tumors and certain pathogens.Citation1,Citation2 In response to antigenic stimulation, IL12 is produced from activated antigen-presenting cells such as dendritic cells, macrophages, and B cells.Citation3 IL12 subsequently stimulates the proliferation and cytotoxicity of activated T cells and natural killer (NK) cells via induction of cytotoxic enzymes and cytokines, mainly interferon-gamma (IFN-γ),Citation2 which are believed to elicit potent antitumor immunity.Citation3
IL12 is a heterodimeric cytokine (70 kDa) composed of two different polypeptide chains: an α-chain (p35 subunit) and a β-chain (p40 subunit), which are covalently linked by a disulfide bond.Citation4 IL12 signals by monovalent binding to IL12 receptor (IL12R) comprised of IL12Rβ1 and IL12Rβ2 subunits via 1:1 complex formation.Citation3 IL12 signaling plays an important role in the proliferation and differentiation of effector CD8+ T cells.Citation5 During the effector phase, both the duration and strength of IL12 signaling determines the fates of effector CD8+ T cells, with respect to whether they differentiate into short-lived effector cells (SLECs) or memory precursor effector cells (MPECs), by modulating the expression levels of transcription factors, mainly T-bet and eomesodermin (Eomes) through the signal transducer and activator of transcription 4 (STAT4) pathway.Citation6 Strong IL12-STAT4 signaling upregulates T-bet expression in effector CD8+ T cells, leading to their subsequent differentiation into SLECs.Citation7,Citation8 Modest signaling induces high Eomes and low T-bet expression to promote the differentiation into MPECs, which are capable of generating long-lived memory CD8+ T cells.Citation7,Citation8
In preclinical studies, the systemic administration of recombinant mouse IL12 (rmIL12) elicited potent antitumor effects against various human tumor mouse models.Citation1,Citation9 To achieve therapeutic effects of recombinant human IL12 (rhIL12), its short serum half-life (5.3–10.3 h) required administration at a high dose and daily injections in clinical trials.Citation10 However, this systemic high-level dosing caused dose-related toxicities, including fatalities in two patients.Citation11 To overcome this limitation, many approaches have been proposed to develop IL12-based antitumor regimens, including intratumoral IL12 injection, IL12-secreting cell injection, and systemic injection of antibody-fused IL12.Citation1,Citation12 However, no agents have been clinically approved to date.
IL12 has been fused to tumor-targeting antibodies, called immunocytokines, based on full-length immunoglobulin G (IgG) or antibody fragments.Citation13 Single-chain variable fragment (scFv)-fused IL12 immunocytokines exhibit substantial antitumor activity owing to the improved tumor targeting of IL12.Citation14-Citation16 However, the potential of scFv-based immunocytokines is hindered by their short serum half-life and manufacturing challenges.Citation12 To extend the serum half-life, IL12 can be constructed in the single-chain form (scIL12), in which the two subunits p35 and p40 connected by a polypeptide linker (p40-linker-p35) are fused to the C-termini or N-termini of the heavy chains of IgG antibodies.Citation17-Citation19 Despite the potent antitumor activity conferred by their good tumor-targeting ability and extended serum half-life, these IgG-fused IL12 immunocytokines were still insufficient to eliminate tumors, particularly large established tumors, in mouse studies. It is noteworthy that such IgG-based immunocytokines contain two molecules of the IL12 heterodimer (i.e., bivalent fusion) due to the characteristics of the symmetric, bivalent architecture of IgG antibody. Thus, we reasoned that the strong bivalent-mediated IL12 signaling of IgG-based IL12 immunocytokines might fail to properly induce protective CD8+ T cell responses. However, transient exposure of CD8+ T cells to IL12 did not efficiently improve the in vivo survival and proliferation of the cells.Citation20 Therefore, we hypothesized that the generation of long-acting IL12 by Ig Fc fusion while maintaining the naturally occurring monovalent, heterodimeric form of IL12 might be a desirable approach to trigger the parental IL12-like instructive signaling for achieving protective innate and adaptive immunity against tumors.
Here, we describe the generation of heterodimeric Ig Fc-fused IL12 in the naturally occurring heterodimeric form of IL12, termed mono-mIL12-Fc, in which the p35 and p40 subunits were fused to the N-terminus of two different Fc variants, respectively. We also generated Fc-fused bivalent IL12 with two IL12 units (bi-mIL12-Fc) by fusion of scIL12 (p40-linker-p35) to the N-terminus of wild-type Fc as a control. We found that mono-mIL12-Fc is more potent than bi-mIL12-Fc to rescue tumor-bearing mice (TBM) owing to augmentation of IFN-γ production and the cytotoxic activity of immune effector cells as well as the generation of memory CD8+ T cells. We further demonstrate that bi-mIL12-Fc can program effector CD8+ T cells into SLECs rather than MPECs by inducing higher levels of pSTAT4 and T-bet expression than mono-mIL12-Fc. These findings provide a foundation for applications of heterodimeric-Fc-fused IL12 to realize protective adaptive immunity against large established tumors that can be used as potential antitumor therapy to surmount the limitations of current IL12-based therapy.
Results
Generation and characterization of mono- and bi-mIL12-Fc proteins
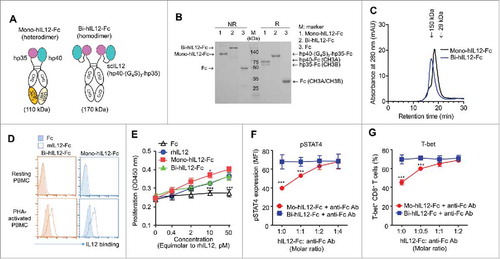
Given that murine T and NK cells do not respond to human IL12 (hIL12),Citation21 we first tested our hypothesis with murine IL12 (mIL12). To generate Ig Fc-fused mIL12 in the naturally occurring heterodimeric form, we exploited the heterodimeric Fc scaffold to separately fuse the two subunits (p35 and p40) of mIL12 to the N-terminus of each heterodimeric Fc chain (). Heterodimeric Fcs have been engineered through modifications to the CH3 domain interface with different mutations on each domain, such that the engineered Fc chains preferentially form heterodimers rather than homodimers.Citation22 We used human IgG4-based heterodimeric Fc with an A107 mutation pair (EW-NVT)Citation23 to minimize the binding to Fcγ receptors on immune cells and to avoid the elimination of IL12R-expressing effector cells.Citation24 The mouse p35 subunit (mp35) with a (G4S)3 linker and the p40 subunit (mp40) alone were fused to the N-terminus of Fc with A107A mutations [hinge-CH2- CH3B (NVT)] and Fc with A107B mutations [hinge-CH2-CH3A (EW)], respectively, to generate mono-mIL12-Fc (). The 15-residue (G4S)3 linker was inserted at the C-terminus of mp35 in order to establish a sufficient distance and conformational flexibility to facilitate the natural heterodimeric assembly of mp35 with mp40. As a bivalent Fc-fusion mIL12 control, we generated wild-type IgG4 Fc-fused single-chain mIL12, termed bi-mIL12-Fc, in the format of mp40-(G4S)3 linker-mp30-(G4S)3 linker-hinge-CH2-CH3 ().
Figure 1. (A) Schematic representation of mono-mIL12-Fc and bi-mIL12-Fc proteins. (B) Purified mIL12-Fc proteins were analyzed by SDS-PAGE under either a reducing ('R') or non-reducing ('NR') condition. The arrows indicate the eluted position of the proteins. Representative images from three independent experiments are shown. (C) Size exclusion elution profiles of purified mIL12-Fc proteins (100 μL of 500 μg/mL) were monitored at 280 nm. Representative chromatogram from three independent experiments are shown. (D) Binding activities of 1 μM of mIL12-Fc proteins on the surface of resting and PHA-activated PBMCs, determined by flow cytometric analysis. Representative histograms from three independent experiments are shown. (E) Proliferation of PHA-activated PBMCs after 72-h culture with the indicated concentrations of proteins, presented in the equimolar concentration of rmIL12. Representative results from three independent experiments performed in triplicates are shown as means ± SD. ***p < 0.001 versus rmIL12 determined by Newman-Keuls multiple comparison post-hoc test. (F) Serum concentrations of mIL12-Fc proteins in non-tumor-bearing Balb/c mice were determined by ELISA following a single intravenous injection of proteins at an equimolar amount to 1 μg of rmIL12 in a total volume of 200 μL. Symbols and error bars represent the mean ± SD (n = 3 per time point). Data shown are representative of two independent experiments. The solid lines represent the fit of a two-compartment pharmacokinetic model to the data to estimate the initial rapid clearance phase (T1/2α) and the later terminal serum clearance phase (T1/2β). The inset table shows the pharmacokinetic parameters.
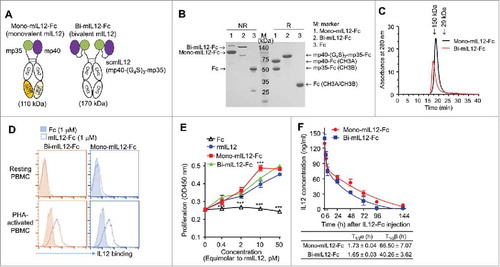
For the expression of mono-mIL12-Fc, two plasmids, one encoding mp35-Fc based on CH3B and the other encoding mp40-Fc based on CH3A, were transiently co-transfected at the equivalent molar ratio in HEK293F cells.Citation25 The secreted proteins were purified and analyzed by reducing and non-reducing sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Mono-mIL12-Fc preferentially favored the formation of heterodimers (86.9 ± 6.2%) over undesirable homodimers (). As expected, bi-mIL12-Fc was expressed as a homodimer (). Both fusion proteins existed as correctly assembled monomers in solution, as judged from size-exclusion chromatography (SEC) analysis (). Given that high-affinity IL12R is induced on human peripheral blood mononuclear cells (PBMCs) by T cell mitogen stimulation and that mIL12 cross-reacts with human IL12R on activated human T cells and NK cells,Citation26 the binding ability of mIL12-Fc proteins for phytohemagglutinin (PHA)-activated PBMCs was analyzed by flow cytometry. Both mono-mIL12-Fc and bi-mIL12-Fc bound to PHA-activated PBMCs, but not to unstimulated PBMCs (), demonstrating the IL12R binding specificity. In addition, the two fusion proteins induced the proliferation of PHA-activated PBMCs in a dose-dependent manner, showing efficacy at the equivalent molar dose similar to that of rmIL12 (), demonstrating in vitro bioactivity comparable to that of rmIL12.
Pharmacokinetics of mono- and bi-mIL12-Fc proteins
To evaluate the pharmacokinetics (PK) of mIL12-Fc proteins, equivalent molar amounts of mono-mIL12-Fc (1.6 μg) and bi-mIL12-Fc (2.4 μg), corresponding to an equivalent molar amount of 1 μg mIL12, were intravenously injected in non-tumor-bearing Balb/c mice for comparison. Mono-mIL12-Fc and bi-mIL12-Fc showed extended terminal serum half-lives (T1/2β) of 66 h and 40 h, respectively (), which are 5–10 times longer than that of recombinant IL12.Citation10,Citation17 The faster plasma clearance of bi-mIL12-Fc relative to mono-mIL12-Fc seems to result from the preferential binding to IL12R-expressing cells due to the bivalency-mediated avidity effects, as previously observed with other immunocytokines,Citation27,Citation28
Twice-weekly systemic injection of mIL12-Fc proteins eliminates small tumors
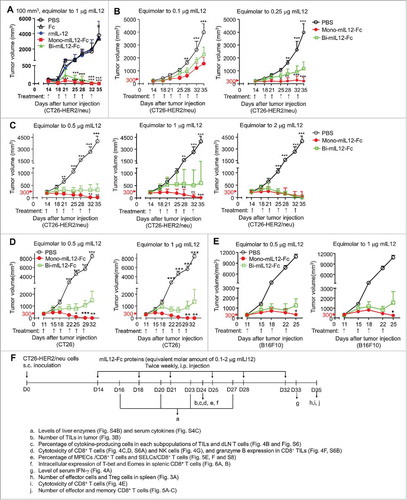
We first evaluated the antitumor activity of mono- and bi-mIL12-Fc proteins against established CT26-HER2/neu tumors in immunocompetent Balb/c mice, which are able to induce adaptive immune responses against human HER2/neu antigen.Citation29 It should be noted that, hereafter, the dose of mIL12-Fc proteins used in this study will be presented as the equivalent molar amount of rmIL12 (μg) to allow for comparisons of antitumor activities at the equivalent molar amounts of each protein. For example, 1 μg mono-mIL12-Fc (actually 1.6 μg administered) and 1 μg bi-mIL12-Fc (actually 2.4 μg administered) denote that the used molar amount of mIL12-Fc proteins is equivalent to that of 1 μg rmIL12. After randomization of mice with similar baseline tumor volumes (∼100–120 mm3), intraperitoneal (i.p.) injection of either phosphate-buffered saline (PBS) or 1 μg of each protein was initiated 11 days after tumor inoculation. Considering the extended serum half-life of mIL12-Fc proteins, they were administered twice weekly. Remarkably, mono-mIL12-Fc and bi-mIL12-Fc completely eradicated CT26-HER2/neu tumors at the end of treatment, whereas rmIL12 and Fc alone did not inhibit tumor growth compared with the PBS control (). This result demonstrated that Fc-fused long-acting mIL12 elicited a potent antitumor effect even with the infrequent and systemic administration schedule for which rmIL12 did not exhibit any antitumor activity. In previous mouse studies, repeated daily injection of rmIL12 (1–5 μg) exerted significant antitumor activity.Citation30,Citation31
Figure 2. (A) In vivo antitumor efficacy of mIL12-Fc proteins compared to that of PBS, Fc, and rmIL12 controls analyzed in syngeneic Balb/c mice bearing CT26-HER2/neu tumors. Twice-weekly i.p. injection of mIL12-Fc proteins (equivalent molar amount of 1 μg IL12) was initiated 11 days after tumor cell inoculation when the tumors were small (∼100 mm3). (B–E) Antitumor effect of mIL12-Fc proteins on large established CT26-HER2/neu tumors (B, C) and CT26 tumors (D) in syngeneic Balb/c mice, and B16F10 tumors (E) in syngeneic C57BL/6 mice. (B, C) Twice-weekly i.p. injection of mIL12-Fc proteins (equivalent molar amount of 0.1–0.25 μg mIL12 (B) and 0.5–2 μg mIL12 (C)) was initiated 14 days after tumor cell inoculation when the tumors were large (∼300 mm3). (D, E) Twice-weekly i.p. injection of mIL12-Fc proteins (equivalent molar amount of 0.5 and 1 μg mIL12) was initiated 11 days after tumor cell inoculation when the tumors were large (∼300 mm3). In (A–E), symbols and error bars represent means ± SEM of 2–3 pooled (A–C) or representative (D, E) experiments with at least 5 mice/group. *p < 0.05, **p < 0.01, ***p < 0.001 versus rmIL12 (A) or bi-mIL12-Fc (B–E) determined by one-way ANOVA with Newman-Keuls post-hoc test. (F) The effect of mIL12-Fc proteins on the phenotypes and functions of effector cells in CT26-HER2/neu TBM were analyzed as described in a-j on the indicated day after tumor inoculation. The arrows indicate each time point for treatment or assay. In (A–F), the dose of mIL12-Fc proteins is presented as the equivalent molar amount of rmIL12 (μg).
Mono-mIL12-Fc more efficiently eliminates large established tumors than bi-mIL12-Fc
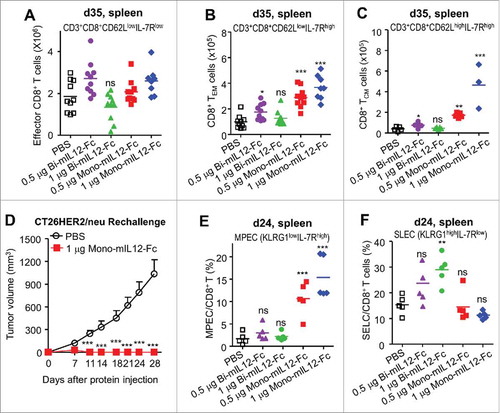
To further evaluate the antitumor activity of mIL12-Fc proteins against large established tumors, Balb/c mice bearing CT26-HER2/neu tumors of approximately 300 mm3 were treated with mIL12-Fc proteins by twice-weekly i.p. injection over a wide range of doses corresponding to the equimolar amount of 0.1–4 μg rmIL12. Mono-mIL12-Fc induced robust tumor regression in a dose-dependent manner and resulted in durable cures in 40% of the mice, even at the low dose of 0.25 μg, whereas bi-mIL12-Fc displayed only modest therapeutic effects under the identical dosing regimen (). Impressively, twice per week, five-times administration of 0.5 μg mono-mIL12-Fc could fully control tumor growth, resulting in a 73% (n = 11/15) rate of tumor-free survival in the mice (, Fig. S1A–C) 35 days after tumor inoculation. Most importantly, even a single injection of 1 μg and 2 μg mono-mIL12-Fc substantially impeded tumor growth in almost 50% of the TBM on day 18 after tumor inoculation, and four-times administration was sufficient to rescue 66% of the TBM (n = 10/15). However, administration of 1 μg bi-mIL12-Fc had limited effects against the large established tumors, resulting in progressive tumor growth in 60% of the mice (, Fig. S1A–C). Even though 1 μg bi-mIL12-Fc was sufficient to control small tumors (), doubling of this dose (2 μg) of bi-mIL12-Fc was required to rescue mice against large established tumors ().
To further test the antitumor activity of mIL12-Fc proteins in other syngeneic tumor models with different immunogenicity, we evaluated them in CT26 tumor in immunocompetent Balb/c mice and B16F10 melanoma in immunocompetent C57BL/C mice. CT26 tumor without expression of HER2/neu is less immunogenic than CT26-HER2/neu tumor and B16F10 is known to poorly immunogenic due to the low major histocompatibility complex (MHC) expression.Citation32 Twice-weekly i.p. injection of mIL12-Fc proteins (equivalent molar amount of 0.5 and 1 μg mIL12) was initiated 11 days after tumor cell inoculation when the tumors were large (∼300 mm3). In CT26 tumor, five-times administration of 0.5 μg or 1 μg mono-mIL12-Fc completely eradicated CT26 tumors, whereas the same regimens with bi-mIL12-Fc only retarded the growth of CT26 tumors without complete tumor rejection (, Fig. S2). In the case of B16F10 tumor, four-times injection of 0.5 μg or 1 μg mono-mIL12-Fc elicited tumor regression. However, bi-mIL12-Fc treatments elicited weaker therapeutic responses than mono-mIL12-Fc, leading to only 40% of sustained protection from B16F10 progression (, Fig. S3). These results demonstrated that antitumor activity of mono-mIL12-Fc could be generalized in various immunogenic tumor models with different immunogenicity.
Notably, despite the high response rates, the regimen of mono-mIL12-Fc and bi-mIL12-Fc appeared to be well tolerated by the CT26-HER2/neu TBM, with all mice maintaining their body weights during treatment (Fig. S4A). Compared to PBS-treated control, no significant differences were noted in the serum levels of liver enzymes (alanine aminotransferase (ALT) and aspartate aminotransferase (AST)) and cytokines (IL2 and IL10) during the mIL12-Fc treatment (Fig. S4B, C), although an acute increase in serum levels of IFN-γ were observed (Fig. S4C). These results demonstrate that mono-mIL12-Fc is more effective for eradicating large established immunogenic tumors than bi-mIL12-Fc over wide dose ranges without noticeable systemic toxicities, indicating that mono-mIL12-Fc expands the therapeutic window of the mIL12 regimen.
Mono-mIL12-Fc induces proliferation of effector cells in the spleen and tumor
To understand the cellular mechanisms underlying the potent antitumor activity of mono-mIL12-Fc in CT26-HER2/neu TBM, we evaluated the contribution of distinct effector cells such as CD4+ T cells, CD8+ T cells, and NK cells as well as immunosuppressive regulatory T (Treg) cells in the spleen three days after the last treatment (day 35) (). Consistent with mono-mIL12-Fc-based tumor protection, both 0.5 μg and 1 μg mono-mIL12-Fc resulted in significant (P < 0.001) increases of CD4+ T and CD8+ T cell populations in dose-dependent manners compared to PBS treatment; by contrast, only 0.5 μg bi-mIL12-Fc was slightly effective to increase CD8+ T cells, but not CD4+ T cells (). Repeated injection of 1 μg bi-mIL12-Fc provided no benefits to increase CD4+ T and CD8+ T cells in the spleens of TBM (). We did not observe significant increases in NK cell numbers on day 35 after six-times administration of mIL12-Fc fusion proteins. This finding is in line with previous works in that no NK cell memory has been observed against tumors under physiological conditions.Citation33,Citation34 Additionally, both mono- and bi-mIL12-Fc proteins triggered slight but significant decrease in the frequency of Treg cells in CD4+ T cells in the spleen of TBM, whereas they did not substantially affect the absolute number of Treg cells, compared to the PBS control ().
Figure 3. (A) Effect of mIL12-Fc proteins (as described in the legend to ) on the expansion of effector cells and reduction in Treg cells with the indicated lineage markers in the spleens of mice bearing CT26-HER2/neu tumors, analyzed by a hemocytometer and flow cytometry 35 days after tumor inoculation. (B) Effect of mIL12-Fc proteins (as described in ) on the number of indicated immune cells and percentage of Treg cells infiltrated into CT26-HER2/neu tumors in mice, analyzed by flow cytometry with the indicated lineage markers 24 days after tumor inoculation. In (A, B), each symbol represents the value obtained from individual mice, and midlines represent the means of two pooled experiments. **p < 0.01, ***p < 0.001 versus the PBS control group or between the indicated groups determined by one-way ANOVA with Newman-Keuls post-hoc test.
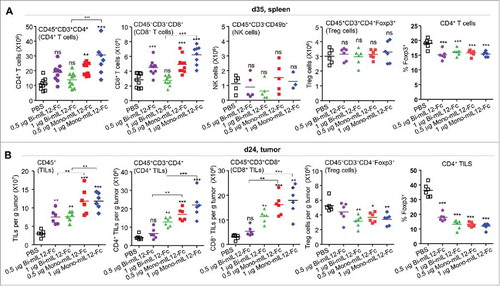
The presence of adaptive immune cells such as CD4+ and CD8+ T cells and the inhibitory Treg cells in tumor sites is important to destroy developing tumors.Citation33 To provide further evidence of the elevated adaptive immunity in mono-mIL12-Fc-treated tumors, we isolated tumor-infiltrating lymphocytes (TILs) from CT26-HER2/neu tumors and quantified the numbers of TILs by flow cytometry. Since five-times administration of 0.5 μg or 1 μg mono-mIL12-Fc abolished CT26-HER2/neu tumors in the mice, we assessed the TILs on day 24 after three-times treatment of mIL12-Fc. Both mono- and bi-mIL12-Fc treatment induced dose-dependent higher tumor infiltration of CD45+CD3+CD4+ T cells and CD45+CD3+CD8+ T cells compared to PBS treatment (). In addition, both mono- and bi-mIL12-Fc proteins caused dramatic decrease in the frequency of Treg cells in CD4+ T cells in tumor, leading to reduction in the number of Treg cells compared to the PBS control group (). Notably, mono-mIL12-Fc exhibited more profound effects on these overall effects than bi-mIL12-Fc at the equivalent dosing. Collectively, these results demonstrate that mono-mIL12-Fc caused greater expansion and tumor infiltration of CD4+/CD8+ T cells while diminishing Treg cell accumulation in the tumor, accounting for the stronger tumor control, compared to bi-mIL12-Fc.
Mono-mIL12-Fc augments the function of effector cells in TBM
Given that the antitumor efficacy of IL12 is concomitant with the endogenous production of IFN-γ, which induces cytostatic effects on tumor cells,Citation2,Citation31 we analyzed the serum levels of IFN-γ in mIL12-Fc-treated CT26-HER2/neu TBM at 24 h after the final treatment (day 33). The increase of serum IFN-γ levels was restricted to the mice receiving the mono-mIL12-Fc treatment in proportion to the dose up to an equivalent molar amount of 1 μg rmIL12 (). Higher doses (at an equimolar dose of 2 μg and 4 μg rmIL12) of mono-mIL12-Fc led to a relative decrease in the level of IFN-γ compared with treatment of 1 μg mono-mIL12-Fc. Intriguingly, bi-mIL12-Fc elicited no significant production of IFN-γ compared to the PBS control (). Analysis of serum IFN-γ levels after a single injection (day 14 after tumor inoculation) of 1 μg bi-mIL12-Fc proteins into mice bearing large established tumors (∼300 mm3) showed a profile of IFN-γ production for several days comparable to that observed in mice receiving the 1 μg mono-mIL12-Fc regimen, suggesting that there were no intrinsic defects in IFN-γ production upon stimulation with bi-mIL12-Fc in vivo (Fig. S4D). However, repeated cycles of bi-mIL12-Fc treatment resulted in the dramatic diminution of IFN-γ level at day 23 after tumor inoculation, whereas mono-mIL12-Fc treatment led to a slight decrease in the level of IFN-γ on day 27 compared to that on day 16 (Fig. S4C).
Figure 4. (A) Serum levels of IFN-γ in CT26-HER2/neu TBM treated with mIL12-Fc protein (as described in ), measured by ELISA 33 days after tumor inoculation. (B) Percentages of cytokine-producing cells in each subpopulation of TILs and dLN T cells isolated from mice bearing CT26-HER2/neu tumors, analyzed by flow cytometry 24 days after tumor inoculation. (C–E) Cytotoxic activity of splenic CD8+ T cells isolated from CT26-HER2/neu TBM treated with mIL12-Fc proteins (as described in ), analyzed 24 days (C, D) and 35 days (E) after tumor inoculation, followed by 4 h of culture with calcein-AM-labeled CT26-HER2/neu and 4T1 (control) cells. The percentage of calcein-AM-negative (dead) CT26-HER2/neu and 4T1 cells was measured by flow cytometry. (C) Representative dot plots from two independent experiments. (F) Granzyme B-expressing CD8+ TILs in CT26-HER2/neu TBM treated with mIL12-Fc proteins, analyzed by flow cytometry 24 days after tumor inoculation. (G) Cytotoxicity of NK cells isolated from the spleens of CT26-HER2/neu TMB treated with the indicated mIL12-Fc proteins (as described in ), measured by a calcein-AM release assay on day 24 after tumor inoculation. In (A, B, D–G), each symbol represents the value obtained from individual mice. Midlines represent the means of data compiled from 2–3 independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001 versus the PBS control or between the indicated groups determined by one-way ANOVA with Newman-Keuls post-hoc test.
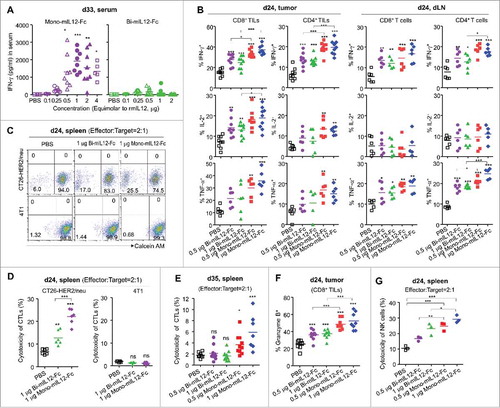
To functionally characterize CD8+ or CD4+ T cells on tumor rejection, we first analyzed cytokines (IFN-γ, IL2 and TNF-α) expression of CD8+ or CD4+ T cells in tumor and dLN isolated from mIL12-Fc-treated CT26-HER2/neu TBM on day 24 after tumor inoculation. CD8+ TILs and CD4+ TILs revealed elevated cytokine expression relative to dLN CD8+ T cells and CD4+ T cells, following mIL12-Fc treatment (, Fig. S5). The frequency of IFN-γ+, IL2+ or TNF-α+ CD8+ TILs was higher than that of IFN-γ+, IL2+ or TNF-α+ CD4+ TILs in tumor, whereas the frequency of these cytokine-producing CD8+ T cells and CD4+ T cells in dLN was similar each other in mIL12-Fc-treated CT26-HER2/neu TBM (). Notably, mono-mIL12-Fc exhibited more profound effects on these overall effects than bi-mIL12-Fc at the equivalent dosing.
To address whether cytotoxic T lymphocytes (CTLs) are responsible for the rejection of tumors,Citation2,Citation18,Citation19 the cytotoxic activity of CTLs from the spleens of mIL12-Fc-treated mice was measured using the calcein AM-release assayCitation35 with either tumor antigen-bearing CT26-HER2/neu cells or antigen-absent 4T1 cells. CTLs from mice treated with 1 μg mIL12-Fc fusion proteins only three-times (day 24) exhibited significant cytotoxic activity against CT26-HER2/neu cells compared to PBS-treated CTLs (, Fig. S6A), explaining the tumor suppression activities of mIL12-Fc (). No killing of control 4T1 breast cancer cells was observed (, Fig. S6A). At 72 h after the final treatment (day 35), however, CTLs from mono-mIL12-Fc-treated TBM, but not those from bi-mIL12-Fc-treated TBM, significantly (p < 0.05) manifested cytolytic activity against CT26-HER2/neu cells (). These results indicated that mono-mIL12-Fc promotes the tumor-specific cytotoxicity of CTLs more efficiently than bi-mIL12-Fc.
To further assess cytotoxic potential of CD8+ TILs, we analyzed granzyme B expression of CD8+ TILs in mIL12-Fc-treated CT26-HER2/neu TBM on day 24 after tumor inoculation. The expression level of granzyme B in CD8+ TILs is positively correlated with the rejection of immunogenic tumors.Citation36 Both mono- and bi-mIL12-Fc treatment caused elevated expression of granzyme B in CD8+ TILs compared to PBS treatment (, Fig. S6B). Remarkably, mono-mIL12-Fc exhibited more profound effects to induce granzyme B-expressing CD8+ TILs than bi-mIL12-Fc at the equivalent dosing (, Fig. S6B). These data, along with the above characterization of CD8+ and CD4+ T cells for cytokine production, suggested that functional CD8+ and CD4+ T cells are responsible for the immunogenic tumor rejection in mono-IL12-Fc-treated CT26-HER2/neu TBM.
We next sought to determine the contribution of mIL12-Fc proteins to the cytotoxic activity of NK cells in the tumor control group.Citation37 Since we did not observe significant increases in NK cell numbers 35 days after tumor inoculation (), we analyzed these activities on day 24. The cytolytic activities of splenic NK cells from mono- and bi-mIL12-Fc protein-treated TBM were significantly elevated in proportion to the dose relative to those of cells from the PBS-treated group (), revealing the partial contribution of NK cells in tumor control. However, mono-mIL12-Fc invigorated NK cells better than bi-mIL12-Fc in the TBM. Taken together, these data suggest that the therapeutic performance of mono-mIL12-Fc in the production of IFN-γ and the augmented cytotoxicity of NK and CD8+ T cells was clearly superior to that of bi-mIL12-Fc.
Mono-mIL12-Fc induces the generation of protective memory CD8+ T cells
Effector memory CD8+ T cells (TEM) and central memory CD8+ T cells (TCM) found in the spleen and blood are the hallmarks of the acquired immune response.Citation38,Citation39 However, the contributions of IL12 to the development of memory CD8+ T cells in TBM have not been well appreciated. To determine how mono- and bi-mIL12-Fc differentially affect the generation of memory CD8+ T cells, we analyzed the subsets of effector and memory CD8+ T cells in the spleen of CT26-HER2/neu TBM at the end of treatment (day 35), including effector CD8+ T (CD62LlowIL-7Rlow), TEM (CD62LlowIL-7Rhigh), and TCM (CD62LhighIL-7Rhigh) cells, based on the expression levels of CD62L and IL7 receptor (IL7R) as their phenotypic markers.Citation39 Neither mono-mIL12-Fc nor bi-mIL12-Fc treatment affected the number of effector CD8+ T cells compared with the PBS-treated control (). This was expected because effector CD8+ T cells that populate during the expansion phase (e.g., day 24 after tumor inoculation, as shown in ) die over the next few days during the contraction phase wherein only a small percentage (5–10%) survive to further differentiate into memory CD8+ T cells.Citation5 Intriguingly, both 0.5 μg and 1 μg mono-mIL12-Fc treatment significantly increased the population of TEM and TCM cells compared to PBS treatment (). For bi-mIL12-Fc treatment, only the 0.5 μg regimen, but not the 1 μg regimen, only slightly enhanced the population of TEM and TCM cells. In CT26 and B16F10 tumor models, these effects followed the similar pattern (Fig. S7). In addition, mono-mIL12-Fc-treated long-term surviving mice rejected a re-challenge with 106 CT26-HER2/neu cells at 3 months after cessation of therapy, confirming the generation of functional memory CD8+ T cells (). Together, these results suggest that mono-mIL12-Fc efficiently generated protective long-lived memory CD8+ T cells, while bi-mIL12-Fc had limited activity in this regard.
Figure 5. (A–C) Phenotypic analysis of CD8+ T cells with indicated lineage markers in the spleens of CT26-HER2/neu TBM treated with mIL12-Fc proteins (as described in ), analyzed by a hemocytometer and flow cytometry 35 days after tumor inoculation. Effector CD8+ T cells, TEM, and TCM were gated based on CD62L versus IL-7R expression. (D) Mice cured with mono-mIL12-Fc treatment were re-challenged with 106 CT26-HER2/neu tumor cells at 20 weeks after initial tumor inoculation. Tumor growth of re-challenged mice was compared with that of age-matched naïve mice without further treatment. Symbols and error bars represent the means ± SEM of 3 pooled experiments with at least 3 mice/group. ***p < 0.001 versus the PBS control determined by Student's t test. (E, F) Percentage of MPECs and SELCs in splenic CD8+ T cells of CT26-HER2/neu TBM treated with mIL12-Fc proteins (as described in ), analyzed by flow cytometry 24 days after tumor inoculation. SELCs and MPECs were gated based on KLRG1 versus IL-7R expression. (A–C, E, F) Each symbol represents the value obtained from individual mice. Midlines represent the means of data compiled from two independent experiments (A, B) or representing two independent experiments (C, E, F). **p < 0.01, ***p < 0.001 versus the PBS control group determined by one-way ANOVA with Newman-Keuls post-hoc test.
Mono-mIL12-Fc programs effector CD8+ T cells to differentiate into MPECs
Our results presented thus far demonstrated that bi-mIL12-Fc-treated TBMs were not able to fully eradicate the tumor with a quantitative deficiency of effector cells and memory CD8+ T cells compared to mono-mIL12-Fc-treated mice. To understand this difference, we compared the relative portions of SLECs and MPECs in splenic CD8+ T cells between mono-mIL12-Fc- and bi-mIL12-Fc-treated CT26-HER2/neu TBMs after three-times treatment of mIL12-Fc (day 24). SLECs (KLRG1highIL-7Rlow) and MPECs (KLRG1lowIL-7Rhigh) were distinguished using the SLEC marker killer cell lectin-like receptor G1 (KLRG1)Citation8 and the MPEC marker IL7R.Citation40 Both 0.5 μg and 1 μg mono-mIL12-Fc treatment resulted in substantial enhancement in the relative population of MPECs, but did not affect the population of SLECs compared to PBS treatment (, Fig. S8). By contrast, bi-mIL12-Fc treatment did not increase the pool of MPECs, and instead augmented the pool of SLECs significantly at 1 μg and slightly at 0.5 μg compared to PBS treatment (, Fig. S8). Together, these data indicated that mono-mIL12-Fc programs effector CD8+ T cells toward further differentiation into MPECs, whereas bi-mIL12-Fc provides them with an instructive signal toward SLEC differentiation during the effector and contraction phases.
Mono-mIL12-Fc induces lower expression levels of pSTAT4 and T-bet in CD8+ T cells than bi-mIL12-Fc
IL12-STAT4 signaling on responding CD8+ T cells determines their differentiation into either SLECs or MPECs by reciprocal modulation of the expression levels of T-bet and Eomes.Citation5,Citation6,Citation8 Therefore, we hypothesized that mono-mIL12-Fc and bi-mIL12-Fc would differentially modulate T-bet and Eomes expression in CD8+ T cells, to exert the distinct effects observed on the populations of SLECs and MPECs in the spleen (). Given that the relative difference in T-bet and Eomes expression is particularly pronounced at the effector phase,Citation7,Citation8 we determined T-bet and Eomes expression levels in splenic CD8+ T cells in the TBM after three-times treatment with mIL12-Fc (day 24). Mono-mIL12-Fc treatment significantly reduced the population of T-bethi CD8+ T cells (), but increased the number of Eomes+T-betlo CD8+ T cells () compared to PBS treatment. Compared to 1 μg mono-mIL12-Fc, 1 μg bi-mIL12-Fc manifested a more profound effect on the increased frequency of T-bethi CD8+ T cells () and reduced the population of Eomes+T-betlo CD8+ T cells (), although this differential effect between mono- and bi-mIL12-Fc at 0.5 μg was relatively weak.
Figure 6. (A, B) Intracellular expression of T-bet and Eomes in splenic CD8+ T cells of CT26-HER2/neu TBM treated with the indicated mIL12-Fc proteins (as described in ), analyzed by flow cytometry 24 days after tumor inoculation. Each symbol represents the value obtained from individual mice. Horizontal bars represent the means of data compiled from two independent experiments. **p < 0.01, ***p < 0.001 versus the PBS control group determined by one-way ANOVA with Newman-Keuls post-hoc test. (C, D) Intracellular expression of pSTAT4 (C) on day 15 and T-bet (D) on day 17 in dLN CD8+ T cells of CT26-HER2/neu TBM after single treatment with the indicated mIL12-Fc proteins (as described in Fig. S4D), analyzed by flow cytometry. Results are depicted as the mean ± SEM compiled from two independent experiments for at least 3 mice/group. ***p < 0.001 versus the PBS control or between the indicated groups by one-way ANOVA with Newman-Keuls post-hoc test. (E, F) Effect of cross-linked mIL12-Fc on the expression of pSTAT4 (E) and T-bet (F) in mouse CD8+ T cells after 2 h (E) or 72 h (F) of culture with plate-bound anti-CD3 antibody (1 μg/mL) and soluble anti-CD28 antibody (0.5 μg/mL) in the presence of 20 pM of mIL12-Fc alone or in combination with anti-Fc antibody at the indicated molar ratios, analyzed by flow cytometry. Results are depicted as the mean ± SEM compiled from two independent experiments. *p < 0.05, ***p < 0.001 versus bi-mIL12-Fc determined by Student's t test.
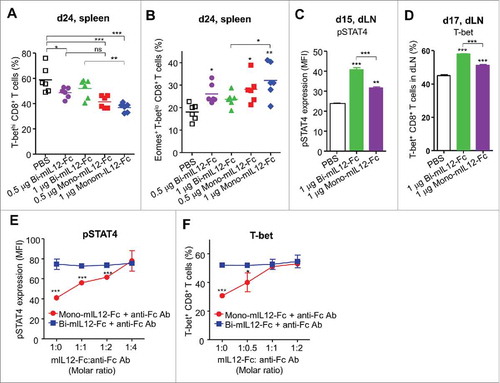
Strong IL12 signaling instigates the terminal differentiation of SLECs by inducing the pSTAT4-mediated upregulation of T-bet expression.Citation6-Citation8 Analysis of the expression levels of pSTAT4 and T-bet in CD8+ T cells from the tumor-draining lymph nodes (dLNs) of the TBM after single injection (day 15 or 17) of 1 μg mIL12-Fc proteins showed that mono-mIL12-Fc resulted in reduced pSTAT4 and T-bet expression in CD8+ T cells compared to bi-mIL12-Fc (), indicating that bi-mIL12-Fc triggers more potent intracellular signaling than mono-mIL12-Fc.
To further determine whether bi-mIL12-Fc triggers strong signaling due to the bivalent IL12-mediated receptor clustering, we compared bi-mIL12-Fc-induced signaling with that of cross-linked mono-mIL12-Fc, using a co-incubation assay with a secondary cross-linking agent of anti-Fc antibody at various molar ratios. Cross-linking of mono-mIL12-Fc progressively augmented the pSTAT4 and T-bet expression in stimulated CD8+ T cells from Balb/c mice in proportion to the increased molar ratio of the cross-linking agent, resulting in an equivalent magnitude at the higher molar ratio to those found under bi-mIL12-Fc treatment (, Fig. S9). In contrast, cross-linking of bi-mIL12-Fc did not exhibit any incremental effects on the levels of pSTAT4 and T-bet in CD8+ T cells. This result indicated that bivalent ligation of IL12R on CD8+ T cells enhances pSTAT4 signaling to cause upregulation of T-bet expression, whereas oligomeric complexes comprising more than two IL12Rs could not be formed or further enhance the downstream signaling. Taken together, the above results indicated that bi-mIL12-Fc transduces strong IL12 signaling to cause strong STAT4 activation and subsequently high T-bet expression in CD8+ T cells, leading to their differentiation toward SLECs rather than memory CD8+ T cells. However, monovalent ligation of mono-mIL12-Fc with IL12R induces only modest STAT4 activation and T-bet expression in CD8+ T cells, directing them toward MPECs and then memory CD8+ T cells.
Human IL12 version of mono-hIL12-Fc also triggers modest expression of pSTAT4 and T-bet in CD8+ T cells
To evaluate whether the observations with mono-mIL12-Fc in TBM are also relevant in humans for possible future clinical application, we generated the hIL12 version of mono- and bi-hIL12-Fc proteins in the same manner as described for mIL12 (). Mono-hIL12-Fc preferentially formed the desirable heterodimer (82 ± 3.3%), and bi-hIL12-Fc was expressed as a homodimer (). Flow cytometry analysis revealed that both mono-hIL12-Fc and bi-hIL12-Fc specifically bound to hIL12R expressed on PHA-activated PBMCs, and not to that on resting PBMCs (). In addition, the two fusion proteins were as potent as rhIL12 in inducing the proliferation of PHA-activated PBMCs (). Furthermore, consistent with the data of mIL12-Fc proteins, mono-hIL12-Fc also led to reduced expression of pSTAT4 and T-bet compared to bi-hIL12-Fc in stimulated human CD8+ T cells. Mono-hIL12-Fc required further cross-linking with anti-Fc antibody to show comparably high levels of pSTAT4 and T-bet expression in CD8+ T cells to those found in bi-hIL12-Fc treatment (, Fig. S10). These data suggest that mono-hIL12-Fc might also be more effective to generate MPECs for long-lived memory CD8+ T cell formation than bi-mIL12-Fc in humans.
Figure 7. (A) Schematic representation of mono-hIL12-Fc and bi-hIL12-Fc proteins. (B) Purified hIL12-Fc proteins were analyzed by SDS-PAGE under either a reducing ('R') or non-reducing ('NR') condition. The arrows indicate the eluted position of the proteins. Representative images from three independent experiments are shown. (C) Size exclusion elution profiles of purified hIL12-Fc proteins (100 μL of 500 μg/mL) were monitored at 280 nm. Representative chromatogram from three independent experiments are shown. (D) Binding activities of 1 μM of hIL12-Fc proteins on the surface of resting and PHA-activated PBMCs, determined by flow cytometric analysis. (E) Proliferation of PHA-activated PBMCs after 72-h culture with the indicated concentrations of proteins, presented in the equimolar concentration of rhIL12. Representative results from three independent experiments performed in triplicates are shown as the means ± SD. ***p < 0.001 versus rmIL12 determined by Newman-Keuls multiple comparison post-hoc test. (F, G) Effect of cross-linked hIL12-Fc on the expression of pSTAT4 (F) and T-bet (G) in human CD8+ T cells after 2 h (F) or 72 h (G) of culture with plate-bound anti-CD3 antibody (1 μg/mL) and soluble anti-CD28 antibody (0.5 μg/mL) in the presence of 20 pM of hIL12-Fc alone or in combination with anti-Fc antibody at the indicated molar ratios, analyzed by flow cytometry. Results are depicted as the mean ± SEM compiled from two independent experiments. ***p < 0.001 versus bi-hIL12-Fc determined by Student's t test.
Discussion
In this study, we exploited heterodimeric Fc technology to develop mono-mIL12-Fc, which presents mIL12 in the naturally occurring heterodimeric form with an Fc-mediated extended serum half-life. We demonstrated that mono-mIL12-Fc possesses a prolonged serum half-life and elicits enhanced antitumor effects of mIL12 to eradicate large established tumors in syngeneic mouse models, as compared with the bivalent fusion format of bi-mIL12-Fc. These effects were associated with the enhanced proliferation and tumor infiltration of CD4+ and CD8+ T cells without significant systemic toxicity. In addition, mono-mIL12-Fc was more efficacious to augment secretion of IFN-γ as well as TNF-α and IL2 from CD8+ and CD4+ effector T cells and potentiate the cytotoxic activities of NK cells and CD8+ T cells than bi-mIL12-Fc. More importantly, mono-mIL12-Fc, but not bi-mIL12-Fc, stimulated the generation of functional memory CD8+ T cells. Notably, our study illustrates that mono-mIL12-Fc triggers modest pSTAT4 activation and T-bet expression in effector CD8+ T cells, resulting in a switch from T-bet to Eomes activation for the differentiation into MPECs and eventually long-lived memory CD8+ T cells (Fig. S11). However, the strong IL12 signaling mediated by bi-mIL12-Fc drives effector CD8+ T cells into terminally differentiated SLECs, thereby excluding the development of MPECs. To our knowledge, this is the first report to show that the antitumor immunity of IL12 can be mediated through the generation of functional memory CD8+ T cells in addition to previously known mechanisms of augmented IFN-γ production and cytotoxic activities of NK cells and effector CD8+ T cells.
Here, we demonstrated that the extension of the serum half-life of IL12 by heterodimeric Fc fusion is critical to exert the potent antitumor effects by allowing for infrequent injections at low dosages. Twice-weekly systemic injection of mono-mIL12-Fc at doses equivalent to 0.5–2 μg mIL12 efficiently eradicated even the large established tumors (∼300 mm3) () without noticeable systemic toxicities (Fig. S4), although they lack tumor-targeting properties. However, the same regimen of rmIL12 (1 μg) did not show any antitumor activity, even for small-sized tumors (∼100 mm3) (). In previous studies, daily injection or injection every two days of rmIL12 (1–5 μg) was required to exert significant antitumor activity in mice.Citation15,Citation30,Citation31 For the large established tumors, mono-mIL12-Fc performed markedly better than bi-mIL12-Fc at an equimolar dose of less than 2 μg mIL12 (, Figs. S1–3). Although the bi-mIL12-Fc protein contains 2 units of mIL12, it could not mediate two-times greater tumor regression compared to mono-mIL12-Fc. By contrast, 2 μg bi-mIL12-Fc was required to rescue 80% of the mice against large established tumors while 0.5 μg mono-mIL12-Fc was sufficient to show the same effect. This result suggests that mono-mIL12-Fc rather than bi-mIL12-Fc is a suitable format for the systemic administration of rmIL12 to enlarge the therapeutic window.
To date, the antitumor activity of IL12 has been attributed to IFN-γ production,Citation30,Citation31 cytotoxicity by NK cells,Citation37 and/or the proliferation and cytotoxicity of effector CD8+ T cells.Citation2,Citation18,Citation19 Surprisingly, however, the effects of IL12 and IL12-based immunocytokines on the differentiation of effector CD8+ T cells into SLECs and/or MPECs have not been assessed in the context of protective antitumor responses. In addition to the known antitumor mechanisms of IL12, our study newly reports that mono-mIL12-Fc primed effector CD8+ T cells to promote the development of MPECs, resulting in a population of long-lived memory CD8+ T cells that elicit recall protection against subsequent tumor challenge (). Bi-mIL12-Fc was also effective in retarding tumor growth via augmenting the proliferation and tumor infiltration of CD4+ and CD8+ T cells at the effector phase (). However, the bi-mIL12-Fc regimen limited the development of memory CD8+ T cells. This was further supported by the observation that bi-mIL12-Fc treatment resulted in the progressive diminution of IFN-γ production (, Fig. S4C) and the cytotoxicity of CTLs at the end of treatment (). Accordingly, our data emphasize the importance of IL12 fusion to heterodimeric Fc in the native heterodimeric form to induce IL12-mediated protective adaptive immune responses.
Recent studies have shown that the strong IL12 signaling during CD8+ T cell priming dictates a gradient of T-bet and Eomes expression, in which high T-bet expression induces SLECs that eventually undergo apoptosis, whereas low T-bet and high Eomes expression promote differentiation to MPECs and then long-lived memory CD8+ T cells.Citation7,Citation8,Citation41,Citation42 Importantly, we found that mono-mIL12-Fc imposed moderate pSTAT4 activation and T-bet expression in effector CD8+ T cells (, 7F, G), thereby favoring MPEC differentiation (, Fig. S11), to a lesser extent than that observed with bi-mIL12-Fc. Signaling of bi-mIL12-Fc, the bivalent fusion of scIL12 to wild-type Fc, imitated the cross-linking of mono-mIL12-Fc to drive maximal STAT4 activation and T-bet expression in CD8+ T cells (, 7F, G), which explains their differentiation toward SLECs (, Fig. S11) while precluding the development of long-lived memory CD8+ T cells (, C). This poor efficacy of bi-mIL12-Fc to induce long-lived memory CD8+ T cells might also account for the failure of IgG-based IL12 immunocytokines with bivalent IL12 fusion to eradicate large established tumors, despite the potent tumor growth inhibition activity.Citation17-Citation19
An important question is whether the potent antitumor immunity of mono-mIL12-Fc observed in a mouse setting is feasible in humans. The observation that mono-hIL12-Fc also triggered lower levels of pSTAT4 and T-bet expression than bi-hIL12-Fc in human CD8+ T cells suggests that mono-hIL12-Fc is likely to also generate protective adaptive immune responses in humans, as seen with mono-mIL12-Fc in mice. Therefore, mono-hIL12-Fc is worthy of further testing in human trials.
Our observation that bi-IL12-Fc or cross-linked mono-IL12-Fc induces enhanced pSTAT4 and T-bet expression bears a resemblance to the findings for other multi-subunit immune recognition receptors that engage clustered rafts during signaling.Citation43 The cross-linking of IgE bound to FcϵRI on mast cells by oligomeric antigens increased the phosphorylation of immune receptor tyrosine-based activation motifs of FcϵRI followed by raft clustering.Citation44 Artificial cross-linking of T cell receptor (TCR) on T cells by a secondary cross-linking antibody resulted in the generation of multiple TCR complexes to amplify the TCR signaling.Citation45 Although evidence of IL12R oligomeric complex formation remains unclear, clustering of IL12R by bi-IL12-Fc seems to magnify the IL12-IL12R signaling in close proximity (Fig. S11). Accordingly, our study indicates that stimulation of IL12-IL12R in a 1:1 complex by mono-IL12-Fc is optimal to repress pSTAT4 and T-bet expression in effector CD8+ T cells for their differentiation into MPECs, followed by the generation of memory CD8+ T cells (Fig. S11). Our approach of mono-IL12-Fc also provides a rationale for exploiting heterodimeric Fc as an attractive scaffold for the generation of Fc-fused cytokines as well as IgG-based immunocytokines with the native structure of monomeric (e.g., IL2, IL4, IL7, IL9, IL12, and IL21) and heterodimeric (e.g., IL12, IL23, IL27, and IL35) cytokines for anticancer therapy.Citation22,Citation46
In conclusion, we have demonstrated that extending the serum half-life of IL12 in a naturally occurring monovalent form by fusion to heterodimeric Fc can lead to complete rejection of large immunogenic tumors by augmenting the IFN-γ production and cytotoxicity of effector cells as well as through the efficient induction of long-lived memory CD8+ T cells. These results suggest that the mono-mIL12-Fc fusion protein can effectively combine the therapeutic potential of IL12 with the Fc-mediated extended serum half-life and may provide a practical alternative to the systemic administration of IL12 for antitumor therapy.
Materials and methods
Construction, expression, and purification of Fc fusion proteins
The heterodimeric Fc, mono-IL12-Fc and bi-IL12-Fc fusion proteins were constructed as described in the Supplement. The plasmids encoding FcCH3A/FcCH3B, mono-IL12-Fc and bi-IL12-Fc were transiently cotransfected in pairs into 30 ml to 100 ml HEK293F cell cultures in FreeStype 293 media following the standard protocol.Citation25 Fusion proteins were purified from the culture supernatants using Protein-A agarose chromatography column (GE Healthcare).Citation25
Mice
Female Balb/c mice and C57BL/6 mice were purchased from NARA Biotech and aged between 5 and 6 weeks before tumor inoculation. All animal experiments were evaluated under approval of the Animal and Ethics Review Committee of Ajou University (Suwon, Korea) in accordance with the guidelines established by the Institutional Animal Care and Use Committee.
Tumor inoculation and treatment
For induction of CT26 or CT-26Her2/neu tumors, 106 CT26 or CT-26Her2/neu cancer cells in 100 μL of PBS were injected subcutaneously into the flanks of BALB/c mice. For induction of B16F10 tumors, 5 × 105 B16F10 cancer cells in 100 μL of PBS were injected subcutaneously into the back of C57BL/6 mice. Intraperitoneal injection of PBS, carrier-free rmIL12 (1 μg, eBioscience), Fc (equivalent molar amount of 1 μg rmIL12), mono-mIL12-Fc (equivalent molar amount of 0.1-2 μg rmIL12), bi-mIL12-Fc (equivalent molar amount of 0.1-2 μg rmIL12) were done twice a week from the indicated day after tumor inoculation for the indicated periods. Additional details are described in the Supplemental Experimental Procedures.
Flow cytometry
Single cell suspensions were prepared, stained, and analyzed as described in the Supplement.
Statistical analysis
Data are presented as the mean ± SEM for pooled data or the mean ± SD for representative data from two to three independent experiments, unless otherwise specified. All statistical analyses were performed using GraphPad Prism 5 software (GraphPad). Student's t tests and one-way analysis of variance with the Newman-Keuls multiple comparison post-hoc test were performed to determine significance for the in vivo tumor growth experiments. A p value less than 0.05 was considered to indicate statistical significance; the significance levels are indicated in the Figs.
Disclosure of potential conflicts of interests
Y.S.K., J.H.H., and K.J. are listed as inventors on pending patent applications related to the technology described in this work. J.A.K., Y.J.K., and C.H.K. declare no competing financial interests.
supp_data.pdf
Download PDF (8 MB)Acknowledgments
We thank Chan-Yuil Kang for providing the CT26-HER2/neu colon cancer cells. This work was supported by grants from the Pioneer Research Center Program (2014M3C1A3051470) and the Global Frontier Project (2013M3A6A4043874) from the National Research Foundation (NRF), and the Korea Health Technology R&D Project (HI16C0992) through the Korea Health Industry Development Institute (KHIDI), funded by the Republic of Korea.
Additional information
Funding
References
- Tugues S, Burkhard SH, Ohs I, Vrohlings M, Nussbaum K, Vom Berg J, Kulig P, Becher B. New insights into IL-12-mediated tumor suppression. Cell Death Differ 2015;22:237–46. doi:10.1038/cdd.2014.134. PMID:25190142.
- Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol 2003;3:133–46. doi:10.1038/nri1001. PMID:12563297.
- Vignali DA, Kuchroo VK. IL-12 family cytokines: immunological playmakers. Nat Immunol 2012;13:722–8. doi:10.1038/ni.2366. PMID:22814351.
- Teng MW, Bowman EP, McElwee JJ, Smyth MJ, Casanova JL, Cooper AM, Cua DJ. IL-12 and IL-23 cytokines: from discovery to targeted therapies for immune-mediated inflammatory diseases. Nat Med 2015;21:719–29. doi:10.1038/nm.3895. PMID:26121196.
- Kaech SM, Cui W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat Rev Immunol 2012;12:749–61. doi:10.1038/nri3307. PMID:23080391.
- Lazarevic V, Glimcher LH, Lord GM. T-bet: a bridge between innate and adaptive immunity. Nat Rev Immunol 2013;13:777–89. doi:10.1038/nri3536. PMID:24113868.
- Takemoto N, Intlekofer AM, Northrup JT, Wherry EJ, Reiner SL. Cutting Edge: IL-12 inversely regulates T-bet and eomesodermin expression during pathogen-induced CD8+ T cell differentiation. J Immunol 2006;177:7515–9. doi:10.4049/jimmunol.177.11.7515. PMID:17114419.
- Joshi NS, Cui W, Chandele A, Lee HK, Urso DR, Hagman J, Gapin L, Kaech SM. Inflammation directs memory precursor and short-lived effector CD8(+) T cell fates via the graded expression of T-bet transcription factor. Immunity 2007;27:281–95. doi:10.1016/j.immuni.2007.07.010. PMID:17723218.
- Colombo MP, Trinchieri G. Interleukin-12 in anti-tumor immunity and immunotherapy. Cytokine Growth Factor Rev 2002;13:155–68. doi:10.1016/S1359-6101(01)00032-6. PMID:11900991.
- Atkins MB, Robertson MJ, Gordon M, Lotze MT, DeCoste M, DuBois JS, Ritz J, Sandler AB, Edington HD, Garzone PD, et al. Phase I evaluation of intravenous recombinant human interleukin 12 in patients with advanced malignancies. Clin Cancer Res 1997;3:409–17. PMID:9815699.
- Leonard JP, Sherman ML, Fisher GL, Buchanan LJ, Larsen G, Atkins MB, Sosman JA, Dutcher JP, Vogelzang NJ, Ryan JL. Effects of single-dose interleukin-12 exposure on interleukin-12-associated toxicity and interferon-gamma production. Blood 1997;90:2541–8. PMID:9326219.
- Lasek W, Zagozdzon R, Jakobisiak M. Interleukin 12: still a promising candidate for tumor immunotherapy? Cancer Immunol Immunother 2014;63:419–35. doi:10.1007/s00262-014-1523-1. PMID:24514955.
- Kiefer JD, Neri D. Immunocytokines and bispecific antibodies: two complementary strategies for the selective activation of immune cells at the tumor site. Immunol Rev 2016;270:178–92. doi:10.1111/imr.12391. PMID:26864112.
- Gafner V, Trachsel E, Neri D. An engineered antibody-interleukin-12 fusion protein with enhanced tumor vascular targeting properties. Int J Cancer 2006;119:2205–12. doi:10.1002/ijc.22101. PMID:16823838.
- Halin C, Rondini S, Nilsson F, Berndt A, Kosmehl H, Zardi L, Neri D. Enhancement of the antitumor activity of interleukin-12 by targeted delivery to neovasculature. Nat Biotechnol 2002;20:264–9. doi:10.1038/nbt0302-264. PMID:11875427.
- Pasche N, Wulhfard S, Pretto F, Carugati E, Neri D. The antibody-based delivery of interleukin-12 to the tumor neovasculature eradicates murine models of cancer in combination with paclitaxel. Clin Cancer Res 2012;18:4092–103. doi:10.1158/1078-0432.CCR-12-0282. PMID:22693354.
- Fallon J, Tighe R, Kradjian G, Guzman W, Bernhardt A, Neuteboom B, Lan Y, Sabzevari H, Schlom J, Greiner JW. The immunocytokine NHS-IL12 as a potential cancer therapeutic. Oncotarget 2014;5:1869–84. doi:10.18632/oncotarget.1853. PMID:24681847.
- Peng LS, Penichet ML, Morrison SL. A single-chain IL-12 IgG3 antibody fusion protein retains antibody specificity and IL-12 bioactivity and demonstrates antitumor activity. J Immunol 1999;163:250–8. PMID:10384123.
- Helguera G, Rodriguez JA, Penichet ML. Cytokines fused to antibodies and their combinations as therapeutic agents against different peritoneal HER2/neu expressing tumors. Mol Cancer Ther 2006;5:1029–40. doi:10.1158/1535-7163.MCT-05-0488. PMID:16648575.
- Starbeck-Miller GR, Xue HH, Harty JT. IL-12 and type I interferon prolong the division of activated CD8 T cells by maintaining high-affinity IL-2 signaling in vivo. J Exp Med 2014;211:105–20. doi:10.1084/jem.20130901. PMID:24367005.
- Schoenhaut DS, Chua AO, Wolitzky AG, Quinn PM, Dwyer CM, McComas W, Familletti PC, Gately MK, Gubler U. Cloning and expression of murine IL-12. J Immunol 1992;148:3433–40. PMID:1350290.
- Ha JH, Kim JE, Kim YS. Immunoglobulin Fc Heterodimer Platform Technology: From Design to Applications in Therapeutic Antibodies and Proteins. Front Immunol 2016;7:394. doi:10.3389/fimmu.2016.00394. PMID:27766096.
- Choi HJ, Kim YJ, Choi DK, Kim YS. Engineering of Immunoglobulin Fc Heterodimers Using Yeast Surface-Displayed Combinatorial Fc Library Screening. PLoS One 2015;10:e0145349. doi:10.1371/journal.pone.0145349. PMID:26675656.
- Overdijk MB, Verploegen S, Ortiz Buijsse A, Vink T, Leusen JH, Bleeker WK, Parren PW. Crosstalk between human IgG isotypes and murine effector cells. J Immunol 2012;189:3430–8. doi:10.4049/jimmunol.1200356. PMID:22956577.
- Choi HJ, Kim YJ, Lee S, Kim YS. A heterodimeric Fc-based bispecific antibody simultaneously targeting VEGFR-2 and Met exhibits potent antitumor activity. Mol Cancer Ther 2013;12:2748–59. doi:10.1158/1535-7163.MCT-13-0628. PMID:24132142.
- Desai BB, Quinn PM, Wolitzky AG, Mongini PK, Chizzonite R, Gately MK. IL-12 receptor. II. Distribution and regulation of receptor expression. J Immunol 1992;148:3125–32.
- Gillies SD, Young D, Lo KM, Roberts S. Biological activity and in vivo clearance of antitumor antibody/cytokine fusion proteins. Bioconjug Chem 1993;4:230–5. doi:10.1021/bc00021a008. PMID:8324014.
- Tzeng A, Kwan BH, Opel CF, Navaratna T, Wittrup KD. Antigen specificity can be irrelevant to immunocytokine efficacy and biodistribution. Proc Natl Acad Sci U S A 2015;112:3320–5. doi:10.1073/pnas.1416159112. PMID:25733854.
- Park S, Jiang Z, Mortenson ED, Deng L, Radkevich-Brown O, Yang X, Sattar H, Wang Y, Brown NK, Greene M, et al. The therapeutic effect of anti-HER2/neu antibody depends on both innate and adaptive immunity. Cancer Cell 2010;18:160–70. doi:10.1016/j.ccr.2010.06.014. PMID:20708157.
- Brunda MJ, Luistro L, Warrier RR, Wright RB, Hubbard BR, Murphy M, Wolf SF, Gately MK. Antitumor and antimetastatic activity of interleukin 12 against murine tumors. J Exp Med 1993;178:1223–30. doi:10.1084/jem.178.4.1223. PMID:8104230.
- Nastala CL, Edington HD, McKinney TG, Tahara H, Nalesnik MA, Brunda MJ, Gately MK, Wolf SF, Schreiber RD, Storkus WJ, et al. Recombinant IL-12 administration induces tumor regression in association with IFN-gamma production. J Immunol 1994;153:1697–706. PMID:7913943.
- Seliger B, Wollscheid U, Momburg F, Blankenstein T, Huber C. Characterization of the major histocompatibility complex class I deficiencies in B16 melanoma cells. Cancer Res 2001;61:1095–9. PMID:11221838.
- Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science 2011;331:1565–70. doi:10.1126/science.1203486. PMID:21436444.
- Cerwenka A, Lanier LL. Natural killer cell memory in infection, inflammation and cancer. Nat Rev Immunol 2016;16:112–23. doi:10.1038/nri.2015.9. PMID:26806484.
- Hong S, Lee H, Jung K, Lee SM, Lee SJ, Jun HJ, Kim Y, Song H, Bogen B, Choi I. Tumor cells loaded with alpha-galactosylceramide promote therapeutic NKT-dependent anti-tumor immunity in multiple myeloma. Immunol Lett 2013;156:132–9. doi:10.1016/j.imlet.2013.10.002. PMID:24148970.
- Cullen SP, Brunet M, Martin SJ. Granzymes in cancer and immunity. Cell Death Differ 2010;17:616–23. doi:10.1038/cdd.2009.206. PMID:20075940.
- Parihar R, Dierksheide J, Hu Y, Carson WE. IL-12 enhances the natural killer cell cytokine response to Ab-coated tumor cells. J Clin Invest 2002;110:983–92. doi:10.1172/JCI0215950. PMID:12370276.
- Ahlers JD, Belyakov IM. Memories that last forever: strategies for optimizing vaccine T-cell memory. Blood 2010;115:1678–89. doi:10.1182/blood-2009-06-227546. PMID:19903895.
- Bachmann MF, Wolint P, Schwarz K, Jager P, Oxenius A. Functional properties and lineage relationship of CD8+ T cell subsets identified by expression of IL-7 receptor alpha and CD62L. J Immunol 2005;175:4686–96. doi:10.4049/jimmunol.175.7.4686. PMID:16177116.
- Huster KM, Busch V, Schiemann M, Linkemann K, Kerksiek KM, Wagner H, Busch DH. Selective expression of IL-7 receptor on memory T cells identifies early CD40L-dependent generation of distinct CD8+ memory T cell subsets. Proc Natl Acad Sci U S A 2004;101:5610–5. doi:10.1073/pnas.0308054101. PMID:15044705.
- Kohlhapp FJ, Zloza A, O'Sullivan JA, Moore TV, Lacek AT, Jagoda MC, McCracken J, Cole DJ, Guevara-Patiño JA. CD8(+) T cells sabotage their own memory potential through IFN-gamma-dependent modification of the IL-12/IL-15 receptor alpha axis on dendritic cells. J Immunol 2012;188:3639–47. doi:10.4049/jimmunol.1101580. PMID:22430740.
- Zhang X, Starnbach MN. An Excess of the Proinflammatory Cytokines IFN-gamma and IL-12 Impairs the Development of the Memory CD8+ T Cell Response to Chlamydia trachomatis. J Immunol 2015;195:1665–75. doi:10.4049/jimmunol.1500457. PMID:26179901.
- Simons K, Toomre D. Lipid rafts and signal transduction. Nat Rev Mol Cell Biol 2000;1:31–9. doi:10.1038/35036052. PMID:11413487.
- Geha RS, Jabara HH, Brodeur SR. The regulation of immunoglobulin E class-switch recombination. Nat Rev Immunol 2003;3:721–32. doi:10.1038/nri1181. PMID:12949496.
- Smith JA, Tso JY, Clark MR, Cole MS, Bluestone JA. Nonmitogenic anti-CD3 monoclonal antibodies deliver a partial T cell receptor signal and induce clonal anergy. J Exp Med 1997;185:1413–22. doi:10.1084/jem.185.8.1413. PMID:9126922.
- Klein C, Waldhauer I, Nicolini VG, Freimoser-Grundschober A, Nayak T, Vugts DJ, Dunn C, Bolijn M, Benz J, Stihle M, et al. Cergutuzumab amunaleukin (CEA-IL2v), a CEA-targeted IL-2 variant-based immunocytokine for combination cancer immunotherapy: Overcoming limitations of aldesleukin and conventional IL-2-based immunocytokines. Oncoimmunology 2017;6:e1277306. doi:10.1080/2162402X.2016.1277306. PMID:28405498.