
ABSTRACT
Prostate cancer (PCa) was estimated to have the second highest global incidence rate for male non-skin tumors and is the fifth most deadly in men thus mandating the need for novel treatment options. MG1-Maraba is a potent and versatile oncolytic virus capable of lethally infecting a variety of prostatic tumor cell lines alongside primary PCa biopsies and exerts direct oncolytic effects against large TRAMP-C2 tumors in vivo. An oncolytic immunotherapeutic strategy utilizing a priming vaccine and intravenously administered MG1-Maraba both expressing the human six-transmembrane antigen of the prostate (STEAP) protein generated specific CD8+ T-cell responses against multiple STEAP epitopes and resulted in functional breach of tolerance. Treatment of mice with bulky TRAMP-C2 tumors using oncolytic STEAP immunotherapy induced an overt delay in tumor progression, marked intratumoral lymphocytic infiltration with an active transcriptional profile and up-regulation of MHC class I. The preclinical data generated here offers clear rationale for clinically evaluating this approach for men with advanced PCa.
Introduction
In 2012 it was estimated that prostate cancer (PCa) had the second highest global incidence rate for male non-skin tumors accounting for approximately 1,100,000 cases and claiming over 250,000 lives that year.Citation1 Over the last decade immunotherapy has been assessed in patients with PCa. Trials using autologous cell vaccines (sipuleucel-T) and poxvirus (rilimogene galvacirepvec) based therapies (targeting the PCa tumor associated antigens (TAAs) prostatic acid phosphatase (PAP) and prostate-specific antigen (PSA) respectively) have increased median survival times in PCa patients.Citation2,Citation3 These vaccine-based studies alongside clinical evidence of CTLA4 blockade activityCitation4 have fuelled interest in the potential for immunotherapy to treat patients with advanced PCa.
Despite recent progress, a significant opportunity remains to optimize PCa immunotherapeutics.Citation5 A major obstacle to cancer immunotherapy is the immunosuppressive tumor microenvironment (TME) resulting in exclusion of tumor infiltrating lymphocytes (TIL),Citation6,Citation7 in PCa low TIL numbers have been associated with poor outcomes.Citation8 When compared to benign nodular prostatic hyperplasia, the density of immune infiltrate is decreased in prostatic adenocarcinoma samples consistent with immunosuppression in more advanced disease.Citation9 Genomic analysis of metastatic tumors revealed that PCa was weakly infiltrated by leukocytes, compared to the majority of other histotypes.Citation10 Therapeutically in late stage PCa patients treated with salvage radiotherapy, high CD8+ TIL scores predicted increased progression free and overall survivals.Citation11 Thus converting an immunologically cold prostatic TME to one that is heavily infiltrated by TIL is a desirable property for prospective medical interventions.
Oncolytic viruses (OVs) may be beneficial in targeting PCa as they induce pro-inflammatory changes within the TME amongst a variety of other anti-neoplastic actions.Citation12 Numerous OVs including vesicular stomatitis virus (VSV), adenovirus, vaccinia and herpes simplex virus (HSV) have been investigated pre-clinically emphasizing the potential of these biologics for PCa. MG1-Maraba virus is a versatile OV that exerts direct oncolytic effects as well as having the capacity to express transgenes, including TAAs resulting in specific anti-tumor T cell immunity as part of a robust heterologous prime: boost platform.Citation13-Citation15 Direct oncolytic effects of MG1-Maraba have been demonstrated in multiple human and murine cell lines, syngeneic and xenografted murine models as well as primary tumor biopsies.Citation13,Citation15,Citation16 Oncolytic vaccination using MG1-Maraba expressing HPV and melanoma TAAs induces specific CD8+ T cell immunity and is efficacious in pre-clinical models of HPV-associated cancer and melanoma,Citation14,Citation15 however in depth analyses of the effects exerted by this therapeutic platform in the TME have not been reported. MG1-Maraba represents an unexplored therapy for patients with advanced PCa.
We predicted that MG1-Maraba virus would exert oncolytic activity against PCa and by engineering MG1 to express six-transmembrane antigen of the prostate (STEAP) (a human.Citation17 and murine.Citation18 prostatic TAA) we would be able to break immunologic tolerance against this antigen, transform an immunologically cold prostatic cancer TME and extend survival in an advanced model of PCa. Data presented here reveal MG1-Maraba is cytotoxic to a range of PCa cell lines and productively infects primary human PCa biopsies. Furthermore the use of an engineered OV, namely MG1-STEAP, as part of a potent heterologous prime: boost vaccination strategy targeting PCa, is described in this study. Treatment of immunocompetent mice bearing large TRAMP-C2 tumors with oncolytic STEAP vaccination resulted in a marked influx of TIL and prolonged survival. Collectively these data introduce a therapeutic approach for prostate cancer supporting future clinical assessment of this strategy.
Methods
Recombinant viruses
A codon-optimized transgene was manufactured encoding human STEAP (GensScript, Piscataway, NJ). Ad-BHG and Ad-STEAP are human serotype 5 replication deficient (E1/E3 deleted) adenoviruses. Ad-BHG contains no transgene Ad-STEAP encodes human STEAP. The GFP and STEAP transgenes were inserted between the G and L viral genes of the attenuated MG1 strain of Maraba virus to produce MG1-GFP and MG1-STEAP respectively.
Cell lines
DU145 and LNCaP were a gift from Dr Dimitroulakos (University of Ottawa, Canada) in 2015. L929 and PC3 cells were a gift from Dr Bell (University of Ottawa, Canada) in 2007 and 2015 respectively. Vero 76 and TRAMP-C1 were purchased from the American Type Culture Collection (ATCC Manassas, VA) in 2012 and 2015 respectively. TRAMP-C2 were a gift from Dr Graham (Queens University, Canada) in 2013. TRAMP-C1 and TRAMP-C2 cell lines were cultured in Dulbecco's Modified Eagle's Medium (DMEM) (Corning Cellgro, Tewksbury, MA) supplemented with 10% fetal bovine serum (FBS) (ThermoScientific, Waltham, MA), 2mmol/l L-glutamine (Invitrogen, Waltham, MA), 0.005 mg/ml human recombinant zinc insulin (Gibco, MA) and 10 nm dehydroisoandrosterone (MP Biomedicals, Santa Ana, CA). DU145, LNCaP, PC3 and Vero76 cell lines were cultured in DMEM supplemented with 10% FBS. L929 cells were cultured in αMEM containing 8% FBS and 2mmol/l L-glutamine. All cell lines were incubated at 37°C in a 5% CO2 humidified incubator. All cell lines were tested for Mycoplasma using Hoescht, TLR2 and PCR assays between 2011 and 2017. TRAMP-C2 had epithelial morphology, grew in adherent monolayers and were positive for murine STEAP at a transcriptional and protein level. L929 had fibroblastic morphology, grew in adherent monolayers and were responsive to type I IFN. Vero 76 had epithelial morphologic features and grew in adherent monolayers exhibiting viral plaques and cellular cytopathia following rhabdoviral infection. TRAMP-C1, DU145, LNCaP and PC3 had epithelial morphology and grew in adherent monolayers.
In vitro cytotoxicity screen
PCa cells were plated in 24-well plates to a confluency of 95%. These cells were infected at various multiplicities of infection (MOIs) (0.00001–10 PFU/cell) with MG1-GFP. Following a 48-hour incubation, 10% alamarBlue® (AbD Serotec, Raleigh, NC) was added. After a 3-hour incubation, absorbance was read at wavelength of 575 nm using Fluoroskan Ascent™ Microplate Fluorometer (ThermoFisher Scientific, Waltham MA). Viability was expressed as the % viable cells in infected wells cf. viable cells in mock treated wells.
Interferon β response test
L929 were plated alongside TRAMP-C2 cells in a 96-well plate and when confluent treated with a dilution series of murine IFNβ overnight. The following day the cells were infected with 5 × 105 PFU per well of wild type VSV expressing GFP. Viral proliferation was determined by detecting fluorescence 24 hours post-infection using a Typhoon Trio Variable Mode Imager (GE Healthcare, Buckinghamshire, U.K.). Fluorescence was quantified with ImageQuant TL software (GE Healthcare, Buckinghamshire, U.K.).
Immunofluoresence
TRAMP-C2 cells were fixed with 4% paraformaldehyde on coverslips (Electron Microscopy Sciences, Hatfield, PA) at 37°C for 10 minutes. Cells were blocked at room temperature with 1% BSA (Equitech-Bio, Kerrville, TX) in PBS. The polyclonal rabbit anti-STEAP antibody (Santa Cruz, Dallas, TX) was applied overnight at 4°C. Secondary fluorescent-conjugated donkey anti-rabbit (Thermo Fisher Scientific, Waltham, MA) was applied for 1 hour at room temperature prior to staining with DAPI (Molecular Probes, Eugene, OR) for 10 minutes. Images were captured using EVOS FL Cell Imaging System (Thermo Fisher Scientific, Waltham, MA).
Patient biopsy samples
Patients with non-metastatic palpable prostate tumors consented for the study. In the operating room multiple core biopsy samples were obtained from prostatectomy specimens using an 18-gauge Tru-cut biopsy needle (approved by the Ottawa Hospital Research Ethics Board Protocol# 20120559-01H). Samples were placed in a 24-well plate containing 1 mL of DMEM supplemented with 10% FBS and 1% penicillin streptomycin and 250 ng/ml amphotericin B (Sigma-Aldrich, St Lois, MO). Samples were treated with MG1-GFP (3 × 104 PFU/core). After 1-hour incubation, cores were washed with DMEM three times and incubated in 1 mL of DMEM (10% FBS and antimicrobials) for 24 hours. Bright field and fluorescent images were captured with EVOS FL Cell Imaging System (ThermoFisher Scientific, Waltham, MA) 24 and 48 hours post-infection. At 48 hours post-infection, samples were collected for standard plaque assay performed on Vero 76 cells to determine ex vivo infectivity and for immunohistochemistry.
Mice
Six to eight week old C57BL/6 mice were purchased from Charles River (Wilmington, MA) and housed in specific pathogen-free conditions. Animals were euthanized when tumor volumes reached 1500 mm.Citation3 Animal studies were approved by McMaster University's Animal Research Ethics Board and complied with Canadian Council on Animal Care guidelines.
In vivo efficacy and vaccination studies
Male mice were engrafted with 2.5 × 106 TRAMP-C2 cells subcutaneously on the left flank under gaseous anesthesia. For direct oncolysis studies tumors were first treated when mean volume reached 500mmCitation3 with three doses, given every 48 hours, of 5 × 108 PFU MG1-GFP in a total volume of 75 μL and 200 μL of 0.9% NaCl (Hospira, Lake Forest, IL) for intra-tumoral and intravenous administration respectively. For prime: boost studies treatment began when mean tumor volumes were 250mm.Citation3 Adenovirus was administered under gaseous anesthesia at a dose of 2 × 108 plaque-forming units (PFU) in 100 μL of 0.9% NaCl for injection, the dose was split in 2– 50 μL was injected intramuscularly in each hind limb. Initial tumor-free immune analysis was performed on female mice boosted with 2 doses of 1 × 109 PFU of MG1-STEAP in 200 μL of 0.9% NaCl given intravenously at 13 and 16 days post-prime. For all other experiments male mice were boosted IV with the same dose of MG1 at 8 and 11 days post-prime.
Intracellular cytokine staining
Blood samples were taken for prime analysis on the day of the first boost. Analyses of boost vaccinations were performed 6 days after the first MG1 dose. Peripheral blood mononuclear cells and splenocytes were incubated in complete RPMI (containing 10% FBS and 2mmol/l L-glutamine) with peptide. Incubations were performed for 5 hours total in a 37°C, 5% CO2 incubator at 95% humidity, 1 μg/ml of brefeldin A (GolgiPlug, BD, Franklin Lakes, NJ) was added for the last 4 hours. Cells were then incubated with anti CD16/CD32 (clone 2.4 G2, Mouse BD Fc Block, BD, Franklin Lakes, NJ). T cell surface staining was performed with antibodies against CD8a (clone 53–6.7, eBiosciences, Inc., SanDiego, CA) and CD4 (clone RM4-5, eBiosciences, Inc., San Diego, CA). Cells were subsequently fixed and permeabilized (Cytofix/Cytoperm, BD, Franklin Lakes, NJ). Intracellular cytokine staining was then performed using anti-IFNγ (clone XMG1.2, BD, Franklin Lakes, NJ). Data were acquired using an LSRFortessa cytometer (BD, Franklin Lakes, NJ) and analyzed with FlowJo Mac software (Treestar, Ashland, OR).
Peptides for immune analyses
Peptides were synthesized by Biomer Technologies (San Francisco, CA) in crude form. The following specific short peptides were used to re-stimulate PBMCs at 5 μg/ml: muSTEAP 5–13 (KDITNQEEL), muSTEAP 186 (RSYRYKLL), muSTEAP 327 (VSKINRTEM) and huSTEAP 327 (VTKINKTEI).
Ex-vivo infection of TRAMP-C2 tumors
Volume endpoint tumors were harvested from untreated male mice. 2 mm cores were obtained using a sterile biopsy punch and were further dissected by scalpel to obtain approximately 2 × 1 mm slices. Individual slices were cultured in a 96-well plate containing 200 μL media and 1 × 106 PFU of MG1-GFP per well. A column of wells contained uninfected tumor in media as a negative control. Fluorescence was detected 24 hours post-infection using a Typhoon Trio Variable Mode Imager (GE Healthcare, Buckinghamshire, U.K.). Fluorescence was quantified with ImageQuant TL software (GE Healthcare, Buckinghamshire, U.K.).
Immunohistochemistry and histopathology
TRAMP-C2 tumors were snap frozen in O.C.T. Compound (Sakura Finetek, Torrance, CA) for MHCI staining, all other immunohistochemical samples were paraffin embedded following formalin fixation. TRAMP-C2 tumors were collected 6 days after the first MG1-STEAP dose for peak-boost and tumors were harvested at the same time point from untreated mice, tumors were also harvested from STEAP vaccinated mice upon reaching end-stage volume. Human core sections were stained using polyclonal anti-VSV rabbit serum (generated at the University of Ottawa, Canada). Staining was visualized with DAB substrate chromogen (Vector Laboratories, Burlingame, CA). The rabbit EnVision system (Agilent, Santa Clara, CA) was used as per the manufacturers instructions after primary incubation with polyclonal anti-Maraba rabbit serum (generated at McMaster Immunology Research Centre, Canada) for detection of MG1 following ex vivo infection of TRAMP-C2 tumors using AEC substrate chromogen (Sigma-Aldrich, St Lois, MO). Antibodies against CD3 (clone SP7, Abcam, Cambridge, U.K.), CD8a (clone 4SM15, eBiosciences, Inc., San Diego, CA), MHCI (clone ER-HR 52 Abcam, Cambridge, U.K.) and STEAP (rabbit polyclonal, Santa Cruz, Dallas, TX) were used with the Bond RX IHC auto-stainer (Leica, Wetzlar, Germany) and the Bond Polymer Refine Detection system (utilizing DAB substrate chromogen and hematoxylin) (Leica, Wetzlar, Germany) as per manufacturers instructions. For assessment of immunopathology 4 male mice received Ad-STEAP IM followed by 2 doses of MG1-STEAP IV and were sacrificed 6 weeks after the first dose of MG1-STEAP. As controls 5 untreated mice and 5 mice receiving Ad-BHG and MG1-GFP were included. Mice were perfused under anesthesia with 10% neutral-buffered formalin (Sigma-Aldrich, St Lois, MO). Prostate, testes, lungs, kidneys and bladder were removed, paraffin-embedded and stained with hematoxylin and eosin. Human biopsy slides ere scanned using ScanScope XT (Aperio Technologies, Visa, CA). Slides for quantification of TRAMP-C2 tumors for CD3, CD8a, STEAP and MG1 staining were scanned using an Aperio Slide Scanner (Aperio Technologies, Visa, CA). Staining was quantified using ImageScope software (Aperio Technologies, Visa, CA). Mouse tissues displayed in Figs. were scanned using an Olympus VS120 Slide Scanner (Olympus, Tokyo, Japan). Contrast and brightness were adjusted uniformly only for displayed STEAP IHC images in Adobe Photoshop CC2017 (Adobe, San Jose, CA). MHCI staining was quantified utilizing an H-score as previously described.Citation19 Immunohistochemical (IHC) staining for MHCI intensity was classified as negative (0), weakly positive (1), moderately positive (2) or strongly positive (3) (supplementary Fig 1).
NanoString analysis
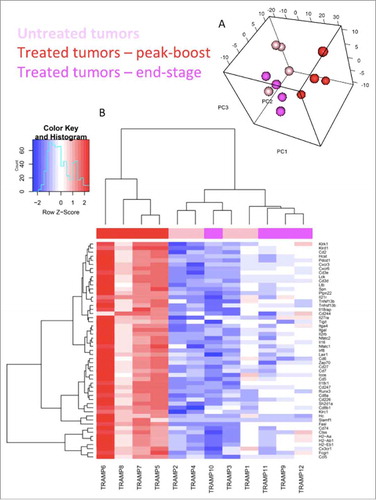
RNA was extracted from formalin-fixed paraffin-embedded TRAMP-C2 tumors used for TRAMP-C2 IHC with RNeasy FFPE kit (Qiagen, Hilden, Germany) and a QIAcube robotic workstation (Qiagen, Hilden, Germany) as per the manufacturers instructions. The nCounter® Mouse Immunology panel was used to examine differential expression of mRNAs. 50 ng of total RNA was hybridized at 65°C for 21 hours and data were acquired using nCounter® Max Analysis System. Each tumor contributed 1 sample. Profiled data were pre-processed following the manufacturer's recommendations, specifically background was subtracted by using geometric mean of negative controls, normalization was performed using positive controls and housekeeping genes using nSolver® software. Obtained values were Log2 transformed. To visualize sample distribution, whole profiles were used for hierarchical clustering (Euclidean distance, complete linkage) and for principal component analysis (PCA). Profiles were analyzed for differential expression by using limma package in R.Citation20
Statistical analyses
Data were displayed and analyzed utilizing GraphPad Prism (GraphPad Software, San Diego, CA). Mean ex vivo tumor fluorescence intensities, tumor volumes, fold changes, immune analyses and IHC quantification were compared using ordinary one-way ANOVA tests. Fluorescence intensities from IFNβ were compared using multiple t tests with Holm-Sidak correction. Survival times were plotted using Kaplan-Meier curves and median survivals were compared using the log-rank test. Statistical significance was defined as p ≤ 0.05 (*p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001).
Results
MG1-Maraba lethally infects and replicates in PCa
To test the oncolytic potential of MG1 in PCa we used a variety of human and murine PCa cell lines and primary PCa biopsy tissues. In culture monolayers, MG1-Maraba was markedly cytotoxic to 3 well-studied human PCa cell lines DU145, PC3 and LNCaP. Cytotoxicity was also observed against the syngeneic murine cell lines TRAMP-C1 and TRAMP-C2 with the latter being more sensitive to oncolysis at lower MOIs (). The ability of MG1-Maraba to replicate in primary human PCa was assessed by infecting core biopsies () from 10 patients with 3 × 104 MG1-GFP ex vivo. Productive infections as determined by plaque assay were demonstrated in 90% of biopsies with virus output greatly exceeding input (). Transgene expression was confirmed by fluorescence (). When quantified immunohistochemically MG1 was present in a mean of 13.2% (range 7–21.8%) of all cells within biopsy specimens (). MG1 therefore exhibits oncolytic properties in a wide variety of PCa cell lines and primary biopsies.
Figure 1. MG1-Maraba lethally infects prostatic cancer. Relative viability of multiple PCa cell lines was assessed using alamar blue 48 hours after infection of PCa cells in monolayer over a range MOIs (mean and SEM displayed, assays performed in triplicate) (A). Representative image of PCa core biopsies being obtained following prostatectomy for the treatment of a patient diagnosed with advanced PCa (B). Plaque assays following ex vivo infection from 10 patient's PCa core biopsies with MG1-GFP (mean and SEM displayed, assays performed in duplicate) (C). Representative bright field, fluorescent and overlaid images following infection of a core biopsy with MG1-GFP (4x magnification) (D). Immunohistochemical quantification of % cell positivity for MG1-Maraba following ex vivo infection of PCa biopsy cores with MG1-GFP (mean and SEM displayed, all sections stained at least in duplicate except patient #7 where biopsy material was limited) (E). Representative image of immunohistochemical staining for MG1-Maraba using DAB substrate chromogen and hematoxylin counterstain (F) (scale bar = 900 μm).
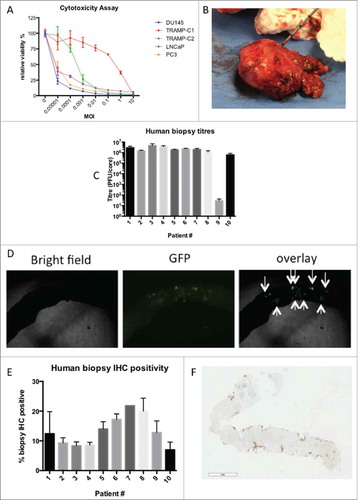
MG1-Maraba exerts oncolytic activity against large TRAMP-C2 tumors
Immunocompetent male mice bearing large subcutaneous TRAMP-C2 tumors (mean volume of 500mmCitation3 at first treatment) were used to assess the in vivo efficacy of direct viral oncolysis, 3 doses of 5 × 108 PFU MG1-GFP were given 48 hours apart administered either intratumorally (IT) or intravenously (IV). IT dosing with MG1-GFP significantly impeded tumor progression compared to untreated mice () and whilst no statistically significant overall effect was observed, subtle but repeatable changes to the trajectory of the tumor volume curves were documented after the first IV treatment with MG1-GFP at the 5 × 108 PFU dose level (, supplementary Fig 2). End-stage TRAMP-C2 tumors were collected from mice and infected ex vivo with MG1-GFP, fluorescence was documented after 24 hours (.). MG1-GFP infection was confirmed immunohistochemically (supplementary Fig 3). Pre-treatment of TRAMP-C2 cells in a monolayer with murine IFN-β led to only partial protection from rhabdoviral infection compared to IFN responsive L929 cells (). These results are consistent with TRAMP-C2 tumors being susceptible to MG1-Maraba infection and displaying deficient innate anti-viral protection. Direct oncolytic treatment of mice bearing advanced TRAMP-C2 tumors resulted in significant but transient anti-neoplastic activity after IT delivery.
Figure 2. MG1-Maraba is oncolytic against TRAMP-C2 tumors. Treatment of male mice with established TRAMP-C2 tumors with 3 intravenous doses of 5 × 108 PFU MG1-GFP (n = 5) results in transient decreases in tumor volumes (A) (arrows denote timings of MG1 treatments, mean tumor volumes and SEM displayed) whereas intra-tumoral (n = 5) dosing (5 × 108 PFU MG1-GFP) exerts a significant anti-neoplastic effect (B) (mean fold change of tumor volume and SEM displayed, comparison performed using ANOVA, *p≤0.05) compared to untreated control mice (n = 5). TRAMP-C2 tumors were harvested from mice (n = 4), processed into uniform cores (biologic replicates n = 8), maintained in tissue culture and infected with MG1-GFP ex vivo, infection was demonstrated by fluorescence as depicted by black signal (C) and fluorescence was quantified (mean and SEM displayed, comparison performed using ANOVA, *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001) (D). Monolayers of TRAMP-C2 and the type I IFN responsive control L929 cells were pre-treated with murine IFN-β (excluding 0 IFN control columns) prior to infection with a WT VSV expressing GFP and infection was demonstrated by black signal indicative of fluorescence (E) and fluorescence was quantified (mean and SEM displayed, comparison performed using multiple t tests with Holm-Sidak correction, *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001) (F).
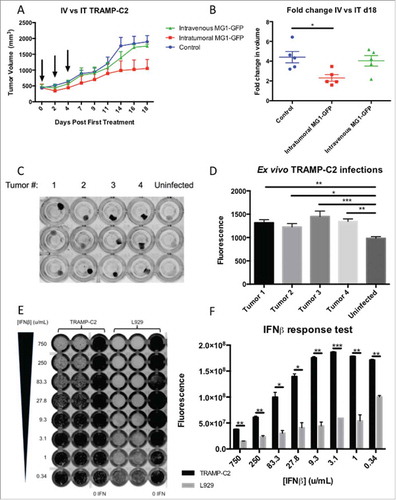
Oncolytic vaccination with the xenoantigen STEAP induces CD8+ immunity
In order to target a PCa antigen as part of an oncolytic vaccination regimen replication-deficient human serotype-5 adenovirus (administered intramuscularly) and MG1-Maraba (given IV) were manufactured encoding human STEAP1 (supplementary Fig 4). For initial screening, 2 groups of tumor-free female mice were vaccinated with the STEAP vectors. ICS was performed on PBMCs re-stimulated with STEAP peptides from blood obtained 13 days after priming and 6 days after the first dose of MG1. Specific CD8+ T cell responses were documented against all 4 STEAP peptides in vaccinated mice with the largest magnitude observed for the human specific epitope (h327) (). Tumor-free male mice were subsequently vaccinated with the STEAP vectors alongside a group of control-vaccinated mice. Only STEAP vaccinated mice had specific immunity and the largest responses were again directed against h327. Responses were observed against the other conserved and murine epitopes with the exception of the 5–13 epitope (). Four STEAP vaccinated tumor-free male mice underwent post-mortem examination 6 weeks post-MG1 for assessment of immunopathology. Mice receiving Ad-BHG: MG1-GFP (n = 5) and untreated mice (n = 5) were included as controls. No gross pathology was identified in any mouse. Histologic examination of H&E sections revealed no evidence of architectural change representative of primary pathology in the testes, lungs, bladder or kidneys of any mouse. No pathology was identified in the genito-urinary tract of untreated and Ad-BHG: MG1-GFP mice. All 4 STEAP vaccinated mice had histologic changes within the prostate ranging from mild polymorphic cellular infiltrate within the glandular lumen to severe pleocellular infiltrate with accompanying epithelial dysplasia and epithelial hypertrophy consistent with prostatic inflammation (). Prominent lymphoid follicles were noted within the bladder wall of the mouse with the most marked prostatic changes and this was consistent with local reactive lymphoid hyperplasia secondary to prostatic inflammation. No pathology was noted in the healthy control mice () and STEAP expression was confirmed on the luminal epithelial surface of healthy murine prostatic tissue (). These data support the ability of oncolytic STEAP vaccination to break immune tolerance resulting in target organ specific inflammation.
Figure 3. Oncolytic vaccination breaches immune tolerance. Tumor-free female C57BL/6 mice were treated with oncolytic vaccination against STEAP (n = 3) to assess the induction of specific immune responses against known STEAP peptide epitopes as indicated by IFNγ production from CD8+ T cells collected after the prime (A) and boost (B) (mean and SEM displayed). Tumor-free male mice were treated with oncolytic vaccination against STEAP (n = 5) as well as a control group of mice treated with Ad-BHG: MG1-GFP (n = 5). ICS was performed after prime (C) and boost (D) (mean and SEM displayed). Prostatic inflammation was observed following STEAP vaccination (E) (hematoxylin and eosin)- note the heavy pleocelluar luminal infiltrate compared to healthy prostatic tissue (F) (hematoxylin and eosin). Luminal epithelial STEAP expression was confirmed immunohistochemically in the healthy prostate (G) (DAB substrate chromogen and hematoxylin counterstain) (scale bars = 100 μm).
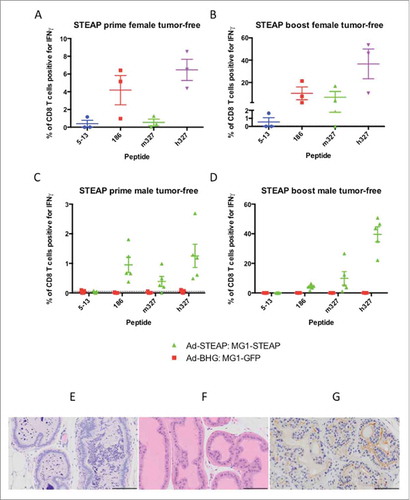
Oncolytic STEAP vaccination prolongs survival in mice bearing advanced TRAMP-C2 tumors
Male mice were engrafted subcutaneously with TRAMP-C2 and treatment was instigated at a mean tumor volume of approximately 250 mm.Citation3 Immunofluorescence () and RT-PCR (Supplementary Fig 5) confirmed STEAP expression in TRAMP-C2 cells. Groups of mice received Ad-BHG: MG1-GFP or Ad-STEAP: MG1-STEAP and an untreated group of mice were included as controls. Immune analysis by ICS on PBMCs re-stimulated with pooled murine STEAP peptides was undertaken after priming and MG1 boosting. Vaccination with the STEAP vectors exerted clear antineoplastic activity (, Supplementary Fig 6) and resulted in a significant survival advantage (). Although treatment with the Ad-BHG: MG1-GFP did not improve survival, significant tumor volume responses were observed 48 hours after IV administration of MG1-GFP at a dose of 1 × 109 PFU (Supplementary Fig 7). Compelling and specific circulating CD8+ immunity was only noted in TRAMP-C2 bearing mice following vaccination with STEAP vectors (). Maraba virus has oncolytic activity against an established murine prostatic carcinoma and oncolytic vaccination against STEAP induces CD8+ immunity and prolongs survival in this model.
Figure 4. STEAP vaccination prolongs survival in a prostate cancer model. Immunofluorescence of TRAMP-C2 cells was performed using a polyclonal STEAP antibody followed by a fluorescent-conjugated secondary antibody and nuclei were stained with DAPI. Images were captured on the DAPI and Texas Red channels and a composite overlay image was made (A) (white bar represents 100μm). Male mice with established TRAMP-C2 tumors were treated with Ad-BHG: MG1-GFP (n = 8), Ad-STEAP: MG1-STEAP (n = 16) and compared to a control group of untreated mice (n = 18) (data combined from 2 experiments). Tumor volumes are plotted for each group from a single experiment (B) (arrows denote timings of MG1 treatments, mean and SEM displayed) and tumor volume fold change was calculated for all mice (mean fold change of tumor volume and SEM displayed, comparison performed using ANOVA, **p ≤ 0.01, ***p ≤ 0.001) and collective median survival times were calculated from the initiation of treatment (C) (log rank test used to compare survival times, ****p ≤ 0.0001). ICS on peripheral blood cells following re-stimulation with pooled murine STEAP peptides was performed at the time of peak-boost responses (E) (mean and SEM displayed, comparison performed using ANOVA, ****p ≤ 0.0001).
STEAP vaccination induces an active T cell gene signature in the TME
RNA was extracted from TRAMP-C2 tumors 6 days after the first dose of IV MG1-STEAP was given (n = 4), untreated control tumors (n = 4) from age-matched mice were collected concurrently and tumors were also collected from mice treated with IV MG1-STEAP vaccination after these tumors had progressed to end-point (n = 4). NanoString analysis using a murine immune panel was performed on all tumors. Unsupervised analyses of peak-boost samples compared to both untreated and end-stage tumors resulted in clustering of peak-boost samples, whereas untreated and end-stage tumor expression profiles exhibited overlap (). Amongst the genes up-regulated at peak-boost were T cell surface genes Cd2, Cd3, Cd5, Cd7 and Cd8 as well as CXCR3- a chemokine receptor expressed primarily on activated T cells. Down-stream markers of T cell activation and effector function including Nfat, Zap70, Lck and Fasl also had increased expression levels as did checkpoint-encoding genes Icos, Tigit, Pdc1 and Klrc1, with the latter having an approximate 40 fold induction at peak-boost over untreated. However, combining oncolytic STEAP vaccination with PD1 blockade did not enhance anti-neoplastic activity (Supplementary Fig 6). A number of H2 antigen-processing genes were also increased at peak-treatment. When looking at genes that were only significantly increased when comparing peak-boost to untreated time points Cxcl9 (a gene encoding for CXCR3 ligand) was increased as were genes encoding for granzyme effector molecules (). Expression of the class-I processing associated genes Ciita, H2-K1 and Tap1 was significantly increased at peak-boost compared to untreated tumors (), when comparing peak-boost to end-stage tumors there were non-significant trends for decreased fold changes of expression in these three genes (−2.62, adjusted p = 0.072; −1.91, adjusted p = 0.074 and −1.66, adjusted p = 0.079 respectively). A number of genes including Ifnar1 had decreased expression only when end-stage tumors were compared to peak-boost. Reactome pathway analysis revealed significant increases in multiple pathways associated with T cell signaling in peak-boost compared to both untreated and end-stage samples (Supplementary Table 1). Overall, significant expansion of circulating anti-STEAP CD8+ T cell responses in the blood was mirrored by an active intra-tumoral T cell transcriptional profile.
Table 1. Alterations to the immune transcriptome after STEAP vaccination. List of all significant differentially expressed genes (adjusted p≤0.05) between peak-boost (n = 4) and untreated (n = 4) TRAMP-C2 tumors and a separate analysis between peak-boost and end-stage (n = 4) STEAP vaccinated tumors using a murine immunology NanoString panel. Increased and decreased expressions depicted by red and green respectively and mean fold change is displayed numerically.
Figure 5. Oncolytic vaccination induces an active T cell gene signature. Sample distribution (principal component analysis (PCA) plot). Visualization of the first 3 components of PCA. The analysis was performed based on all 561 genes profiled on the NanoString platform (A). Genes differentially expressed within TRAMP-C2 tumors. mRNA was isolated from TRAMP-C2 tumors of 4 mice receiving STEAP vaccination at peak-boost (6 days after the first dose of MG1-STEAP) and compared to 4 mice with untreated tumors at the same point. Peak-boost tumors were also compared to 4 end-stage tumors that were previously treated with STEAP vaccination. NanoString analysis using a murine immunology panel was undertaken and genes with significantly altered expressions between groups (adjusted p≤0.05) are displayed using a heat map with increased and decreased expressions depicted by red and blue respectively. Unsupervised clustering was performed based on genes that were significantly differentially expressed in both comparisons (B).
Oncolytic vaccination results in T cell infiltration and up-regulation of MHCI
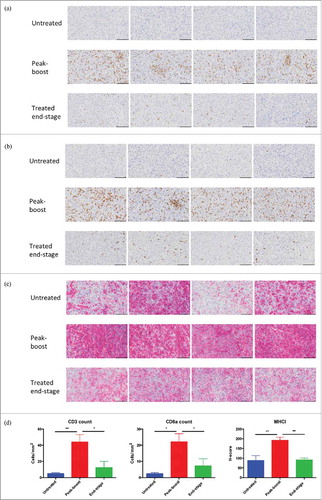
TRAMP-C2 tumors that were analyzed by NanoString also underwent IHC staining. All sets were stained for CD3 and CD8a cell surface antigens. A marked CD3+ () and CD8a+ () cell infiltrate consistent with an influx of TILs was noted at peak-boost vaccination relative to a sparse population of such cells in untreated controls, end-stage tumors exhibited intermediate TIL numbers. Tumors were also stained for MHCI () variable expression was seen in untreated tumors and increased MHCI intensity was noted at peak-boost, end-stage MHCI staining was patchy and less intense. Quantification of IHC revealed significantly increased staining when comparing peak-boost samples to both untreated and end-stage tumors (). Finally tumors were stained for STEAP expression and although the highest intensity staining was observed in untreated tumors and areas of low intensity staining were seen in treated tumors, significant changes in STEAP staining were not documented following vaccination (Supplementary Fig 8). These results are consistent with oncolytic STEAP vaccination inducing pronounced intra-tumoral lymphocytic infiltration and immunologic transformation of the TME.
Figure 6. Oncolytic vaccination converts the tumor microenvironment. Immunohistochemical staining for CD3 (A), CD8a (B) and MHCI (C) of untreated tumors (n = 4), peak-boost (n = 4) (6 days after first dose of MG1-STEAP) and end-stage (STEAP vaccinated) tumors (n = 4). Tumors stained immunohistochemically for the CD3 and CD8a cell surface antigens using DAB substrate chromogen and for MHCI using AEC substrate chromogen, nuclei were counterstained with hematoxylin (scale bars = 100μm). Immunohistochemical quantification of CD3, CD8a and MHCI staining from untreated (n = 4), peak-boost (n = 4) and end-stage (n = 4) TRAMP-C2 tumors (D) (mean and SEM displayed, comparison performed using ANOVA, *p ≤ 0.05, **p ≤ 0.01,).
Discussion
Heterologous prime: boost vaccination strategies are a relatively new potential therapeutic option for men suffering from advanced PCa. PROSTVAC is the best characterized example and uses a vaccinia prime and fowlpox boost encoding PSA and induces targeted immunity against PSA and enhancement in overall survival.Citation21 Although such results are promising, much room for improvement exists as treatment with PROSTVAC alone does not improve progression free survival and most patients succumb to disease within 5 years of vaccination(2). Here we report the use of an oncolytic virus as part of a heterologous prime: boost vaccination strategy in the setting of prostate cancer. The ability of this multi-faceted combination therapy to induce marked, specific anti-tumor immunity and significantly enhance survival in an immunocompetent murine model of advanced prostate carcinoma supports the future clinical evaluation of this approach.
Incorporating an OV into a vaccination strategy broadens the anti-neoplastic mechanisms of the treatment over conventional vaccine platforms. Infection of tumors by OVs induces pro-inflammatory changes to the TMECitation12 resulting in a tumor that is more likely to respond to immunotherapy.Citation22 All cell lines tested were susceptible to killing by MG1-Maraba and ex vivo infection of 90% of human PCa samples resulted in MG1 replication. Debate exists as to whether systemic or intra-tumoral administration will best facilitate OV delivery, however systemic delivery is advantageous as it enables potential infection of multiple distant lesions with a single OV dose.Citation23,Citation24 Intravascular viral dilution, viral sequestration in phagocytic cells and restricted intra-tumoral vascular permeability all potentially limit delivery of systemically administered OVs to the tumor compared to IT treatment.Citation24 In the TRAMP-C2 direct oncolysis studies, IT delivery resulted in significant anti-neoplastic activity against large neoplasms, and although IV delivery of MG1-GFP was not effective at the equivalent IT dose of 5 × 108 PFU, MG1-GFP given IV at a dose of 1 × 109 PFU induced significant but transient tumor volume responses. These findings support the theory that high-level viremia is required for effective systemic OV therapy(24). Early phase clinical trials determining maximal tolerated doses will be fundamental for the success of OVs.
Defects in type I IFN are frequently encountered in tumors.Citation25 We demonstrated that TRAMP-C2 cells have defective type I interferon responses and are susceptible to infection in monolayer and in a heterogeneous ex vivo setting from advanced neoplasms. Defects in IFN responses have been demonstrated in human PCa predicting susceptibility to oncolysis by MG1, in normal diploid cells oncolytic rhabdoviruses are exquisitely sensitive to interferon and therefore selectively infect tumors.Citation25,Citation26 Such defects in interferon signaling have also been recognized in tumor-associated endothelial cells facilitating infection of tumor vasculature mediated by VEGF signaling.Citation27 Multiple tumor suppressor genes are also key players in type I IFN pathways.Citation28 PTEN mutation is frequently encountered in PCa.Citation29 and enhances oncolytic apoptosis induced by another rhabdovirus, VSV, by exploiting IFN defects in a murine PCa modelCitation30 Clinically we anticipate that MG1-Maraba will exert significant oncolytic activity against PCa.
By designing an oncolytic vaccination strategy to express a human xenoantigen we were able to break tolerance against the murine form of the protein as demonstrated by responses against multiple CD8+ epitopes. In the context of the prime: boost, direct infection of splenic follicular B cells following IV MG1-Maraba facilitates antigen transfer to DCs in the follicular region of the spleen.Citation31 In the case of a primed immune response central memory (TCM) cells specific for the TAA against which they were primed reside in these follicles, however effector T cells are absent from this niche(31). This key functional and anatomic feature affords IV MG1-STEAP the privilege of directly boosting STEAP-specific TCM cells (via follicular DCs that are unimpeded by T effector cells) resulting in boosting of specific anti-tumor CD8+ T cells(31). By encoding the entire antigen we induced responses against various conserved, human and murine epitopes in vivo. Predicting immunogenic epitopes in silico is becoming more popular however, such approaches rely on complex bioinformatic procedures and as yet are not universally reliable.Citation32 Utilizing a vaccine platform that encodes an entire antigen is of potential therapeutic value to all individuals irrespective of MHC haplotype.
Post-mortem examination of tumor-free mice vaccinated against STEAP detected only expected, on-target inflammation and provided preclinical evidence of safety and specificity. Normally murine STEAP expression is highest in prostatic tissue with low level expression reported in the kidneys and testes.Citation33 Concordant with this we observed the induction of prostatic inflammation reminiscent of prostatitis following STEAP vaccination. No gross or microscopic pathology was noted at six weeks post-vaccination in the kidneys, testes, lungs or urinary bladder. Prostatic inflammation following STEAP vaccination highlights the potency of our platform, as indicated by a functional breach in immune tolerance. STEAP is over-expressed in various human malignancies including but not limited to prostatic, lung, renal and urinary bladder cancers.Citation34-Citation38 Early stage clinical trials of oncolytic vaccination using MG1-Maraba will provide further information regarding the safety of this approach for PCa and other indications. Oncolytic STEAP vaccination generates specific immunity leading to prostatic inflammation and this coupled with the absence of any other documented pathology emphasizes the potential utility of directing cancer immunotherapies capable of inducing anti-STEAP CD8+ immunity.
In advanced TRAMP-C2 tumors oncolytic STEAP vaccination impeded tumor progression. Other groups have employed adenoviral STEAP primes followed by boosting with either DCs pulsed with tumor cell lysates(39) or a modified vaccinia Ankara (virus)(40) and were able to demonstrate therapeutic efficacy in the TRAMP-C1 prostate model once tumors became palpable. Here TRAMP-C2 tumors were treated at a more advanced stage (mean tumor volume of 250mmCitation3) and we documented larger peripheral blood immune responses and more extensive TIL infiltrate than the alternative STEAP based prime: boost platforms.Citation39,Citation40 Boosting with MG1-STEAP compares favorably with other vaccines targeting STEAP, as not only is MG1 oncolytic but immune responses of a greater magnitude than previously described approaches are generated.
The exclusion of CD8+ T cells from the TME is considered a roadblock to immunotherapy in many solid tumors including prostatic cancer(7). Marked and widespread CD8+ infiltration of TRAMP-C2 tumors following oncolytic STEAP vaccination supports the ability of our therapeutic strategy to overcome this barrier. Low expression of MHCI by TRAMP-C2 cells act as a mechanism for evasion of immune attack by cytotoxic T cells in this model.Citation41 and approaches to recover loss of tumor MHCI expression are sought afterCitation42 Oncolytic STEAP vaccination therefore exhibits two highly desirable properties for prospective cancer immunotherapeutics.
A variety of possible explanations exist for tumor progression in the TRAMP-C2 model. Decreased Ifnar1 expression was detected in our NanoString analysis and intra-tumoral loss of the type I interferon receptor (IFNAR1) has been implicated in tumor progression.Citation43 By treating advanced tumors that had been engrafted at least 5 weeks prior to treatment it is expected that such tumors had undergone significant immune remodeling and escape mechanisms.Citation44 Significant loss of STEAP was not seen following treatment suggesting immunoediting and antigenic loss following vaccination was not a major factor in tumor progression. Patchy and decreased MHCI protein expression in end-stage tumors is incompatible with complete CD8+ mediated tumor clearance and may depict a significant selection pressure exerted on TRAMP-C2 tumors by CD8+ T cells. Native STEAP is primarily expressed within plasma membranes therefore generation of humoral immunity may also exert anti-neoplastic activity(37,38). STEAP targeted antibodies have demonstrated anti-PCa activity in human trials.Citation45 Future studies of combination therapies that target STEAP both humorally and via the induction of CD8+ immunity are indicated.
The lack of improvement in therapeutic efficacy when PD1 blockade was combined with oncolytic vaccination could indicate that anti-STEAP T cells were not functionally exhausted and another group has demonstrated that combining checkpoint blockade does not necessarily enhance the function of anti-tumor T cells induced by OVs.Citation46 Increased transcript levels of immune checkpoints do not always indicate functionally exhausted T cells because such transcriptional changes can simply reflect increased numbers of infiltrating CD8+ T cells,Citation47 a similar phenomenon for PD1 may be at play here given our Reactome pathway data. Alternatively combination with multiple checkpoint blocking agents may improve anti-tumor T cell activity as demonstrated in other murine models.Citation48 A significant increase in the Klrc1 gene expression encoding for the inhibitory NGK2A receptor was documented at peak-boost. Increased NKG2A expression on CD8+ T cells has been associated with suppression of effector function therefore NKG2A can be considered an immune checkpoint.Citation49 A humanized NKG2A blocking antibody has documented immune-mediated anti-neoplastic activity when used in combination with PD1 blockade.Citation50 Future studies characterizing the role of NKG2A in the TRAMP-C2 model and the potential blockade of this checkpoint alongside oncolytic STEAP vaccination are warranted. Oncolytic STEAP vaccination is ideally placed for future combinatorial investigations with other immunotherapeutics.
Current clinical and preclinical data support the value of vaccine-based strategies for the treatment of PCa but optimization of such modalities are required to improve efficacy(5). This study reports the use of a replicating oncolytic virus in a prime: boost regimen with a relevant prostatic antigen. MG1-Maraba is able to boost CD8+ T cell immune responses, convert an immunologically “cold” TME to a “hot” TME as well as exerting direct oncolytic activity against multiple PCa models. Oncolytic STEAP vaccination represents a readily translatable medical avenue worthy of clinical appraisal in the on-going quest to develop safe and effective therapies for aggressive prostate cancer.
Conflict of interest
DFS, JCB and BDL are named as inventors on patents covering Maraba virus as an oncolytic vaccine.
supp_data.docx
Download MS Word (2.8 MB)Additional information
Funding
References
- Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA. Cancer J Clin. 2015;65:87–108. doi:10.3322/caac.21262.
- Kantoff PW, Schuetz TJ, Blumenstein BA, Glode LM, Bilhartz DL, Wyand M, Manson K, Panicali DL, Laus R, Schlom J, et al. Overall Survival Analysis of a Phase II Randomized Controlled Trial of a Poxviral-Based PSA-Targeted Immunotherapy in Metastatic Castration-Resistant Prostate Cancer. J Clin Oncol. 2010;28:1099–105. doi:10.1200/JCO.2009.25.0597. PMID:20100959.
- Small EJ, Schellhammer PF, Higano CS, Redfern CH, Nemunaitis JJ, Valone FH, Verjee SS, Jones LA, Hershberg RM. Placebo-controlled phase III trial of immunologic therapy with sipuleucel-T (APC8015) in patients with metastatic, asymptomatic hormone refractory prostate cancer. J Clin Oncol. 2006;24:3089–94. doi:10.1200/JCO.2005.04.5252. PMID:16809734.
- Kwek SS, Cha E, Fong L. Unmasking the immune recognition of prostate cancer with CTLA4 blockade. Nat Rev Cancer. 2012;12:289–97. doi:10.1038/nrc3223. PMID:22378189.
- Ren R, Koti M, Hamilton T, Graham CH, Nayak JG, Singh J, Drachenberg DE, Siemens DR. A primer on tumour immunology and prostate cancer immunotherapy. Can Urol Assoc J. 2016;10:60–5. doi:10.5489/cuaj.3418. PMID:26977209.
- Gajewski TF. The Next Hurdle in Cancer Immunotherapy: Overcoming the Non-T-Cell-Inflamed Tumor Microenvironment. Semin Oncol. 2015;42:663–71. doi:10.1053/j.seminoncol.2015.05.011. PMID:26320069.
- Joyce JA, Fearon DT. T cell exclusion, immune privilege, and the tumor microenvironment. Science. 2015;348:74–80. doi:10.1126/science.aaa6204. PMID:25838376.
- Vesalainen S, Lipponen P, Talja M, Syrjänen K. Histological grade, perineural infiltration, tumour-infiltrating lymphocytes and apoptosis as determinants of long-term prognosis in prostatic adenocarcinoma. Eur J Cancer. 1994;30A:1797–803. doi:10.1016/0959-8049(94)E0159-2.
- Hussein M-RA, Al-Assiri M, Musalam AO. Phenotypic characterization of the infiltrating immune cells in normal prostate, benign nodular prostatic hyperplasia and prostatic adenocarcinoma. Exp Mol Pathol. 2009;86:108–13. doi:10.1016/j.yexmp.2008.11.010. PMID:19111537.
- Robinson DR, Wu Y-M, Lonigro RJ, Vats P, Cobain E, Everett J, Cao X, Rabban E, Kumar-Sinha C, Raymond V, et al. Integrative clinical genomics of metastatic cancer. Nature. 2017;548:297–303. doi:10.1038/nature23306. PMID:28783718.
- Nardone V, Botta C, Caraglia M, Martino EC, Ambrosio MR, Carfagno T. Tumor infiltrating T lymphocytes expressing FoxP3, CCR7 or PD-1 predict the outcome of prostate cancer patients subjected to salvage radiotherapy after biochemical relapse. Cancer Biol Ther. 2016;17:1213–20. doi:10.1080/15384047.2016.1235666. PMID:27791459.
- Lichty BD, Breitbach CJ, Stojdl DF, Bell JC. Going viral with cancer immunotherapy. Nat Rev Cancer. 2014;14:559–67. doi:10.1038/nrc3770. PMID:24990523.
- Brun J, McManus D, Lefebvre C, Hu K, Falls T, Atkins H, Bell JC, McCart JA, Mahoney D, Stojdl DF, et al. Identification of genetically modified Maraba virus as an oncolytic rhabdovirus. Mol Ther. 2010;18:1440–9. doi:10.1038/mt.2010.103. PMID:20551913.
- Pol JG, Zhang L, Bridle BW, Stephenson KB, Rességuier J, Hanson S, Chen L, Kazdhan N, Bramson JL, Stojdl DF, et al. Maraba Virus as a Potent Oncolytic Vaccine Vector. Mol Ther. 2014;22:420–9. doi:10.1038/mt.2013.249. PMID:24322333.
- Atherton MJ, Stephenson KB, Pol J, Wang F, Lefebvre C, Stojdl DF, Nikota JK, Dvorkin-Gheva A, Nguyen A, Chen L, et al. Customized Viral Immunotherapy for HPV-Associated Cancer. Cancer Immunol Res. 2017;5:847–59. doi:10.1158/2326-6066.CIR-17-0102. PMID:28912369.
- Le Boeuf F, Selman M, Son HH, Bergeron A, Chen A, Tsang J, Butterwick D4 Arulanandam R, Forbes NE, Tzelepis F, et al. Oncolytic Maraba Virus MG1 as a Treatment for Sarcoma. Int J Cancer. 2017;141:1257–64. doi:10.1002/ijc.30813. PMID:28568891.
- Hubert RS, Vivanco I, Chen E, Rastegar S, Leong K, Mitchell SC, Madraswala R, Zhou Y, Kuo J, Raitano AB, et al. STEAP: A prostate-specific cell-surface antigen highly expressed in human prostate tumors. Proc Natl Acad Sci. 1999;96:14523–8. doi:10.1073/pnas.96.25.14523. PMID:10588738.
- Yang D, Holt GE, Velders MP, Kwon ED, Kast WM. Murine Six-Transmembrane Epithelial Antigen of the Prostate, Prostate Stem Cell Antigen, and Prostate-specific Membrane Antigen Prostate-specific Cell-Surface Antigens Highly Expressed in Prostate Cancer of Transgenic Adenocarcinoma Mouse Prostate Mice. Cancer Res. 2001;61:5857–60. PMID:11479226.
- Walter A, Barysch MJ, Behnke S, Dziunycz P, Schmid B, Ritter E, Gnjatic S, Kristiansen G, Moch H, Knuth A, et al. Cancer-testis antigens and immunosurveillance in human cutaneous squamous cell and basal cell carcinomas. Clin Cancer Res. 2010;16:3562–70. doi:10.1158/1078-0432.CCR-09-3136. PMID:20519358.
- Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3:Article3. doi:10.2202/1544-6115.1027. PMID:16646809.
- Singh P, Pal SK, Alex A, Agarwal N. Development of PROSTVAC immunotherapy in prostate cancer. Future Oncol. 2015;11:2137–48. doi:10.2217/fon.15.120. PMID:26235179.
- Devaud C, John LB, Westwood JA, Darcy PK, Kershaw MH. Immune modulation of the tumor microenvironment for enhancing cancer immunotherapy. Oncoimmunology. 2013;2:e25961. doi:10.4161/onci.25961. PMID:24083084.
- Breitbach CJ, Lichty BD, Bell JC. Oncolytic Viruses: Therapeutics With an Identity Crisis. EBioMedicine. 2016;9:31–6. doi:10.1016/j.ebiom.2016.06.046. PMID:27407036.
- Russell SJ, Peng K-W. Oncolytic Virotherapy: A Contest between Apples and Oranges. Mol Ther. 2017;25:1107–16. doi:10.1016/j.ymthe.2017.03.026. PMID:28392162.
- Stojdl DF, Lichty BD, tenOever BR, Paterson JM, Power AT, Knowles S, Marius R, Reynard J, Poliquin L, Atkins H, et al. VSV strains with defects in their ability to shutdown innate immunity are potent systemic anti-cancer agents. Cancer Cell. 2003;4:263–75. doi:10.1016/S1535-6108(03)00241-1. PMID:14585354.
- Shou J, Soriano R, Hayward SW, Cunha GR, Williams PM, Gao W-Q. Expression profiling of a human cell line model of prostatic cancer reveals a direct involvement of interferon signaling in prostate tumor progression. Proc Natl Acad Sci. 2002;99:2830–5. doi:10.1073/pnas.052705299. PMID:11880635.
- Arulanandam R, Batenchuk C, Angarita FA, Ottolino-Perry K, Cousineau S, Mottashed A, Burgess E, Falls TJ, De Silva N, Tsang J, et al. VEGF-Mediated Induction of PRD1-BF1/Blimp1 Expression Sensitizes Tumor Vasculature to Oncolytic Virus Infection. Cancer Cell. 2015;28:210–24. doi:10.1016/j.ccell.2015.06.009. PMID:26212250.
- Muñoz-Fontela C, Mandinova A, Aaronson SA, Lee SW. Emerging roles of p53 and other tumour-suppressor genes in immune regulation. Nat Rev Immunol. 2016;16:741–50. doi:10.1038/nri.2016.99. PMID:27667712.
- Cairns P, Okami K, Halachmi S, Halachmi N, Esteller M, Herman JG, Jen J, Isaacs WB, Bova GS, Sidransky D, et al. Frequent Inactivation of PTEN/MMAC1 in Primary Prostate Cancer. Cancer Res. 1997;57:4997–5000. PMID:9371490.
- Moussavi M, Fazli L, Tearle H, Guo Y, Cox M, Bell J, Ong C, Jia W, Rennie PS. Oncolysis of prostate cancers induced by vesicular stomatitis virus in PTEN knockout mice. Cancer Res. 2010;70:1367–76. doi:10.1158/0008-5472.CAN-09-2377. PMID:20145134.
- Bridle BW, Nguyen A, Salem O, Zhang L, Koshy S, Clouthier D, Chen L, Pol J, Swift SL, Bowdish DM, et al. Privileged Antigen Presentation in Splenic B Cell Follicles Maximizes T Cell Responses in Prime-Boost Vaccination. J Immunol. 2016;196:4587–95. doi:10.4049/jimmunol.1600106. PMID:27183620.
- Soria-Guerra RE, Nieto-Gomez R, Govea-Alonso DO, Rosales-Mendoza S. An overview of bioinformatics tools for epitope prediction: implications on vaccine development. J Biomed Inform. 2015;53:405–14. doi:10.1016/j.jbi.2014.11.003. PMID:25464113.
- Garcia-Hernandez Mde la L, Gray A, Hubby B, Kast WM. In vivo effects of vaccination with six-transmembrane epithelial antigen of the prostate: a candidate antigen for treating prostate cancer. Cancer Res. 2007;67:1344–51. doi:10.1158/0008-5472.CAN-06-2996. PMID:17283172.
- Challita-Eid PM, Morrison K, Etessami S, An Z, Morrison KJ, Perez-Villar JJ, Raitano AB, Jia XC, Gudas JM, Kanner SB, et al. Monoclonal antibodies to six-transmembrane epithelial antigen of the prostate-1 inhibit intercellular communication in vitro and growth of human tumor xenografts in vivo. Cancer Res. 2007;67:5798–805. doi:10.1158/0008-5472.CAN-06-3849. PMID:17575147.
- Zhuang X, Herbert JMJ, Lodhia P, Bradford J, Turner AM, Newby PM, Thickett D, Naidu U, Blakey D, Barry S, et al. Identification of novel vascular targets in lung cancer. Br J Cancer. 2015;112:485–94. doi:10.1038/bjc.2014.626. PMID:25535734.
- Azumi M, Kobayashi H, Aoki N, Sato K, Kimura S, Kakizaki H, Tateno M. Six-transmembrane epithelial antigen of the prostate as an immunotherapeutic target for renal cell and bladder cancer. J Urol. 2010;183:2036–44. doi:10.1016/j.juro.2009.12.094. PMID:20303532.
- Moreaux J, Kassambara A, Hose D, Klein B. STEAP1 is overexpressed in cancers: A promising therapeutic target. Biochem Biophys Res Commun. 2012;429:148–55. doi:10.1016/j.bbrc.2012.10.123. PMID:23142226.
- Gomes IM, Maia CJ, Santos CR. STEAP proteins: from structure to applications in cancer therapy. Mol Cancer Res. 2012;10:573–87. doi:10.1158/1541-7786.MCR-11-0281. PMID:22522456.
- Kim S, Lee J-B, Lee GK, Chang J. Vaccination with recombinant adenoviruses and dendritic cells expressing prostate-specific antigens is effective in eliciting CTL and suppresses tumor growth in the experimental prostate cancer. The Prostate. 2009;69:938–48. doi:10.1002/pros.20942. PMID:19267351.
- Cappuccini F, Stribbling S, Pollock E, Hill AVS, Redchenko I. Immunogenicity and efficacy of the novel cancer vaccine based on simian adenovirus and MVA vectors alone and in combination with PD-1 mAb in a mouse model of prostate cancer. Cancer Immunol Immunother. 2016;65:701–13. doi:10.1007/s00262-016-1831-8. PMID:27052571.
- Martini M, Testi MG, Pasetto M, Picchio MC, Innamorati G, Mazzocco M, Ugel S, Cingarlini S, Bronte V, Zanovello P, et al. IFN-gamma-mediated upmodulation of MHC class I expression activates tumor-specific immune response in a mouse model of prostate cancer. Vaccine. 2010;28:3548–57. doi:10.1016/j.vaccine.2010.03.007. PMID:20304037.
- Garrido F, Aptsiauri N, Doorduijn EM, Garcia Lora AM, van Hall T. The urgent need to recover MHC class I in cancers for effective immunotherapy. Curr Opin Immunol. 2016;39:44–51. doi:10.1016/j.coi.2015.12.007. PMID:26796069.
- Katlinski KV, Gui J, Katlinskaya YV, Ortiz A, Chakraborty R, Bhattacharya S, Carbone CJ, Beiting DP, Girondo MA, Peck AR, et al. Inactivation of Interferon Receptor Promotes the Establishment of Immune Privileged Tumor Microenvironment. Cancer Cell. 2017;31:194–207. doi:10.1016/j.ccell.2017.01.004. PMID:28196594.
- Hanahan D, Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell. 2011;144:646–74. doi:10.1016/j.cell.2011.02.013. PMID:21376230.
- Danila DC, Szmulewitz RZ, Baron AD, Higano CS, Scher HI, Morris MJ. A phase I study of DSTP3086S, an antibody-drug conjugate (ADC) targeting STEAP-1, in patients (pts) with metastatic castration-resistant prostate cancer (CRPC). J Clin Oncol. 2014;32:5024-.
- Shim KG, Zaidi S, Thompson J, Kottke T, Evgin L, Rajani KR, Schuelke M, Driscoll CB, Huff A, Pulido JS, et al. Inhibitory Receptors Induced by VSV Viroimmunotherapy Are Not Necessarily Targets for Improving Treatment Efficacy. Mol Ther. 2017;25:962–75. doi:10.1016/j.ymthe.2017.01.023. PMID:28237836.
- Ganesan A-P, Clarke J, Wood O, Garrido-Martin EM, Chee SJ, Mellows T, Samaniego-Castruita D, Singh D, Seumois G, Alzetani A, et al. Tissue-resident memory features are linked to the magnitude of cytotoxic T cell responses in human lung cancer. Nat Immunol. 2017;18:940–950. doi:10.1038/ni.3775. PMID:28628092.
- Benci JL, Xu B, Qiu Y, Wu TJ, Dada H, Twyman-Saint\sVictor C, Cucolo L, Lee DSM, Pauken KE, Huang AC, et al. Tumor Interferon Signaling Regulates a Multigenic Resistance Program to Immune Checkpoint Blockade. Cell. 2016;167:1540–1554.e12. doi:10.1016/j.cell.2016.11.022. PMID:27912061.
- Sheu B-C, Chiou S-H, Lin H-H, Chow S-N, Huang S-C, Ho H-N. Up-regulation of inhibitory natural killer receptors CD94/NKG2A with suppressed intracellular perforin expression of tumor-infiltrating CD8+ T lymphocytes in human cervical carcinoma. Cancer Res. 2005;65:2921–9. doi:10.1158/0008-5472.CAN-04-2108. PMID:15805295.
- Sola C, Arnoux T, Chanuc F, Fuseri N, Rossi B, Gauthier L. Abstract 2342: NKG2A immune checkpoint blockade enhances the anti-tumor efficacy of PD1/PD-L1 inhibitors in a preclinical model. Cancer Res. 2016;76:2342-. doi:10.1158/1538-7445.AM2016-2342.