
ABSTRACT
Purpose: Tumor development has been closely linked to tumor microenvironment, particularly in terms of myeloid-derived suppressive cells (MDSCs), a heterogeneous population of immature myeloid cells that protect tumors from elimination by immune cells. Approaches aimed at blocking MDSC accumulation could improve cancer clinical outcome.
Experimental Design: We investigated that metformin suppressed MDSC migration to inhibit cancer progression. Primary tumor tissues were incubated with metformin, and proinflammatory chemokine production was measured. To study MDSC chemotaxis in vivo, BALB/C nude mice were injected subcutaneously with TE7 cells and treated with metformin. Migration of adoptively transferred MDSCs was analyzed using flow cytometry and immunohistochemistry.
Results: The frequency of tumor-infiltrated polymorphonuclear (PMN)-MDSCs was increased compared to their circulating counterparts. There was a significant correlation between PMN-MDSCs accumulation in tumors and ESCC prognosis. Moreover, PMN-MDSCs displayed immunosuppressive activity in vitro. Treatment with metformin reduced MDSC migration in patients. Metformin inhibited CXCL1 secretion in ESCC cells and tumor xenografts by enhancing AMPK phosphorylation and inducing DACH1 expression, leading to NF-κB inhibition and reducing MDSC migration. Knockdown of AMPK and DACH1 expression blocked the effect of metformin on MDSC chemotaxis.
Conclusions: A novel anti-tumor effect of metformin, which is mediated by reducing PMN-MDSC accumulation in the tumor microenvironment via AMPK/DACH1/CXCL1 axis.
Introduction
Recent therapeutic approaches that use negative checkpoint inhibitors (antibodies against PD-1/PD-L1 or CTLA4) and CAR-T (Chimeric Antigen Receptor T-Cell) or TCR-T (T cell receptor T-Cell) cell-based adoptive transfer have brought new optimism to tumor therapy. However, some patients with advanced solid tumors often do not respond to these treatments and/or the tumor relapses.Citation1,Citation2 A number of studies have suggested that this could be due to the development of an immunosuppressive microenvironment, consisting of various lymphoid and myeloid cells. In particular, persistence of myeloid derived suppressive cells (MDSCs), the heterogeneous immature myeloid cells which consist of a monocytic (M-MDSC) subset and a polymorphonuclear (PMN-MDSC) subset have been shown to support tumor growth and metastasis.Citation3,Citation4
Chemokines have been shown to play a significant role in MDSC migration.Citation5-Citation7 In particular, CXCL12/CXCR4 and CCL2/CCR2 have been reported to recruit M-MDSCs in colorectal cancer, prostate cancer, and breast cancer.Citation8-Citation10 Recent studies demonstrated that CXCL5/CXCR2, CXCL8/CXCR2, CXCL6/CXCR6, and CCL15 drive PMN-MDSC migration in melanoma, non-small cell lung cancer, head and neck squamous cell carcinoma, and colorectal cancer.Citation11-Citation14 Abnormal expression of specific genes in tumor cells create a characteristic patterns of chemokine secretion in tumor cells. In colorectal cancer, absence of SMAD4 promotes CCL15 expression and induces accumulation of CCR1-expressing MDSCs, which in turn promotes cancer progression.Citation13 A correlation between MDSC infiltration, tumor stage, overall survival and therapeutic effectiveness has been described in some solid tumors such as breast cancer, pancreatic ductal adenocarcinoma, colorectal cancer, liver cancer and bladder cancer.Citation10,Citation11,Citation15-Citation18 Our previous studies revealed a strong correlation between MDSCs and poor prognosis in lung cancer cases.Citation19 Importantly, MDSC accumulation has been shown to reduce the therapeutic effect of adoptive transferred cells, and promote resistance to target therapies in melanoma.Citation20 Therefore, reducing MDSC accumulation in the tumor microenvironment may help to reset the immunosuppressive tumor microenvironment and improve the response to immunotherapy.
In the present study, we investigated the relationship between MDSCs and esophageal squamous cell carcinoma (ESCC) prognosis. We demonstrated that metformin inhibits the accumulation of MDSCs in tumors, and acts via the AMPK-DACH1-CXCL1 axis. Our results suggest that combining metformin with current ESCC treatments could improve the prognosis of patients by remodeling the tumor microenvironment.
Materials and methods
Clinical samples and cell lines
Tumor and peripheral blood samples were collected from patients with ESCC with confirmed histopathological results during surgical resection at the department of Thoracic Surgery, the First Affiliated Hospital of Zhengzhou University. Formalin-fixed, paraffin-embedded tissue sections (5-μm thick) were provided by the department of Pathology, the First Affiliated Hospital of Zhengzhou University. All patients enrolled had not received any treatment such as chemotherapy, radiotherapy and others for esophageal cancer before surgery. Of the 75 patients who applied clinical samples in the experiments of flow-cytometry, 7 had been suffering from diabetes with 5 of them being treated with metformin and 13 had been suffering from hypertension and treated with aspirin or nifedipine. All the patients mentioned above had received sustained treatment before surgery except the tuberculous pleurisy patient, however, no one had been given any method to cure esophageal carcinoma. Human esophageal squamous cancer cells were cultured in RMPI-1640 supplemented with 10% fetal bovine serum (both from Hyclone, Logan, UT, USA).
Study approved : Informed consent was obtained for each patient, and sample collection was approved by the local ethics committee (Approval number: 2014–10).
Establishment of knock down ESCC cells
Small interfering RNA (si-RNA) was used to temporarily downregulate AMPK expression. si-RNA sequences are referenced to the BLOCK-iT™ RNAi Designer in Thermo Fisher Scientific (http://rnaidesigner.thermofisher.com/rnaiexpress/). Oligonucleotides encoding short hairpin RNAs (shRNAs) targeting DACH1 and a non-target control were inserted into a GV248 plasmid using EcoRI and AgeI restriction sites. Plasmids were stably transfected into TE7 cell lines using lenti-viral infection.
Antibodies and human recombinant proteins
The following anti-human monoclonal antibodies were used for flow cytometry: CD33, CD11b, CD14, 7-AAD, granzyme-B and perforin were obtained from Biolegend, USA. And HLA-DR, CD3 and CD8 were from BD Biosciences. Cell fluorescent dye CFSE was obtained from Sigma-Aldrich (#21888). The following anti-human mAbs were used for immunohistochemistry: CD33 (ZSGB-BIO, #ZM-0045), CXCL1 (Proteintech, #12335-1-AP), DACH1 (Proteintech, #10914-1-AP), and phosphorylated AMPK (CST, #2535). The NF-κB inhibitor QNZ (EVP4593) was purchased from Selleck (S4902), CXCL1 recombinant protein from Peprotech (300–11), and metformin from Sigma-Aldrich (D150959).
Animal studies
TE7 cells (1× 106) were injected subcutaneously into BALB/c nude mice. After seven days, a dose of 50 mg/kg metformin or PBS was administered orally daily.Citation42–Citation44 PMN-MDSCs were injected into the animals intravenously. Forty-eight hours later, tumors and spleens were harvested for immunohistology and flow cytometry analysis. Tumor size was measured with a caliper and calculated using the formula: (length × width)Citation2 × 0.5 (mm3).
Study approved : Animal protocols were approved by the Review Board of the First Affiliated Hospital of Zhengzhou University and Henan Academy of Medical Sciences (Approval number:00013150).
Quantitative real-time PCR (qRT-PCR)
Total RNA was extracted from clinical samples and cell lines using Trizol reagent (Invitrogen Life Technologies, Carlsbad, CA, USA). cDNA was prepared using a PrimeScript RT reagent kit with gDNA Eraser (Takara, Shiga, Japan). qRT-PCR was performed using specific primers and SYBR Green qPCR Master Mix (Takara). GAPDH was used as an internal control. We compared expression levels of clinical samples and cell lines using the 2-ΔΔCt method.
Immunohistochemistry
Formalin-fixed, paraffin-embedded tissue sections were treated with xylene and rehydrated through a series of decreasing alcohol concentrations. Antigen retrieval was performed by boiling the sections in 0.1mol/L citrate buffer. Endogenous peroxidase activity was suppressed by incubating in 3% H2O2 for 30 minutes. Sections were stained overnight with primary antibodies at 4°C. Samples were further stained with horseradish peroxidase-conjugated secondary antibodies for 30 minutes at room temperature. Visualization was performed using substrate 3,3-Diaminobenzidine (DAB). Samples were counter-stained with hematoxylin (DingGUO, China), rinsed in running tap water, and dehydrated through an alcohol series followed by two consecutive xylene washes. Sections were prepared for microscopic examination and scored using Leica Application Suite Version 3 software.
Isolation of PMN-MDSCs and CD8+ T cells
Isolation of CD14−CD11b+ PMN-MDSCs from the peripheral blood of ESCC patients was performed using CD14 and CD11b magnetic beads according the manufacturer's instructions (Miltenyi Biotech). CD14− cells were collected and prepared for CD11b+ cell isolation. The remainder of the cell suspension from the same patients was used to purify CD8+ T cells.
PMN-MDSC immunosuppressive assay
Isolated CD8+ T cells, numbered as 105, were labeled with CFSE and washed twice with precooled PBS. CFSE-labeled CD8+ T cells were stimulated with CD3/CD28 beads and IL-2, and co-cultured with purified PMN-MDSCs for 48 hours. Analysis was performed using flow cytometry. And the ratio of PMN-MDSCs versus CD8+ T cells was 0:1, 1:1, and 2:1 respectively. In the function experiments, 5 × 105 CD8+ T cells were incubated with different numbers of MDSCs for 48 hours. In addition, Granzyme-B and perforin production was detected using flow cytometry.
PMN-MDSC migration assay
Freshly isolated PMN-MDSCs from peripheral blood were seeded onto the upper chamber of a 24-well plate. Tumor cell TE7, recombinant CXCL1, metformin or Compound-C were added to the lower chamber. PMN-MDSCs that had migrated into the lower chamber were counted after 6, 12 or 24 hours.
ELISA and western blot
CXCL1 protein was detected in conditional supernatants from TE-7 cells and serum from clinical samples using a Quantikine Human CXCL1/GRO alpha ELISA kit (R&D Systems). Whole lysates from cells or tumor tissues were extracted using RIPA lysis buffer supplemented with a protease inhibitor cocktail (Sigma-Aldrich, P8340) and phosphatase inhibitor cocktail 2 (Sigma-Aldrich, P5726). Lysates from nucleus and cytoplasm were extracted with the kit from Beyotime (P0028). Samples were incubated with appropriate antibodies as phosphorylated AMPK (CST, #2535), total AMPK (CST, #2532), phosphorylated NF-κB (CST, #3033), total NF-κB (CST, #3034), DACH1 (Abcam, #ab77234), TBP (CST, #44059) and imaged using ECL (Thermo Fisher, #34079) according to the manufacturer's protocols.
Multi-factor detection assay
A LEGENDplex™ Human Proinflammatory Chemokine Panel (13-plex, BioLegend, #740003) kit was utilized in these experiments; supernatants from tumor tissues were collected and centrifuged prior to analysis. Results were normalized to standards.
TCGA database analysis
RNA-sequence files related to esophageal squamous cancer samples were downloaded from the The Cancer Genome Atlas (TCGA) data portal (2015). RNA sequencing of 93 esophageal squamous cell carcinoma cases in were obtained from TCGA,(https://gdc-portal.nci.nih.gov/exploration?filters=∼%28op∼%27and∼content∼%28∼%28op∼%27in∼content∼%28field∼%27cases.primary_site∼value∼%28∼%27Esophagus%29%29%29%29%29) and clinical data were obtained from cBioPortal (http://www.cbioportal.org/study?id = esca_tcga#clinical).
Statistics
Data were analyzed using paired or unpaired student's t-test and expressed as mean ± SD. Correlation analysis was performed with parametric or non-parametric tests. A P-value less than 0.05 was considered significant. Survival was evaluated using Kaplan Meier analysis. All statistics and graph preparations were performed using Prism6 (Graph Pad Software Inc.).
Results
Immunological and clinical impact of PMN-MDSCs in ESCC
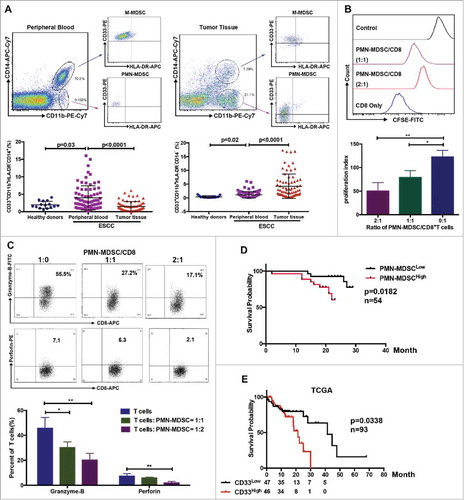
Using multicolor flow cytometry, we observed a significant increase of M-MDSCs and PMN-MDSCs in the peripheral blood of patients with ESCC compared to healthy donors (). Importantly, the infiltration of PMN-MDSCs, but not M-MDSCs, increased in tumor tissues compared to peripheral blood samples (). Further functional analysis showed that purified PMN-MDSCs suppressed CD8+ T cell proliferation as well as granzyme-B and perforin expression in a dose-dependent manner ( and ). Next, we investigated the clinical importance of PMN-MDSCs in patients with ESCC. Using flow cytometry, we demonstrated a significant correlation between PMN-MDSC accumulation in tumor tissues and clinical parameters (Supplementary Table 1). We found that overall survival was reduced in patients with high PMN-MDSC infiltration (, P = 0.0182, n = 54). Moreover, TCGA database demonstrated that patients with high CD33 expression had a worse prognosis (, P = 0.038, n = 93).
Figure 1. Distribution of MDSCs in peripheral blood and tumor samples from patients with ESCC. (A) Single cell suspensions were prepared from peripheral blood and tumor tissues from patients with ESCC (n = 75) or healthy donors (n = 15), and analyzed using flow cytometry. (B, C) Inhibition of T cell proliferation and function were assessed using flow cytometry. (D) Clinical samples were divided into two groups according to accumulation of PMN-MDSCs in tumor tissue, and overall survival was analyzed (n = 54). (E) Survival of patients correlated with CD33 expression from TCGA database (n = 93), as a PMN-MDSC marker. *P < 0.05; **P < 0.01.
Reprogramming of the tumor microenvironment by metformin via suppression of PMN-MDSC accumulation
We performed a comparative analysis of PMN-MDSCs in tumors versus peripheral blood from patients with ESCC, and identified eight patients with lower PMN-MDSC frequencies in tumor tissues compared to peripheral blood samples (). Interestingly, five of the eight patients suffered from diabetes and were being treated with metformin (). We therefore proposed that metformin may be able to reset the tumor microenvironment by inhibiting PMN-MDSC accumulation.
Figure 2. PMN-MDSC accumulation regulated by metformin. (A) Paired analysis of PMN-MDSC frequency in peripheral blood and tumor tissues. (B) Clinical characteristics of eight patients showing higher PMN-MDSC frequency in peripheral blood than in tumor samples. (C) Comparative analysis of PMN-MDSC distribution in patients with ESCC treated with metformin. Results presented as ratio between tumor-infiltrating and circulating MDSCs. Expression of CD33 (D), Arg-1, iNOS, and NOX (E) in tumor tissues from patients treated with metformin was measured using qRT-PCR. (F) CD33 expression in tumor samples was analyzed using immunohistochemistry. (G) Overall survival of patients with ESCC, with or without diabetes, treated or not-treated with metformin shown as a Kaplan-Meier curve. Statistical significance determined by a log-rank test. *P < 0.05; **P < 0.01.
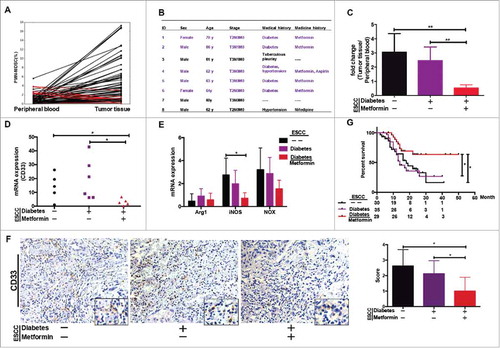
To test this hypothesis, we reanalyzed PMN-MDSC infiltration in these patients. The ratio of PMN-MDSCs in tumor tissue versus peripheral blood was indeed significantly lower in patients with ESCC and diabetes and were undergoing metformin treatment than in other two ESCC groups, which included patients with ESCC but without diabetes and those with diabetes who were not receiving metformin (). Moreover, we showed that CD33 and iNOS expression was significantly lower in tumor samples from patients being treated with metformin than in the other groups of ESCC (, and ). A follow-up of 94 patients with ESCC revealed that the overall survival of patients with ESCC and diabetes and being treated with metformin before surgery was significantly longer than the ESCC only or suffered to diabetes (). Therefore, we speculated that metformin may improve the prognosis of patients with ESCC by reducing the accumulation of PMN-MDSCs in the tumor microenvironment.
Metformin suppresses PMN-MDSC migration by reducing CXCL1 production in ESCC cells
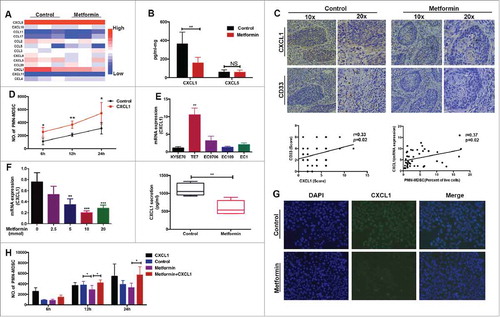
To elucidate the mechanism by which metformin reprograms the tumor microenvironment, we investigated its effect on chemokine secretion in supernatants from esophageal tumor tissues using a human proinflammatory chemokine assay. Production of CXCL1 and CXCL5, which drive PMN-MDSC recruitment,Citation11,Citation21 was decreased following treatment with metformin (). However, only CXCL1 secretion was significantly reduced in tumor tissues after metformin treatment (). Therefore, we further investigated the mechanism underlying the metformin-induced reduction in CXCL1 production and inhibition of PMN-MDSC migration into the tumor microenvironment. Using consecutive sections of ESCC tissues, we found a positive correlation between CXCL1 expression and PMN-MDSC infiltration (). In addition, migration assays demonstrated that CXCL1 increased PMN-MDSC recruitment in vitro (). Furthermore, we measured CXCL1 expression in five ESCC cell lines and found that TE7 cells expressed the highest levels (). Similar to tumor tissues, treatment of TE7 cells with metformin resulted in a decrease in CXCL1 expression in a dose-dependent manner (). These data were confirmed using immunofluorescence staining (). Migration experiments demonstrated that metformin suppressed PMN-MDSC recruitment, which could be reversed by the addition of CXCL1 (). Thus, metformin resets the tumor microenvironment by suppressing CXCL1 secretion, leading to a reduction in PMN-MDSC accumulation in tumor tissues.
Figure 3. Metformin inhibits PMN-MDSC recruitment via CXCL1. (A) Chemokine profiles was performed in supernatants of esophageal tumors from patients treated or not treated with metformin. Results were normalized to tumor weight. (B) CXCL1 and CXCL5 production in tumor samples detected by ELISA, expressed in pg/ml-mg. (C) Expression of CXCL1 and CD33 in consecutive tumor sections was detected using immunohistochemistry (top). CD33 expression plotted against CXCL1 expression (both detected by immunohistochemistry) or CXCL1 measured using qRT-PCR plotted against frequency of PMN-MDSCs detected by flow cytometry (bottom). Correlation was evaluated using a linear regression analysis. (D) Migration of PMN-MDSCs was assessed using a transwell assay after incubation with human recombinant CXCL1 protein. Data are expressed as number of migrated cells. (E) CXCL1 expression was measured in different ESCC cell lines using qRT-PCR. (F, G) TE-7 tumor cells were incubated with metformin and CXCL1 expression was measured by qRT-PCR, ELISA (F), and immunofluorescence (G). (H) TE-7 cells were cultured in the lower chamber with metformin and CXCL1. Migration of PMN-MDSCs was assessed using a transwell assay. *P < 0.05; **P < 0.01.
p-AMPK activation mediates the metformin-induced reduction in PMN-MDSC migration
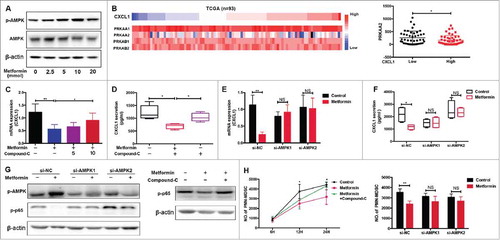
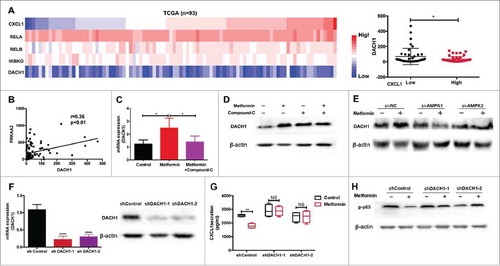
It has been reported that activation of AMP-activated protein kinase (AMPK) plays a key role in the anti-tumor effects of metformin, through modulation of CXCL1 and CXCL12 production.Citation22-Citation24. In the current study, we showed that metformin promotes AMPK phosphorylation in a dose-dependent manner (). Immunohistochemical analysis confirmed enhancement of AMPK phosphorylation in TE7 cells by metformin (Supplementary Fig. 1A). PRKAA1/2 and PRKAB1/2, as the catalytic subunit of AMPK, play a key role in the activation of AMPK signaling pathway.Citation25 We analyzed the TCGA database (ESCC, n = 93) and found that PRKAA2 expression was significantly decreased in the group with high CXCL1 expression levels (), and had a negative correlation with CXCL1 expression (Supplementary Fig. 1B). Therefore, we suggested that p-AMPK activation could have important implications for CXCL1 expression. When we added Compound C, an AMPK pathway inhibitor, to TE7 cells treated with metformin we observed a significant reversal of CXCL1 expression ( and ). To further confirm the effect of AMPK on CXCL1 secretion, we blocked AMPK activity in TE7 cells using siRNA targeting PRKAA2 (Supplementary Fig. 1C). Consistent with our hypothesis, the inhibitory effect of metformin was markedly restored upon treatment with si-AMPK ( and ). A significant downregulation in CXCL1 expression was also observed when cells were treated with QNZ, an inhibitor of the AMPK downstream molecule, NF-κB (Supplementary Fig. 1D). Furthermore, we found that metformin prevented activation of the NF-κB pathway in vitro, which was reversed by AMPK inhibition (). Migration assays demonstrated that metformin inhibition of PMN-MDSC recruitment is dependent on the AMPK pathway (). Therefore, metformin suppresses the accumulation of PMN-MDSCs by activating AMPK and suppressing CXCL1 secretion.
Figure 4. Metformin downregulates CXCL1 expression through AMPK activation. Dose-dependent effect of metformin on AMPK phosphorylation in TE-7 cells was detected by western blotting. (B) TCGA data showing CXCL1 expression together with PRKAA1/2, PRKAB1/2, and PRKAA2. (C-F) After treated with metformin and Compound-C or siRNA-AMPK, CXCL1 expression was measured by qRT-PCR and ELISA. (G) Cells were incubated with metformin, Compound C or siRNA-AMPK, the activity of pAMPK and NF-κB were measured by western blotting. (H) Treatment with Compound-C and si-AMPK reversed the inhibition of PMN-MDSC migration by metformin, shown in a migration assay. And si-AMPK1 or si-AMPK2 alone did not have significantly inhibitory effects on PMN-MDSCs migration compared with si-NC (P = 0.1278 and P = 0.1136 respectively). *P < 0.05; **P < 0.01.
Metformin prevents PMN-MDSC migration by enhancing DACH1 expression
It was previously shown that DACH1, an important regulator of cell proliferation, directly alters the growth pattern of tumor cells by inhibiting CXCL1, IL-6, and IL-8 production.Citation26,Citation27 Using TCGA data, we found a negative correlation between the expression of DACH1 and CXCL1 expression and NF-κB activation (, Supplementary Fig. 2A), as well as with AMPK pathway activation (). Therefore, we speculated that metformin might inhibit PMN-MDSC accumulation in the tumor microenvironment by upregulating DACH1 expression in tumor cells. As shown in and Supplementary Fig. 2B, metformin enhanced the expression of DACH1 in tumor cells, and this was reversed by blocking AMPK. Most importantly, we found that metformin increased the expression of DACH1 in nucleus (Supplementary Fig. 2C, 2D). To further validate the role of DACH1 in the regulation of the tumor microenvironment by metformin, we knocked down DACH1 expression in TE7 cells using short hairpin RNAs (sh-RNA) (). The inhibitory effect of metformin on CXCL1 secretion was significantly reversed in the sh-DACH1 group compared to controls (). A similar trend was observed in levels of NF-κB phosphorylation (). Thus, in ESCC, metformin suppresses CXCL1 expression in tumor cells, thereby preventing PMN-MDSC migration via AMPK activation and DACH1 upregulation.
Figure 5. DACH1 is required for inhibition of PMN-MDSC migration by metformin. (A) TCGA analysis showing expression of CXCL1, DACH1 and NF-κB (B) Expression of PRKAA2 plotted against DACH1. Correlation was evaluated using a linear regression analysis. (C-E) Cells were treated with metformin, Compound C, or AMPK si-RNA. Expression of DACH1 was measured by qRT-PCR or western blotting. (F-H) TE-7 cells were transfected with short hairpin RNAs to inhibit DACH1 expression. Efficiency of transfection was determined by qRT-PCR and western blotting (F). CXCL1 secretion was measured by ELISA (G) and NF-κB activation by western blotting (H). *P < 0.05; **P < 0.01.
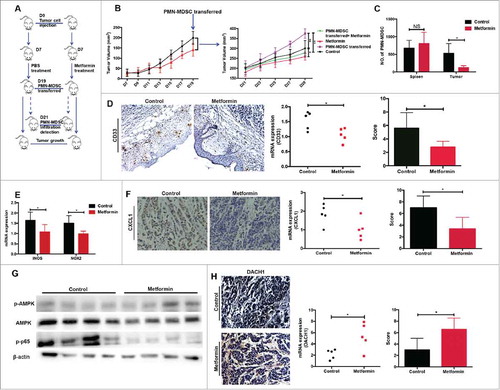
Metformin blocks PMN-MDSC recruitment in vivo
To further investigate the role of metformin in PMN-MDSC migration in vivo, we used a transplanted tumor model of esophageal carcinoma. Tumor-bearing mice were treated with either metformin or PBS for two weeks, followed by intravenous injection of purified PMN-MDSCs from patients with ESCC (). Tumor growth was significantly accelerated in mice injected with PMN-MDSCs compared to animals that did not receive MDSCs (). Moreover, metformin significantly inhibited the stimulating effect of PMN-MDSCs on tumor progression (). Importantly, metformin strongly suppressed PMN-MDSC infiltration into tumor tissues but not into the spleen (). Immunohistochemistry and qRT-PCR analysis showed a significant decrease in CD33 expression in the metformin-treated group (). In addition, metformin significantly reduced the expression of iNOS and NOX, which are associated with PMN-MDSC function (). CXCL1 expression was also decreased in tumor samples from the metformin-treated group (). Finally, in tumor tissues, metformin treatment resulted in an upregulation of AMPK activity and NF-κB inhibition, as detected by western blotting (), as well as an increase in DACH1 expression, as measured using immunohistology (). These data support our model for the mechanism that underlies the therapeutic effects of metformin.
Figure 6. Metformin exerts anti-tumor effects in vivo by inhibiting PMN-MDSC accumulation via the AMPK/DACH1/ CXCL1 axis. (A) Experiment design in vivo. (B) Kinetics of tumor growth in vivo. Tumor volume was measured every second day and expressed in mm3. (C) Accumulation of transferred PMN-MDSCs in tumor-bearing mice treated with metformin was measured using flow cytometry. (D, E) Expression of CD33, iNOS, and NOX2 was measured by qRT-PCR and immunohistochemistry. (F-H) Expression of CXCL1, DACH1 and activation of AMPK or NF-κB in tumor tissues was measured by qRT-PCR, immunohistochemistry and western blotting. *P < 0.05; **P < 0.01.
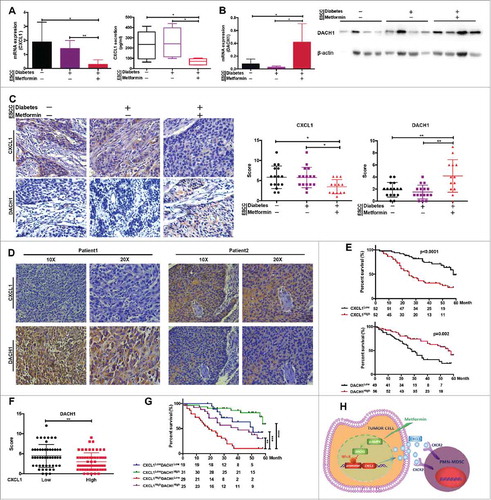
Blocking of PMN-MDSCs by metformin is associated with increased patient survival
To explore the clinical relevance of metformin in resetting the tumor microenvironment and inhibiting tumor progression, we collected tumor samples from patients with ESCC and diabetes that were or were not receiving metformin treatment. These were compared to a separate group comprising tumor samples from patients with ESCC but without diabetes. Treatment with metformin led to a significant decrease in the expression of CXCL1, while DACH1 expression was upregulated (, and ). Importantly, regions of the samples that showed higher CXCL1 expression correlated with areas of lower DACH1 expression, and vice versa (). Furthermore, CXCL1 expression was significantly higher in patients of advanced stage and with lymph node metastasis, while DACH1 expression was markedly decreased (Supplementary Fig. 2). Overall survival was significantly reduced in patients displaying higher CXCL1 expression levels, whereas patients with high levels of DACH1 expression had a better prognosis (). Statistical analysis confirmed that DACH1 expression was significantly inhibited in the group with high CXCL1 expression levels (). The best prognosis was found in patients with low CXCL1 and high DACH1 expression, whereas high CXCL1 and low DACH1 expression was associated with the poorest prognosis (). We therefore suggest that metformin could improve treatment success and prolong the overall survival of patients with ESCC by inhibiting PMN-MDSC accumulation into the tumor microenvironment via regulation of DACH1 expression ().
Figure 7. DACH1/CXCL1 regulation of PMN-MDSC accumulation in patients with ESCC undergoing metformin treatment could improve prognosis. CXCL1 expression was verified in tumor tissues and serum from patients with ESCC using qRT-PCR and ELISA. (B) DACH1 expression in patients with ESCC treated with metformin was measured by qRT-PCR and western blotting. (C) Paired analysis of CXCL1 and DACH1 expression in patients receiving metformin treatment. (D) Two representative ESCC samples showing expression patterns of CXCL1 and DACH1. (E) Overall survival of patients with ESCC showing high and low CXCL1 (n = 104; top) and DACH1 expression (n = 105, bottom), presented as a Kaplan-Meier curve. (F)) Expression of DACH1 and CXCL1 was detected using immunohistochemistry. Data show distribution of DACH1 expression in CXCL1-high and CXCL1-low groups. (G) Overall survival of patients with ESCC displaying differential expression of DACH1 and CXCL1, presented as a Kaplan-Meier curve. (H) Schematic representation of the mechanisms of the anti-tumor effects of metformin. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Discussion
MDSCs promote tumor progression by protecting tumor cells from immune-mediated death and by enhancing their proliferation ability.Citation28 It has been shown that augmented MDSC infiltration in tumor samples from patients with different types of cancer correlates with a bad clinical outcome.Citation11,Citation12,Citation16,Citation29 However, there are only a few reports on the relationship between MDSCs and disease progression in patients with esophageal cancer.Citation30-Citation32 Here, we revealed the mechanism by which MDSCs are recruited into the esophageal tumor site, and found that the hypoglycemic drug metformin prevents MDSC accumulation and resets the tumor microenvironment.
By comparing the distribution of two MDSC subsets in the circulation and tumor tissue from the same 80 patients with ESCC, we found that the frequency of PMN-MDSCs in tumors is significantly higher than in the peripheral blood. These data are in agreement with previous reports for lung, breast, and colorectal cancer.Citation13,Citation33 This could be explained by the possible differentiation of M-MDSCs into dendritic cells or macrophages in the tumor tissue.Citation34,Citation35 Consistent with previous reports,Citation34,Citation36 our experiments demonstrated that PMN-MDSCs inhibit proliferation and cytokine secretion in activated T cells. Subsequent analysis using immunohistochemistry and multicolor flow cytometry revealed that a higher frequency of tumor-infiltrating PMN-MDSCs correlates with a poorer prognosis.
It was recently demonstrated that sunitinib inhibits MDSC recruitment to the tumor site.Citation37,Citation38 In addition, TRAIL-R2 antibody and BMP4 were found to have a considerable inhibitory effect on MDSC accumulation in the tumor microenvironment.Citation10,Citation39 In our investigation, eight patients from the study group showed an opposite effect on PMN-MDSC distribution, in that their frequency in tumor tissue was less than in the circulation. Retrospective information from these patients revealed that five of them had diabetes and were receiving metformin therapy. We therefore hypothesized that metformin could be interfering with the accumulation of MDSCs in the tumor microenvironment. Further examination of the clinical samples revealed that tumor samples from the patients with ESCC that were being treated with metformin contained significantly fewer PMN-MDSCs than those who were not receiving metformin. In addition, overall survival was significantly prolonged in those patients with ESCC that were taking metformin. Therefore, we inferred that metformin could reset the tumor microenvironment and improve the prognosis of patients with ESCC by reducing the accumulation of MDSCs.
The anti-tumor effects of metformin in breast, pancreatic, and ovarian cancer have been reported to be linked to the inhibition of tumor cell growth.Citation40,Citation41 Other studies report the modulation of CD8+ T cells and macrophages following metformin treatment.Citation42-Citation44 In the current study, we demonstrated an important role for metformin blocking CXCL1 production by tumor cells in PMN-MDSC recruitment.
Furthermore, we showed that metformin reprograms the tumor microenvironment by activating AMPK, which inhibits the secretion of CXCL1 and blocks MDSC migration to the tumor site. In addition, we found that AMPK activation inhibits phosphorylation of NF-κB. However, the exact molecular mechanism by which NF-κB activity can suppress PMN-MDSC migration must be clarified. It was recently reported that DACH1, which plays a key role in organ development, can also regulate CXCL1 transcription in combination with NF-κB to promote tumor progression.Citation26,Citation27 Since we found no reports concerning DACH1 regulation of the tumor microenvironment, we explored the role of DACH1 in MDSC recruitment by metformin. Interestingly, metformin did increase the expression of DACH1 by activating AMPK, thereby inhibiting the activity of NF-κB, reducing CXCL1 secretion and blocking MDSC migration to the tumor site. Therefore, our data revealed for the first time a significant role for DACH1 in resetting the tumor microenvironment. The investigation of more than one hundred clinical samples suggested that the combined expression levels of CXCL1 and DACH1 in tumors could be utilized as an important indicator of the prognosis of patients with ESCC. Due to the lack of related reports concerning on the correlation between AMPK and DACH1, our further study will clarify the underlying mechanisms how DACH1 is regulated by AMPK, either via binding the promoter region of DACH1 directly or through activating other signaling pathways.
Taken together, our data have uncovered the mechanism by which metformin regulates MDSC recruitment and resets the tumor microenvironment. Neutralization of the immunosuppressive tumor microenvironment by metformin could be combined with current immunotherapeutic strategies (such as inhibitors of negative immune check-points or adoptive transfer of tumor-reactive T cells) to enhance their therapeutic effects and greatly improve the prognosis of patients.
Disclosures
The authors declare no potential conflicts of interest.
supp_data.zip
Download Zip (1.3 MB)Acknowledgments
This study was supported by the National Natural Science Foundation of China (Grant No. 81171985, 81171986), the Basic and Advanced Technology Research Foundation from Science and Technology Department of Henan Province (Grant No.112300410153, Grant No.122300410155).
Additional information
Funding
References
- Fesnak AD. CHJaBLL. Engineered T cells: the promise and challenges of cancer immunotherapy. Nat Rev Cancer. 2016;23(16):16.
- McAllister SS1 WR. The tumour-induced systemic environment as a critical regulator of cancer progression and metastasis. Nat Cell Biol. 2014;16(8):11.
- Junttila MR, Sauvage FJ. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature. 2013;501(7467):346–54. doi:10.1038/nature12626. PMID:24048067.
- Fridman WH, Pages F, Sautes-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer. 2012;12(4):298–306. doi:10.1038/nrc3245. PMID:22419253.
- Griffith JW, Sokol CL, Luster AD. Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annu Rev Immunol. 2014;32:659–702. doi:10.1146/annurev-immunol-032713-120145. PMID:24655300.
- Ottensmeier CH, Perry KL, Harden EL, Stasakova J, Jenei V, Fleming J, Wood O, Woo J, Woelk CH, Thomas GJ, et al. Upregulated Glucose Metabolism Correlates Inversely with CD8+ T-cell Infiltration and Survival in Squamous Cell Carcinoma. Cancer Res. 2016;76(14):13. doi:10.1158/0008-5472.CAN-15-3121.
- Sawanobori Y, Ueha S, Kurachi M, Shimaoka T, Talmadge JE, Abe J, Shono Y, Kitabatake M, Kakimi K, Mukaida N, et al. Chemokine-mediated rapid turnover of myeloid-derived suppressor cells in tumor-bearing mice. Blood. 2008;111(12):5457–66. doi:10.1182/blood-2008-01-136895. PMID:18375791.
- Obermajer N, Muthuswamy R, Odunsi K, Edwards RP, Kalinski P. PGE(2)-induced CXCL12 production and CXCR4 expression controls the accumulation of human MDSCs in ovarian cancer environment. Cancer Res. 2011;71(24):7463–70. doi:10.1158/0008-5472.CAN-11-2449. PMID:22025564.
- Chun E, Lavoie S, Michaud M, Gallini CA, Kim J, Soucy G, Odze R, Glickman JN, Garrett WS. CCL2 Promotes colorectal carcinogenesis by enhancing polymorphonuclear myeloid-derived suppressor cell population and function. Cell Rep. 2015;12(2):244–57. doi:10.1016/j.celrep.2015.06.024. PMID:26146082.
- Cao Y, Slaney CY, Bidwell BN, Parker BS, Johnstone CN, Rautela J, Eckhardt BL, Anderson RL. BMP4 inhibits breast cancer metastasis by blocking myeloid-derived suppressor cell activity. Cancer Res. 2014;74(18):5091–102. doi:10.1158/0008-5472.CAN-13-3171. PMID:25224959.
- Wang G LX, Dey P, Deng P, Wu CC, Jiang S, Fang Z, Zhao K, Konaparthi R, Hua S, Zhang J, et al. Targeting YAP-Dependent MDSC Infiltration Impairs Tumor Progression. Cancer Discov. 2016;6(1):16. doi:10.1158/2159-8290.CD-15-0224.
- Huang W, Chen Z, Zhang L, Tian D, Wang D, Fan D, Wu K, Xia L. Interleukin-8 induces expression of FOXC1 to promote transactivation of CXCR1 and CCL2 in hepatocellular carcinoma cell lines and formation of metastases in mice. Gastroenterology. 2015;149(4):1053–67 e14. doi:10.1053/j.gastro.2015.05.058. PMID:26065367.
- Inamoto S, Itatani Y, Yamamoto T, Minamiguchi S, Hirai H, Iwamoto M, Hasegawa S, Taketo MM, Sakai Y, Kawada K. Loss of SMAD4 promotes colorectal cancer progression by accumulation of myeloid-derived suppressor cells through the CCL15-CCR1 chemokine axis. Clin Cancer Res. 2016;22(2):492–501. doi:10.1158/1078-0432.CCR-15-0726. PMID:26341919.
- Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol. 2016;37(3):208–20. doi:10.1016/j.it.2016.01.004. PMID:26858199.
- Li L, Xu L, Yan J, Zhen ZJ, Ji Y, Liu CQ, Lau WY, Zheng L, Xu J. CXCR2-CXCL1 axis is correlated with neutrophil infiltration and predicts a poor prognosis in hepatocellular carcinoma. J Exp Clin Cancer Res. 2015;34(129). doi:10.1186/s13046-015-0247-1.
- Zhang H, Ye YL, Li MX, Ye SB, Huang WR, Cai TT, He J, Peng JY, Duan TH, Cui J, et al. CXCL2/MIF-CXCR2 signaling promotes the recruitment of myeloid-derived suppressor cells and is correlated with prognosis in bladder cancer. Oncogene. 2017;36(15):2095–2104.
- Ene-Obong A, Clear AJ, Watt J, Wang J, Fatah R, Riches JC, Marshall JF, Chin-Aleong J, Chelala C, Gribben JG, et al. Activated pancreatic stellate cells sequester CD8+ T cells to reduce their infiltration of the juxtatumoral compartment of pancreatic ductal adenocarcinoma. Gastroenterology. 2013;145(5):1121–32. doi:10.1053/j.gastro.2013.07.025. PMID:23891972.
- Hoechst B, Ormandy LA, Ballmaier M, Lehner F, Kruger C, Manns MP, Greten TF, Korangy F. A new population of myeloid-derived suppressor cells in hepatocellular carcinoma patients induces CD4(+)CD25(+)Foxp3(+) T cells. Gastroenterology. 2008;135(1):234–43. doi:10.1053/j.gastro.2008.03.020. PMID:18485901.
- Li J, Wang L, Chen X, Li L, Li Y, Ping Y, Huang L, Yue D, Zhang Z, Wang F, et al. CD39/CD73 up-regulation on myeloid-derived suppressor cells via TGF-β-mTOR-HIF-1 signaling in patients with non-small cell lung cancer. OncoImmunology. 2017;21(6). doi:10.1080/2162402X.2017.1320011.
- Sade-Feldman M, Kanterman J, Klieger Y, Ish-Shalom E, Olga M, Saragovi A, Shtainberg H, Lotem M, Baniyash M. Clinical significance of circulating CD33+CD11b+HLA-DR- myeloid cells in Stage-IV melanoma patients treated with ipilimumab. Clin Cancer Res. 2016;22(23):12. doi:10.1158/1078-0432.CCR-15-3104.
- Seifert L, Werba G, Tiwari S, Giao Ly NN, Alothman S, Alqunaibit D, Avanzi A, Barilla R, Daley D, Greco SH, et al. The necrosome promotes pancreatic oncogenesis via CXCL1 and Mincle-induced immune suppression. Nature. 2016;532(7598):245–9. doi:10.1038/nature17403. PMID:27049944.
- Sujobert P, Poulain L, Paubelle E, Zylbersztejn F, Grenier A, Lambert M, Townsend EC, Brusq JM, Nicodeme E, Decrooqc J, et al. Co-activation of AMPK and mTORC1 induces cytotoxicity in acute myeloid leukemia. Cell Rep. 2015;11(9):1446–57. doi:10.1016/j.celrep.2015.04.063. PMID:26004183.
- Zhao Y, Tan Y, Xi S, Li Y, Li C, Cui J, Yan X, Li X, Wang G, Li W, et al. A novel mechanism by which sdf-1b protects cardiac cells from palmitate-induced endoplasmic reticulum stress and apoptosis via CXCR7. Diabetes. 2013;62(7):14. doi:10.2337/db12-1233.
- Wang Y, Viollet B, Terkeltaub R, Liu-Bryan R. AMP-activated protein kinase suppresses urate crystal-induced inflammation and transduces colchicine effects in macrophages. Ann Rheum Dis. 2016;75(1):286–94. doi:10.1136/annrheumdis-2014-206074. PMID:25362043.
- Wu Y, Song P, Zhang W, Liu J, Dai X, Liu Z, Lu Q, Ouyang C, Xie Z, Zhao Z, et al. Activation of AMPKα2 in adipocytes is essential for nicotine-induced insulin resistance in vivo. Nat Med. 2015;21(4):10. doi:10.1038/nm.3826.
- Chen K, Wu K, Jiao X, Wang L, Ju X, Wang M, Di Sante G, Xu S, Wang Q, Li K, et al. The endogenous cell-fate factor dachshund restrains prostate epithelial cell migration via repression of cytokine secretion via a cxcl signaling module. Cancer Res. 2015;75(10):1992–2004. doi:10.1158/0008-5472.CAN-14-0611. PMID:25769723.
- Wu K, Katiyar S, Li A, Liu M, Ju X, Popov VM, Jiao X, Lisanti MP, Casola A, Pestell RG. Dachshund inhibits oncogene-induced breast cancer cellular migration and invasion through suppression of interleukin-8. Proc Natl Acad Sci U S A. 2008;105(19):6924–9. doi:10.1073/pnas.0802085105. PMID:18467491.
- Condamine T, Ramachandran I, Youn JI, Gabrilovich DI. Regulation of tumor metastasis by myeloid-derived suppressor cells. Annu Rev Med. 2015;66:97–110. doi:10.1146/annurev-med-051013-052304. PMID:25341012.
- Horikawa N, Abiko K, Matsumura N, Hamanishi J, Baba T, Yamaguchi K, Yoshioka Y, Koshiyama M, Konishi I. Expression of vascular endothelial growth factor in ovarian cancer inhibits tumor immunity through the accumulation of myeloid-derived suppressor cells. Clin Cancer Res. 2017;23(2):13. doi:10.1158/1078-0432.CCR-16-0387. PMID:27742793.
- Stairs DB, Bayne LJ, Rhoades B, Vega ME, Waldron TJ, Kalabis J, Klein-Szanto A, Lee JS, Katz JP, Diehl JA, et al. Deletion of p120-catenin results in a tumor microenvironment with inflammation and cancer that establishes it as a tumor suppressor gene. Cancer Cell. 2011;19(4):470–83. doi:10.1016/j.ccr.2011.02.007. PMID:21481789.
- Chen PT, Hsieh CC, Wu CT, Yen TC, Lin PY, Chen WC, Chen MF. 1alpha,25-dihydroxyvitamin d3 inhibits esophageal squamous cell carcinoma progression by reducing il6 signaling. Mol Cancer Ther. 2015;14(6):1365–75. doi:10.1158/1535-7163.MCT-14-0952. PMID:25824337.
- Chen MF, Yen TC, Lu MS, Lin PY, Chung YH, Chen WC, Lee KD. IL-6-stimulated CD11b+CD14+HLA-DR− myeloid-derived suppressor cells, are associated with progression and poor prognosis in squamous cell carcinoma of the esophagus. Oncotarget. 2014;5(18):13. doi:10.18632/oncotarget.2368.
- Benevides L, da Fonseca DM, Donate PB, Tiezzi DG, De Carvalho DD, de Andrade JM, Martins GA, Silva JS. IL17 Promotes mammary tumor progression by changing the behavior of tumor cells and eliciting tumorigenic neutrophils recruitment. Cancer Res. 2015;75(18):3788–99. doi:10.1158/0008-5472.CAN-15-0054. PMID:26208902.
- Marvel D, Gabrilovich DI. Myeloid-derived suppressor cells in the tumor microenvironment: expect the unexpected. J Clin Invest. 2015;125(9):3356–64. doi:10.1172/JCI80005. PMID:26168215.
- Talmadge JE, Gabrilovich DI. History of myeloid-derived suppressor cells. Nat Rev Cancer. 2013;13(10):739–52. doi:10.1038/nrc3581. PMID:24060865.
- Ham B, Wang N, D'Costa Z, Fernandez MC, Bourdeau F, Auguste P, Illemann M, Eefsen RL, Hoyer-Hansen G, Vainer B, et al. TNF Receptor-2 facilitates an immunosuppressive microenvironment in the liver to promote the colonization and growth of hepatic metastases. Cancer Res. 2015;75(24):5235–47. doi:10.1158/0008-5472.CAN-14-3173. PMID:26483205.
- Ko JS, Zea AH, Rini BI, Ireland JL, Elson P, Cohen P, Golshayan A, Rayman PA, Wood L, Garcia J, et al. Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clin Cancer Res. 2009;15(6):2148–57. doi:10.1158/1078-0432.CCR-08-1332. PMID:19276286.
- Eksioglu EA, Chen X, Heider KH, Rueter B, McGraw KL, Basiorka AA, Wei M, Burnette A, Cheng P, Lancet J, et al. Novel therapeutic approach to improve hematopoiesis in low risk MDS by targeting MDSCs with the Fc-engineered CD33 antibody BI 836858. Leukemia. 2017;31(10):2172–80. doi:10.1038/leu.2017.21. PMID:28096534.
- Dominguez G, Condamine T, Mony S, Hashimoto A, Wang F, Liu Q, Forero A, Bendell JC, Witt R, Hockstein N, et al. Selective targeting of myeloid-derived suppressor cells in cancer patients using DS-8273a, an agonistic TRAIL-R2 antibody. Clin Cancer Res. 2017;23(12):2942–50. PMID:27965309.
- Cifarelli V, Lashinger LM, Devlin KL, Dunlap SM, Huang J, Kaaks R, Pollak MN, Hursting SD. Metformin and rapamycin reduce pancreatic cancer growth in obese prediabetic mice by distinct microRNA- regulated mechanisms. Diabetes. 2015;64(5):11. doi:10.2337/db14-1132.
- Wu L, Zhou B, Oshiro-Rapley N, Li M, Paulo JA, Webster CM, Mou F, Kacergis MC, Talkowski ME, Carr CE, et al. An ancient, unified mechanism for metformin growth inhibition in c. elegans and cancer. Cell. 2016;167(7):1705–18. e13. doi:10.1016/j.cell.2016.11.055. PMID:27984722.
- Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang LS, Jones RG, Choi Y. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature. 2009;460(7251):103–7. doi:10.1038/nature08097. PMID:19494812.
- Vasamsetti SB, Karnewar S, Kanugula AK, Thatipalli AR, Kumar JM, Kotamraju S. Metformin inhibits monocyte-to-macrophage differentiation via AMPK-mediated inhibition of STAT3 activation: potential role in atherosclerosis. Diabetes. 2015;64(6):14. doi:10.2337/db14-1225.
- Eikawa S, Nishida M, Mizukami S, Yamazaki C, Nakayama E, Udono H. Immune-mediated antitumor effect by type 2 diabetes drug, metformin. Proc Natl Acad Sci U S A. 2015;112(6):6. doi:10.1073/pnas.1417636112. PMID:25538304.