
ABSTRACT
Multiple non-redundant immunosuppressive pathways co-exist in the tumor microenvironment and their co-targeting can increase clinical responses. Indeed, concurrent blockade of CTLA-4 and PD-1 in patients with advanced melanoma increased clinical responses over monotherapy alone although the frequency and severity of immune related adverse events (irAEs) also increased. Nevertheless, a substantial number of patients still display an innate resistance phenotype and are unresponsive to current approved immunotherapies even when utilized in combination. In this study, we generated Pdcd1−/−CD96−/− and Tigit−/−CD96−/− mice to investigate how loss of CD96 in combination with PD-1 or TIGIT impacts on immune homeostasis and hence the potential of inducing immune related toxicities following co-targeting of these pairs of receptors. The ability of Pdcd1−/−CD96−/− and Tigit−/−CD96−/− mice to suppress primary tumor growth was also assessed using the MC38 colon carcinoma and SM1WT1 BRAF-mutated melanoma tumor models. Both Pdcd1−/−CD96−/− or Tigit−/−CD96−/− mice displayed no overt perturbations in immune homeostasis over what was previously reported with Pdcd1−/− or Tigit−/− mice even when aged for 22 months. Interestingly, increased suppression of subcutaneous tumor growth and complete responses was seen in Pdcd1−/−CD96−/− mice compared to Pdcd1−/− or CD96−/− mice depending upon the tumor model. In contrast, in these models, growth suppression in Tigit−/−CD96−/− were similar to Tigit−/− or CD96−/− . This enhanced anti-tumor efficacy of Pdcd1−/−CD96−/− appeared to be due to favorable changes in the ratio of CD8+ T cells to T regulatory cells or CD11b+GR-1hi myeloid cells in the tumor microenvironment. Co-targeting CD96 and PD-1 may increase anti-tumor immunity over targeting PD-1 alone and potentially not induce serious immune-related toxicities and thus appears a promising strategy for clinical development.
Introduction
Antibodies targeting cytotoxic T lymphocyte associated antigen 4 (CTLA-4), programmed cell death protein 1 (PD-1) or its ligand PD-L1 have demonstrated clinical activity in more than 15 cancer types, including melanoma and non-small-cell lung carcinoma (NSCLC).Citation1 More recently, concurrent blockade of CTLA-4 and PD-1 in patients with advanced melanoma further increased clinical response over monotherapy alone.Citation2 Overall, these data suggest that multiple non-redundant immunosuppressive pathways co-exist in the tumor microenvironment and that their co-targeting can increase clinical responses. However, a substantial number of patients display an innate resistance phenotype and are unresponsive to current approved immunotherapies. Furthermore, a considerable number of patients treated with anti-CTLA-4 or PD-1/PD-L1, particularly in combination can develop a unique spectrum of toxicities termed immune related adverse events (irAEs), which can result in serious morbidities and can sometimes lead to fatalities.Citation3 Therefore, there is strong interest in identifying new combination therapies that have better therapeutic indices (increased anti-tumor efficacy and reduced irAEs).
The development of irAEs in patients given immunotherapy is not unexpected since immune checkpoint receptors are key in maintaining self-tolerance to minimize tissue damage.Citation4 Gene-targeted mice have been useful to assess how lack of specific immune checkpoint receptors or ligands impact on immune homeostasis and autoimmunity. CTLA-4−/− mice display severe lymphoproliferative disease with lymphocytic infiltration in several tissues including the heart, spleen and lungs causing the mice to become moribund at three to four weeks of age.Citation5, Citation6 Pdcd1−/− mice display strain specific autoimmune phenotypes which are generally quite mild. Loss of PD-1 in C57BL/6 mice was reported to cause late-onset lupus-like glomerulonephritis and arthritis.Citation7 In contrast, loss of PD-1 in BALB/c mice results in their development of dilated cardiomyopathy which leads to their premature death.Citation8 In contrast, C57BL/6 mice lacking other immune checkpoint receptors/ligands such as PD-L1,Citation9,Citation10 LAG-3 (Lymphocyte-activation gene 3)Citation11 or B7-H412 display minimal or subtle immunopathology. However, C57BL/6 Lag3−/−Pdcd1−/− mice develop lethal systemic autoimmunity with most mice becoming moribund by 10 weeks of age.Citation11 Similarly, when bred onto the 2D2 T-cell receptor (TCR) transgenic mice, which were predisposed to developing spontaneous experimental autoimmune encephalomyelitis (EAE), double deficiency in PD-1 and VISTA (V-domain immunoglobulin suppressor of T-cell activation) significantly accelerated the level of disease penetrance compared to similar 2D2-TCR transgenic mice lacking only VISTA or PD-113.
CD96 (TACTILE) and TIGIT (T-cell immunoglobulin and ITIM domain) belong to an emerging family of cell surface receptors that bind to ligands of the nectin and nectin-like family.Citation14 The expression patterns of CD96 and TIGIT are broadly similar between mouse and humans, where they are mainly found on peripheral T cells including regulatory T cells (Tregs) and NK cells, particularly following activation.Citation15-Citation19 CD155 (necl-5; PVR) is the main ligand that binds CD96 and TIGIT in both humans and mice.Citation17,Citation18,Citation20 CD155 also binds the activating receptor DNAM-1 (CD226), which like CD96 and TIGIT, is expressed on T and NK cells.Citation15,Citation21,Citation22 In mice, CD96 also binds CD111 (nectin-1), which has been demonstrated to enhance T cell and NK cell adhesionCitation17,Citation20 while TIGIT binds CD112 (PVRL2, nectin-2) and CD113 (PVRL3, nectin-3).Citation18,Citation19 Recently it was reported that CD112R, a novel co-inhibitory receptor which is preferentially expressed on human T cells binds CD112 with high affinity and competes with CD226 to bind CD112.Citation23
The function of CD96 on T cells is still largely unknown but its role as an inhibitory receptor was recently demonstrated in mice lacking CD96. NK cells from CD96−/− mice produced greater IFNγ in response to LPS and they also displayed enhanced resistance to 3´-methylcholanthrene (MCA)-induced fibrosarcoma and experimental lung metastases.Citation24 Subsequently, in mouse models of experimental and spontaneous lung metastases, blocking antibodies against CD96 (anti-CD96) increased NK cell effector function, resulting in suppression of metastases and this anti-tumor activity was dependent on NK cells, IFNγ, and DNAM-125. This study also demonstrated that anti-CD96 in combination with anti-CTLA-4 or anti-PD-1 further suppressed experimental lung metastases compared to monotherapy alone. In contrast, the inhibitory function of TIGIT on T cells is well described. Increased effector T cell function was reported in Tigit−/− mice or anti-TIGIT treated miceCitation19,Citation26-Citation28 while Tregs lacking TIGIT reportedly displayed reduced suppressive function.Citation29,Citation30 Similar to CD96, dual blockade of TIGIT with either PD-1 or PD-L1 significantly increased anti-tumor immunity against mouse tumors.Citation27 Although Tigit−/− mice displayed no increased protection against experimental or spontaneous lung metastases, anti-CD96 treated Tigit−/− mice displayed further reduction in tumor metastases compared to anti-CD96 treated WT mice suggesting that there could be merit in co-targeting CD96 and TIGIT.Citation25
Here, we have generated two novel strains of double deficient Pdcd1−/−CD96−/− and Tigit−/−CD96−/− mice to investigate whether loss of CD96 in combination with PD-1 or TIGIT impacts immune homeostasis and reveals anything about the potential safety of co-targeting these receptors. The ability of Pdcd1−/−CD96−/− and Tigit−/−CD96−/− mice to suppress primary tumor growth was also assessed using the MC38 colon carcinoma and SM1WT1 BRAF-mutated melanoma tumor models. Both Pdcd1−/−CD96−/− or Tigit−/−CD96−/− mice displayed no overt perturbations in immune homeostasis beyond that previously reported for Pdcd1−/− or Tigit−/− mice, even when aged for 22 months. Interestingly, increased tumor suppression was observed in Pdcd1−/−CD96−/− mice compared to Pdcd1−/− or CD96−/− mice bearing SM1WT1 but not MC38 tumor. The enhanced tumor growth suppression of SM1WT1 in Pdcd1−/−CD96−/− mice appeared to be due to favorable changes in CD8+ T cells to Treg or CD11b+GR-1hi myeloid cell ratios in the tumor microenvironment. The lack of serious immune-related toxicities developing in aged Pdcd1−/−CD96−/− mice study supports the potential safety in co-blockade of CD96 and PD-1 clinically.
Results
Pdcd1−/−CD96−/− and Tigit−/−CD96−/− mice do not display increased autoimmune phenotype
Both Pdcd1−/−CD96−/− and Tigit−/−CD96−/− strains of mice were born fertile and produced normal litter sizes. As C57BL/6 Pdcd1−/− mice were previously reported to develop late-onset autoimmunity,Citation7 we aged a cohort of wildtype (WT), single or double-deficient PD-1, CD96, or TIGIT mice for 22 months (). At 12 months of age, no overt changes were observed in survival between WT mice compared to mice that lacked PD-1 or CD96 alone or in combination and these mice displayed no overt signs of autoimmunity (). However, as these mice were further aged for another 10 months, both Pdcd1−/− and Pdcd1−/−CD96−/− mice displayed significantly decreased survival compared to WT mice (). However, there were no significant differences in survival between Pdcd1−/−CD96−/− mice and Pdcd1−/− mice suggesting the reduced survival effect is most likely due to host PD-1 loss (). By contrast, long-term survival of Tigit−/−CD96−/−, Tigit−/− , and CD96−/− mice was similar to WT mice and no overt signs of autoimmunity were detected ().
Figure 1. Pdcd1−/−CD96−/− or Tigit−/−CD96−/− mice display no overt signs of autoimmunity. (A, B) Groups of female C57BL/6 WT or the indicated gene-targeted mice (n = 14–29) were aged for a period of 22 months and monitored for survival. (C, D) At 12 months of age, some WT or gene-targeted mice from the cohort above (n = 4–5/group) were bled and the level of anti-dsDNA antibodies in sera determined by ELISA. Sera from aged gld−/− mice were used as a positive control. (E) From the cohort in (A), some WT or gene-targeted mice (n = 4–9/group) were euthanized at the age of 21–25 months. Livers were collected, fixed and stained with H&E. Histological liver score for each group is shown. (F) From the cohort in (A), mice at 25–28 weeks of age were weighed and the data shown. Data in C, D, E and F are presented as mean ± SEM with each symbol representing an individual mouse. Statistical differences between the indicated genotypes were determined by (A, B) Log-Rank sum test (*p<0.05, ****p<0.0001) or by (C, D, E and F) one-way ANOVA with Bonferroni's post-test analysis (*p<0.05, **p<0.01). All experiments were performed once.
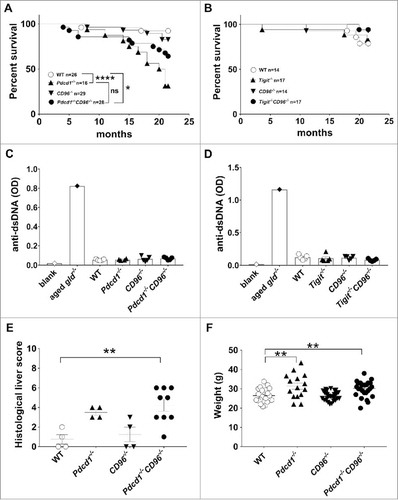
The initial characterization of C57BL/6 Pdcd1−/− mice reported that these mice developed arthritis and some features of late onset lupus-like disease characterized by the presence of autoantibodies and mild glomerulonephritis.Citation7 In contrast, a recent study by Woo et al., reported no significant increase in the level of autoantibodies in C57BL/6 Pdcd1−/− mice.Citation11 When we measured the level of anti-dsDNA antibodies in the sera of 12-month old WT and gene-targeted mice lacking PD-1, CD96, or TIGIT alone or PD-1/CD96 or TIGIT/CD96 in combination (, ), we did not observe any increase in the level of anti-dsDNA antibodies from Pdcd1−/− compared to WT mice similar to the study from Woo et al.Citation11 Furthermore, the levels of anti-dsDNA, total serum IgG and anti-nuclear antibodies in single or double deficient mice were similar to WT mice (, )(data not shown).
From the aging cohort in , a group of WT or gene-targeted mice were euthanized and various organs (liver, colon, lung, and kidney) assessed for histological evidence of immune changes. Similar to a previous study,Citation13 an increase in immune cell infiltration was observed in Pdcd1−/− mice as reflected by an increased histological score in livers (). However, there was no significant difference in the histological liver scores between Pdcd1−/−CD96−/− and Pdcd1−/− mice (). By contrast, we did not see an increase in immune cell infiltrates in other organs assessed (data not shown). We also observed no difference in immune cell infiltration in the livers, colons and lungs of Tigit−/−CD96−/− and Tigit−/− mice when compared to WT mice (data not shown). Loss of immune checkpoint receptors can result in lymphoproliferation and inflammation, which may manifest as weight loss. A cohort of 25–28 week old WT mice was compared to those same aged mice lacking PD-1 or CD96 alone or both receptors. Surprisingly, Pdcd1−/−CD96−/− and Pdcd1−/− mice both displayed increased weight compared to WT mice at this age (), and this difference was also observed in 7–9 week old mice (data not shown). Similarly, 24 week old Tigit−/−CD96−/− and Tigit−/− mice also displayed increased weight compared to age-matched WT mice (data not shown). This suggests that some immune checkpoint receptors may also regulate metabolic pathways.
Minor changes in Treg homeostasis in Pdcd1−/−CD96−/− and Tigit−/−CD96−/− mice
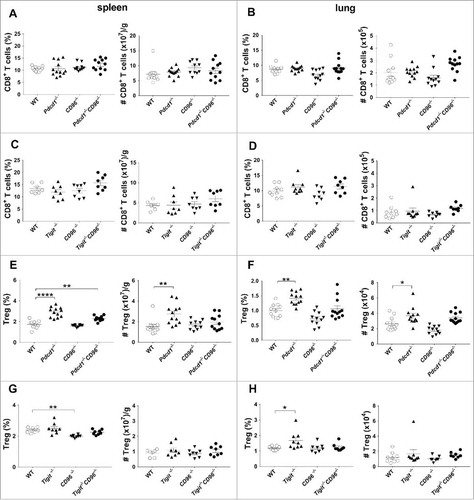
We next examined younger Pdcd1−/−CD96−/− (13–15 weeks old) and Tigit−/−CD96−/− mice (8–10 weeks old) for any changes in major immune cell populations in the spleen and lung (). There were no significant changes in the numbers and proportions of CD45.2+ hematopoietic cells (data not shown). Similarly, no gross changes were seen in the proportion and numbers of CD8+ T cells () or other major immune cell populations such as CD4+ effector T cells, NK cells, B cells and myeloid cells (data not shown) between Pdcd1−/−CD96−/− or Tigit−/−CD96−/− mice compared to age-matched WT mice or age matched mice that were deficient for PD-1, CD96 or TIGIT. In concordance with previous reportsCitation31,,Citation32 we observed a significant increase in CD4+Foxp3+ T regulatory cells (Treg) proportion and number in the spleen and lung of Pdcd1−/− compared to WT mice (, ). Interestingly, Pdcd1−/−CD96−/− mice also displayed an increase in Tregs compared to WT mice, but this increase in Treg proportion and number was similar to that observed in Pdcd1−/− mice (, ). In contrast, WT and Tigit−/−CD96−/− mice had similar levels of Tregs (, ). We next, assessed the activation status of T cells (CD8+, CD4+ Teff, Tregs) in the spleen and lungs of Pdcd1−/−CD96−/− or Tigit−/−CD96−/− mice using the markers CD44 and CD62L (Supp. Fig. 1, 2). Similar to previous reports, Pdcd1−/− mice displayed an increased proportion of T cells with an effector memory (TEM) phenotype in their spleen and lung compared to WT mice and this was also observed for Pdcd1−/−CD96−/− mice (Supp. Fig. 1). The activation status of T cells was generally similar between Tigit−/−CD96−/− mice and WT mice (Supp. Fig. 2). In 12–13 month old mice, we still observed this increase in proportions of TEM phenotype in Pdcd1−/−CD96−/− or Pdcd1−/− mice compared to WT mice in the CD8+ T cell population (Supp. Fig. 3) although this phenotype was not observed in 24 month old mice (data not shown).
Figure 2. No major differences in the frequency and number of CD8+ T cells and Tregs between Pdcd1−/−CD96−/− or Tigit−/−CD96−/− and their corresponding single gene-targeted mice. The proportions and numbers of CD8+ T cells or Tregs from the spleens and lungs of male C57BL/6 WT or the indicated gene-targeted mice (13–15 weeks) (A, B, E, F) or from female C57BL/6 WT or the indicated gene-targeted mice (8–10 weeks) (C, D, G, H) (n = 3–6/group) were analysed by flow cytometry, gating on live CD45.2 cells of leukocyte morphology. Cell numbers are represented as per gram (g) of spleen or per total lung. Data presented as mean ± SEM with each symbol representing an individual mouse. Statistical differences in cell proportions and numbers between different genotypes were determined by a one-way ANOVA with Bonferroni's post-test analysis (*p<0.05; **p<0.01, **** p<0.0001). Data pooled from two independent experiments.
Enhanced tumor resistance of Pdcd1−/−CD96−/- mice is tumor model dependent and requires CD8+ T cells, NK cells and IFNγ
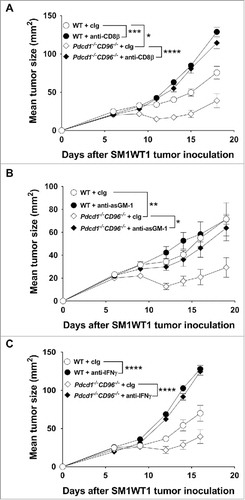
We have previously demonstrated that the anti-metastatic efficacy of anti-CD96 mAbs was enhanced when combined with anti-PD-1 in WT mice or when administered in Tigit−/− mice.Citation25 However, the effects of combined loss of host PD-1 and CD96 or TIGIT and CD96 on subcutaneous tumor growth has not been examined. We used two models, the MC38 colon adenocarcinoma that is responsive to anti-PD-1Citation33 and the SM1WT1 Braf-mutated melanoma that is non-responsive to anti-PD-134 (, Supp. Fig. 4, 5). Growth of SM1WT1 in Pdcd1−/−CD96−/− mice was significantly suppressed compared to WT, Pdcd1−/− or CD96−/− mice ( and Supp. Fig. 4A, B). Additionally, a small but significant proportion of Pdcd1−/−CD96−/− mice completely rejected their tumors compared to Pdcd1−/− mice (). Although MC38 tumor growth was strongly suppressed, no additional resistance was observed in Pdcd1−/−CD96−/− compared with Pdcd1−/− mice (, Supp. Fig. 4C) and similar complete rejection rates were observed between these strains (). Interestingly, in the SM1WT1 and MC38 tumor models, modest growth suppression was seen in Tigit−/− or CD96−/− mice compared to WT mice, but no further tumor resistance was observed in Tigit−/−CD96−/− mice (Supp. Fig. 5). Nevertheless, a greater suppression of B16F10 tumor growth was observed in Tigit−/−CD96−/− mice compared with Tigit−/− or CD96−/− mice (Supp. Fig. 6), and the co-blockade of TIGIT and CD96 using monoclonal antibodies is now being assessed in this and other tumor models (Mittal et al. manuscript in preparation). We next determined the cell type and mechanism that was suppressing SM1WT1 tumor growth in Pdcd1−/−CD96−/− mice (). Depletion of CD8+ T cells entirely abolished the ability of Pdcd1−/−CD96−/− mice to suppress tumor growth (). While NK cells generally do not play a role in control of subcutaneous tumor growth such as MC3835, we have previously shown that NK cells were involved in suppressing SM1WT1 subcutaneous tumor growth.Citation36,Citation37 We therefore set up a similar experiment as and depleted NK cells and we observed a partial loss of tumor suppression (). Neutralization of IFNγ also abrogated tumor suppression in this strain of mice (). Overall, these results demonstrate that CD8+ T cells, NK cells and IFN-γ were critical mediators of anti-tumor efficacy in SM1WT1 tumor-bearing Pdcd1−/−CD96−/− mice.
Figure 3. Enhanced tumor growth suppression in Pdcd1−/−CD96−/− mice is tumor model dependent. Groups of C57BL/6 WT or gene-targeted mice (n = 5–9/group) were injected s.c with either (A) 1 × 106 SM1WT1 melanoma or (C) 1 × 106 MC38 colon carcinoma cells. Tumor sizes were determined with caliper square measurements with data represented as means ± SEM. Data representative of three independent experiments. Proportion and number of mice from the indicated genotype that rejected (B) SM1WT1 or (D) MC38 tumors are shown. Data in (B, D) pooled from three independent experiments. Statistical differences in tumor sizes at (A) day 21 or (C) day 19 between different genotypes were determined by one-way ANOVA with Bonferroni's post-test analysis (*p<0.05, ***p<0.001). (B, D) Pairwise comparison of complete response rates between Pdcd1−/−CD96−/− and Pdcd1−/− mice was performed using Fisher's exact test (****: p<0.0001).
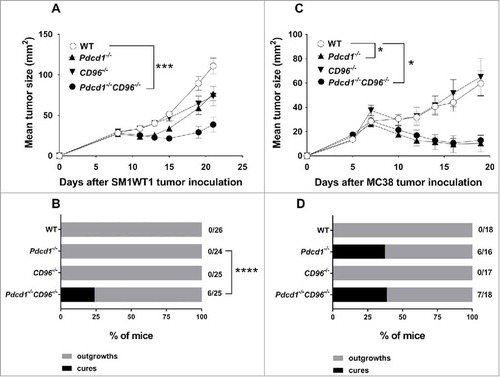
Figure 4. Suppression of SM1WT1 tumor growth in Pdcd1−/−CD96−/− mice requires CD8+ T cells, NK cells and IFNγ. Groups of C57BL/6 WT and Pdcd1−/−CD96−/− mice (n = 7–9/group) were injected s.c with 1 × 106 SM1WT1 melanoma cells on day 0. Some groups of mice were treated i.p on days -1, 0, 7 and 14 with either (A) anti-CD8β or cIg (each 100 µg/mouse) or (B) anti-asGM-1 or cIg (each 50 µg/mouse) or (C) anti-IFNγ or cIg (each 250 µg/mouse). Tumor sizes were determined with caliper square measurements with data represented as mean ± SEM. Experiments were all performed once. Significant differences between tumor sizes at day (A) 18, (B) 19 and (C) 16 were determined by one-way ANOVA with Bonferroni's post-test analysis (*p<0.05; **p<0.01; ***p<0.001; ****: p<0.0001).
Enhanced suppression of SM1WT1 in Pdcd1−/−CD96−/− mice correlates with increased CD8+T cell:Treg and CD8+T cell:CD11b+GR-1hi ratios
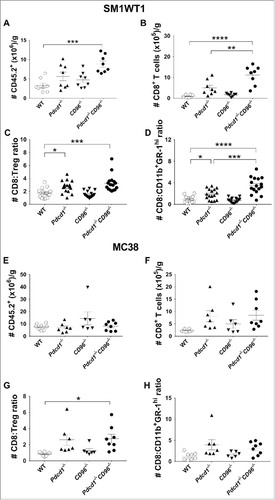
To understand why SM1WT1 tumors, and not MC38 tumors, were better controlled in Pdcd1−/−CD96−/− compared to Pdcd1−/− or CD96−/− mice, we next used flow cytometry to investigate the tumor infiltrating leukocytes (TILs) in these tumor-bearing mice at an early time point (day 8) () (Supp. Fig. 7, 8). This time point was chosen since all the tumors were equal in size, thus eliminating any confounding effects of tumor size on the analysis. We observed a significant increase in the number and proportion of CD45.2+ hematopoietic cells (, Supp. Fig. 7A) and CD8+ T cells (, Supp. Fig. 7B) in SM1WT1 tumor-bearing Pdcd1−/− CD96−/− mice compared to WT mice, whereas such an increase was not as obvious in tumor-bearing Pdcd1−/− or CD96−/− mice. Compared to WT mice, both SM1WT1 tumor-bearing Pdcd1−/−CD96−/− and Pdcd1−/− mice displayed an increase in the number of CD4+ T effector cells and Tregs, while NK cells and myeloid CD11b+GR-1hi cell numbers were generally not changed (Supp. Fig. 8A-D). Despite the increase in Tregs, we still observed a significant increase in the CD8+ T cell:Treg ratio and CD8+ T cell:CD11b+Gr-1hi ratio between Pdcd1−/−CD96−/− mice and other control strains of mice (, , Supp. Fig. 7 C, D). In contrast to SM1WT1, we did not observe differences in the number of CD45.2+ cells in MC38 tumor-bearing WT Pdcd1−/−CD96−/−, Pdcd1−/− or CD96−/− mice (, Supp. Fig. 7E). Compared to WT mice, the number and proportion of CD8+ T cells generally increased similarly for Pdcd1−/−CD96−/−, Pdcd1−/− or CD96−/− mice but there were no major differences between these gene-targeted strains (, Supp. 7F). Similarly, we observed no major changes in CD4+ effector T cells, Tregs, NK cells and myeloid CD11b+Gr-1hi cells (Supp. Fig. 8E-H) or CD8+T cell:Treg ratio and CD8+ T cell:CD11b+Gr-1hi ratio (, , Supp. Fig. 7G, H) between these 4 genotypes.
Figure 5. Increased CD8+ T cell:Treg and CD8+ T cell:CD11b+GR-1hi ratio in SM1WT1 tumor-bearing Pdcd1−/−CD96−/− mice compared to WT, Pdcd1−/− and CD96−/− mice. Groups of C57BL/6 WT and gene-targeted mice (n = 6–8/group) were injected s.c with either (A-D) 1 × 106 SM1WT1 melanoma or (E-H) 1 × 106 MC38 colon carcinoma cells. On day 8 following tumor inoculations, tumors were harvested and single cell suspensions generated and were analysed by flow cytometry, gating on live CD45.2+ cells of leukocyte morphology. The number of CD45.2+ cells (A, E) or CD8+ T cells (B, F) are shown. Cell numbers are represented as per gram (g) of tumor. Data representative of two experiments for (A, B) while experiment was performed once for (E, F). The CD8:Treg (C, G) and CD8:CD11b+GR-1hi (D, H) ratios in SM1WT1 and MC38 tumors are pooled from two or one independent experiment respectively. Data presented as mean ± SEM with each symbol representing an individual mouse. Statistical differences between different genotypes were determined by one-way ANOVA with Bonferroni's post-test analysis (*p<0.05, **p<0.01; ***p<0.001; ****p<0.0001).
In addition to these changes, we also investigated whether the expression of PD-1/CD96/TIGIT/DNAM-1 on tumor-infiltrating lymphocytes or their ligand CD155 and PD-L1 differed between SM1WT1 and MC38 as an explanation for the differences in anti-tumor responses (Supp. Fig. 9, 10). Using FACS analysis, we profiled CD155 and PD-L1 expression on both SM1WT1 and MC38 tumors that were grown in culture or ex vivo derived (Supp. Fig. 9). In vitro, both cell lines expressed CD155 at similar levels (Supp. Fig. 9A, B), while PD-L1 expression was lower in SM1WT1 (Supp. Fig. 9C) compared to MC38 tumors (Supp. Fig. 9D). For the ex vivo analysis, single cell suspensions were generated from early-stage (day 8) SM1WT1 or MC38 tumors. We next assessed PD-L1 and CD155 expression on CD45 negative cells as a surrogate definition of tumors. Similar to the in vitro cultured parental cells, CD155 expression was similar between ex vivo derived SM1WT1 and MC38 tumor cells while PD-L1 expression was again much higher on MC38 compared to SM1WT1 tumor cells (Supp. Fig. 9E, F). Next, we evaluated the expression of PD-1, TIGIT, CD96 and DNAM-1 on CD8+ T cells and NK cells from end-stage MC38 and SM1WT1 tumors (Supp. Fig. 10). Again we observed no major differences as generally the expression of DNAM-1, CD96 and TIGIT on NK cells and CD8+ T cells from SM1WT1 (Supp. Fig. 10A) and MC38 (Supp. Fig. 10B) tumors were similar. The most noticeable difference was the level of PD-1 expression on CD8+ T cells obtained from MC38 compared to SM1WT1 tumors (80% vs 50%). This most likely reflects the more immunogenic nature of MC38 tumors compared to SM1WT1 tumors to elicit a CD8+ T cell response.
Discussion
Given that multiple non-redundant immunosuppressive pathways co-exist in the tumor microenvironment, immunotherapies targeting novel checkpoint receptors will most likely be tested in combination with the aim to further increase clinical efficacy. However, this may potentially increase the frequency and severity of irAEs. The generation of gene-targeted mice deficient for single or double immune checkpoint receptors is a useful first step to assess how their loss impacts on peripheral tolerance and immune homeostasis and whether compound gene loss is synergistic. In this study, we generated gene-targeted mice that were double deficient for either PD-1 and CD96 or TIGIT and CD96. Long-term aging of Pdcd1−/−CD96−/− and Tigit−/−CD96−/− mice demonstrated the potential of co-targeting these pathways without inducing serious immune-related toxicities, since these mice did not display increased disease or pathology beyond that observed in Pdcd1−/−, Tigit−/− or CD96−/− mice. Interestingly, increased anti-tumor immunity and tumor resistance was observed in Pdcd1−/−CD96−/− mice and this enhanced resistance to primary tumor implantation was prominent in the anti-PD-1-insensitive SM1WT1 melanoma.
It was encouraging that even at 22 months of age, Pdcd1−/−CD96−/− mice did not display dysregulated immune homeostasis or enhanced immune-based disease compared to Pdcd1−/− mice. To our knowledge, this represents the longest period where Pdcd1−/− mice have been aged, suggesting that chronic mAb co-targeting these two checkpoint receptors may be safe and not exacerbate the development of irAEs caused by anti-PD-1. Nevertheless, Pdcd1−/−CD96−/− mice should be generated on a permissive genetic background that predisposes them to development of autoimmunity to exclude the potential exacerbation of disease caused by CD96 loss. For example while C57BL/6 mice deficient for LAG-3 did not display any autoimmune toxicities,Citation11 LAG-3 deficiency on the NOD background accelerated their development of type 1 diabetes compared to age-matched wild type controls.Citation38,Citation39 Given that loss of PD-1 on a NOD background can accelerate the development of type I diabetesCitation40 and the recent studies linking new-onset insulin-dependent diabetes in anti-PD-1-treated patients,Citation41,Citation42 it may be of utility to breed Pdcd1−/−CD96−/− on to a NOD background and compare disease development with Pdcd1−/− NOD mice. Alternatively, the development of specific autoimmune diseases such as Experimental Autoimmune Encephalomyelitis (EAE) could be assessed in our C57BL/6 Pdcd1−/−CD96−/− mice given the susceptibility of this genetic background to disease induction.
The immune compartments in Tigit−/−CD96−/− mice appeared normal, similar to WT mice, with no overt changes in their major immune cell populations, suggesting the importance of these two immune checkpoint receptors is lower than PD-1, in terms of maintaining peripheral tolerance. Similarly, 22 month old Tigit−/−CD96−/− mice did not display dysregulated immune homeostasis or enhanced immune-based disease suggesting co-targeting of these receptors clinically may not induce serious immune-related toxicities. In contrast to co-loss of PD-1 and CD96, loss of PD-1 in combination with LAG-3 strongly affected immune homeostasis as T cells in PD-1/LAG-3 double deficient mice were highly activated with enhanced production of pro-inflammatory cytokines such as an IFNγ, which ultimately resulted in most mice becoming moribund by 10 weeks of age.Citation11 Similarly, mice that lacked PD-1 and VISTA displayed an increased frequency of activated T cells although this did not impact on their survival.Citation13 While the expression pattern of co-inhibitory receptors on activated T cells can overlap, the use of gene-targeted mice to delete one or more of these receptors in combination allows their hierarchy of dominance and their redundancy in regulating T cell activation to be examined.
Mice that lacked PD-1 and CD96 displayed better growth suppression of SM1WT1 subcutaneous melanomas compared to mice that lacked PD-1 alone. This was also supported by a higher complete rejection response rate observed in Pdcd1−/−CD96−/− compared with Pdcd1−/− mice. In TILs generated from SM1WT1-bearing Pdcd1−/−CD96−/− mice, we observed a significant increase in CD8+ T cells compared to TILs generated from WT, Pdcd1−/− or CD96−/− mice. This resulted in an increased ratio of CD8+ T cells to Treg or CD11b+GR-1hi myeloid cells, and potentially contributed to their enhanced tumor resistance in Pdcd1−/−CD96−/− mice. In addition, in the TILs of SM1WT1 tumor-bearing Pdcd1−/−CD96−/− or Pdcd1−/− mice, CD4+ effector T cells significantly increased compared to tumors from WT mice. Similarly, a trend for an increase in NK cell numbers was also observed in both SM1WT1 tumor-bearing Pdcd1−/−CD96−/− and CD96−/− mice. However, this same loss of host PD-1 and CD96 did not further improve growth suppression of MC38 primary tumors compared to mice that lacked PD-1 alone. One explanation may be the effector cell type(s) that are present in each of the tumor microenvironment that is critical to control tumor growth and the hierarchy of dominance and redundancy mediated by various immune checkpoint receptors and their ligands. In MC38 tumors, CD8+ T cells were the main effector cell type controlling subcutaneous tumor growthCitation35 and the PD-1/PD-L1 axis has been shown to be the dominant pathway suppressing CD8+ T cells.Citation43 Maximal tumor suppression may already be occurring in the MC38 tumor model with no further efficacy seen with loss of CD96 as NK cells generally did not express PD-1 and were not involved in control of this tumor. In contrast, both CD8+ T cells and NK cells play a role in the control of SM1WT136, 37 and their effector function can be suppressed by PD-L1 and CD155.
Similarly, an explanation for why loss of TIGIT and CD96 did not seem to impact greatly on either SM1WT1 or MC38 tumor growth is likely due to the dominance of the PD-1/PD-L1 axis in the suppression of effector functions on T cells in these tumors. Nevertheless, in other subcutaneous tumor models such as B16F10, loss of TIGIT and CD96 was able to synergistically impact on its growth (Supp. Fig. 6). Previous studies have also demonstrated that antibody co-targeting of TIGIT and PD-1/PD-L1 can synergistically suppress subcutaneous tumor growthCitation26,Citation27 and future experiments could be set up to treat tumor-bearing Tigit−/−CD96−/− mice with anti-PD-1 to determine if any inhibitory effect of CD96 on T cells can be unmasked. Finally, given our previous study demonstrating that anti-CD96 treated Tigit−/− mice displayed further reduction in lung metastases compared to anti-CD96 treated WT miceCitation25 co-targeting of CD96 and TIGIT as a combination may be more useful in the treatment of metastases or haematological malignancies where NK cells are critical for control. Overall, our studies suggest the potential safety of co-targeting PD-1 and CD96 or TIGIT and CD96 and future experiments will aim to determine in which tumor microenvironment these respective combination therapies will be most effective.
Material and methods
Mice
C57BL/6 wild-type (WT) mice were purchased from the Walter and Eliza Hall Institute for Medical Research or bred in house at QIMR Berghofer Medical Research Institute. Pdcd1−/−CD96−/− and Tigit−/−CD96−/− mice were generated by intercrossing C57BL/6 Pdcd1−/− and C57BL/6 CD96−/− or C57BL/6 Tigit−/− and C57BL/6 CD96−/− mice. CD96−/− mice have been described.Citation24 Pdcd1−/− miceCitation44 were kindly provided by M. Wykes (QIMR Berghofer). Tigit−/− mice were kindly provided by Bristol Myers Squibb. In experiments assessing the immune homeostasis and anti-tumor responses in mice lacking PD-1, CD96 or both, the C57BL/6 WT mice used were either bred in house or derived from the intercrossing of C57BL/6 Pdcd1−/− and C57BL/6 CD96−/− mice. All mice were bred and maintained at QIMR Berghofer Medical Research Institute. Eight- to sixteen-week old mice were used for immune phenotyping and tumor challenge experiments. The investigators were not blinded to group allocation during the experiments and/or when assessing the outcomes. All experiments were approved by the QIMR Berghofer Medical Research Institute Animal Ethics Committee.
Tumor models
MC38 colon adenocarcinoma and SM1WT1, a BRAF-mutated melanoma, were maintained, injected and monitored as described previously.Citation45,Citation46 SM1WT1 or MC38 (both 1 × 106) tumor cells were injected subcutaneously (s.c) into WT or gene-targeted mice. All cell lines were routinely tested as negative for mycoplasma, but cell line authentication was not routinely performed. Male mice were always used for the SM1WT1 tumor model, while both female and male mice were used for the MC38 tumor model. Tumor sizes were determined by caliper square measurements of two perpendicular diameters with data represented as mean ± SEM (mm2) for each group. In some experiments, mice were treated with either anti-CD8β (53.5.8, BioXCell), anti-asialoGM-1 (anti-asGM-1) (Wako Chemicals) or anti-IFNγ (H22, Leinco) or control IgG (cIg)(1–1, BioXcell) as indicated.
Anti-dsDNA antibody detection
Anti-dsDNA antibody levels in sera of mice were determined as previously described.Citation47 Briefly sera were incubated on ELISA plates (Nunc) coated with methylated BSA and sonicated calf thymus DNA (Sigma-Aldrich). Plates were then washed and incubated with AP conjugated goat anti mouse IgG (H+L)(Life Technologies) before substrate pNPP was added (Sigma-Aldrich) and absorbance determined. Data is presented as Absorbance (405–620 nm) at a 1:100 serum dilution. Sera pooled from aged gld−/ − mice were used as a positive control.
Histology
Livers of WT, Pdcd1−/−, CD96−/− and Pdcd1−/−CD96−/− mice were fixed in 10% neutral buffered formalin, processed routinely, and embedded in paraffin. Four micrometer tissue sections were cut and stained with H&E and imaged with an Aperio Scanscope AT (Leica) and analysed with an Aperio ImageScope (Leica). Mouse liver pathology was scored by referring to Sparwasser's standards.Citation48 The liver score is the sum of individual scores for inflammatory cell infiltration in portal tracts and the parenchyma, and the amount of necrosis.
Flow cytometry
Single cell suspensions were generated from spleen, tumors and PBS-perfused lungs as previously described.Citation25,Citation49 All samples were resuspended in FACS buffer containing 2.4G2 (anti-CD16/32) to block Fc receptors. Cell surface staining was performed using the following antibodies: anti-CD45.2 (104), anti-TCRβ (H57–597), anti-CD4 (RM4–5), anti-CD8α (53–6.7), anti-NK1.1 (PK136), anti-CD11b (M1/70), anti-GR-1 (RB6–8C5) anti-CD44 (IM7), anti-CD62 L (MEL-14) anti-PD-1 (J43), anti-TIGIT (9D9), anti-CD96 (3.3), anti-DNAM-1 (480.1), anti-CD155 (4.24.1), anti-PD-L1 (10 F.9 G2) and live/dead dye Zombie Aqua (all from BioLegend or eBioscience). BD Liquid Counting Beads (BD Biosciences, 335925) were added directly before samples were run on a flow cytometer. For intracellular staining, cells were first surface stained and then fixed and permeabilized with Foxp3/Transcription Factor Fixation/Permeabilization kit (eBioScience, 005521–00) and stained with anti-Foxp3 (FJK-16 s). CD8+ T cells were defined as TCRβ+CD8α+, CD4+ effector T cells (Teff) as TCRβ+CD4+Foxp3−, Tregs as TCRβ+CD4+Foxp3+ and NK cells as TCRβ−NK1.1+. All data were collected using a Fortessa 4 flow cytometer (Becton, Dickinson and Company) and analyzed with FlowJo v10 (Tree Star).
Statistical analyses
Statistical analyses were carried out using GraphPad Prism software except for the pooled tumor growth curves. In this setting, JMP Pro 13.2.0 (SAS) was used to pool three independent tumor growth experiments for MC38 and SM1WT1 and represented using random effects repeated measures regression. Comparison of different groups was done by using one-way ANOVA followed by Bonferroni's post-test analysis. Log-rank sum test was used to compare survival curves in the aging experiments. Fisher's exact test was used to compare complete response rates between Pdcd1−/−CD96−/− and Pdcd1−/− mice. Data were considered to be statistically significant where the p value was equal to or less than 0.05.
Financial support
The project was funded by a National Health and Medical Research Council of Australia (NH&MRC) Project Grant (1098960) and Development Grant (1093566), a Cancer Council of Queensland (CCQ) Project Grant (1083776), and a Cancer Research Institute CLIP Grant. M.J. Smyth is supported by a Senior Principal Research Fellowship (1078671). C. Guillerey is supported by an NH&MRC Peter Doherty Early Career Fellowship (1107417). E.Ahern was supported by a University of Queensland (UQ) Australian Postgraduate Award (APA). H. Harjunpää was supported by a UQ International Postgraduate Research Scholarship, a UQ APA, and a QIMR Berghofer Top-Up award. J Yan was supported by a UQ International Scholarship (UQI) and a QIMR Berghofer International PhD Scholarship.
Disclosure and potential conflict of interest
M.J. Smyth declares scientific research agreements with Bristol Myers Squibb, Corvus Pharmaceuticals, Tizona Therapeutics and Aduro Biotech. W.C Dougall declares a scientific research agreement with Bristol Myers Squibb. No potential conflicts of interest were disclosed by the other authors.
supp_data.zip
Download Zip (646.3 KB)Acknowledgments
The authors thank Liam Town and Kate Elder for mouse genotyping and maintenance during this study. The authors would also like to thank the QIMR Berghofer Medical Research Institute animal, flow cytometry and histology facilities.
References
- Smyth MJ, Ngiow SF, Ribas A, Teng MW. Combination cancer immunotherapies tailored to the tumour microenvironment. Nat Rev Clin Oncol. 2016;13:143–58. doi:10.1038/nrclinonc.2015.209. PMID:26598942.
- Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, Schadendorf D, Dummer R, Smylie M, Rutkowski P, et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med. 2015;373:23–34. doi:10.1056/NEJMoa1504030. PMID:26027431.
- Boutros C, Tarhini A, Routier E, Lambotte O, Ladurie FL, Carbonnel F, Izzeddine H, Marabelle A, Champiat S, Berdelou A, et al. Safety profiles of anti-CTLA-4 and anti-PD-1 antibodies alone and in combination. Nat Rev Clin Oncol. 2016;13:473–86. doi:10.1038/nrclinonc.2016.58. PMID:27141885.
- Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–64. doi:10.1038/nrc3239. PMID:22437870.
- Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. 1995;3:541–7. doi:10.1016/1074-7613(95)90125-6. PMID:7584144.
- Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, Thompson CB, Griesser H, Mak TW. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science. 1995;270:985–8. doi:10.1126/science.270.5238.985. PMID:7481803.
- Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity. 1999;11:141–51. doi:10.1016/S1074-7613(00)80089-8. PMID:10485649.
- Nishimura H, Okazaki T, Tanaka Y, Nakatani K, Hara M, Matsumori A, Sasayama S, Mizoguchi A, Hiai H, Minato N, et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science. 2001;291:319–22. doi:10.1126/science.291.5502.319. PMID:11209085.
- Latchman YE, Liang SC, Wu Y, Chernova T, Sobel RA, Klemm M, Kuchroo VK, Freeman GJ, Sharpe AH. PD-L1-deficient mice show that PD-L1 on T cells, antigen-presenting cells, and host tissues negatively regulates T cells. Proc Natl Acad Sci U S A. 2004;101:10691–6. doi:10.1073/pnas.0307252101. PMID:15249675.
- Dong H, Zhu G, Tamada K, Flies DB, van Deursen JM, Chen L. B7-H1 determines accumulation and deletion of intrahepatic CD8(+) T lymphocytes. Immunity. 2004;20:327–36. doi:10.1016/S1074-7613(04)00050-0. PMID:15030776.
- Woo SR, Turnis ME, Goldberg MV, Bankoti J, Selby M, Nirschl CJ, Bettini ML, Gravano DM, Vogel P, Liu CL, et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 2012;72:917–27. doi:10.1158/0008-5472.CAN-11-1620. PMID:22186141.
- Zhu G, Augustine MM, Azuma T, Luo L, Yao S, Anand S, Rietz AC, Huang J, Xu H, Flies AS, et al. B7-H4-deficient mice display augmented neutrophil-mediated innate immunity. Blood. 2009;113:1759–67. doi:10.1182/blood-2008-01-133223. PMID:19109567.
- Liu J, Yuan Y, Chen W, Putra J, Suriawinata AA, Schenk AD, Miller HE, Guleria I, Barth RJ, Huang YH, et al. Immune-checkpoint proteins VISTA and PD-1 nonredundantly regulate murine T-cell responses. Proc Natl Acad Sci U S A. 2015;112:6682–7. doi:10.1073/pnas.1420370112. PMID:25964334.
- Martinet L, Smyth MJ. Balancing natural killer cell activation through paired receptors. Nat Rev Immunol. 2015;15:243–54. doi:10.1038/nri3799. PMID:25743219.
- Blake SJ, Dougall WC, Miles JJ, Teng MW, Smyth MJ. Molecular Pathways: Targeting CD96 and TIGIT for Cancer Immunotherapy. Clin Cancer Res. 2016;22:5183–8. doi:10.1158/1078-0432.CCR-16-0933. PMID:27620276.
- Wang PL, O'Farrell S, Clayberger C, Krensky AM. Identification and molecular cloning of tactile. A novel human T cell activation antigen that is a member of the Ig gene superfamily. J Immunol. 1992;148:2600–8. PMID:1313846.
- Seth S, Maier MK, Qiu Q, Ravens I, Kremmer E, Forster R, Bernhardt G. The murine pan T cell marker CD96 is an adhesion receptor for CD155 and nectin-1. Biochem Biophys Res Commun. 2007;364:959–65. doi:10.1016/j.bbrc.2007.10.102. PMID:17971293.
- Yu X, Harden K, Gonzalez LC, Francesco M, Chiang E, Irving B, Tom I, Ivelja S, Refino CJ, Clark H, et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat Immunol. 2009;10:48–57. doi:10.1038/ni.1674. PMID:19011627.
- Levin SD, Taft DW, Brandt CS, Bucher C, Howard ED, Chadwick EM, Johnston J, Hammond A, Bontadelli K, Ardourel D, et al. Vstm3 is a member of the CD28 family and an important modulator of T-cell function. Eur J Immunol. 2011;41:902–15. doi:10.1002/eji.201041136. PMID:21416464.
- Fuchs A, Cella M, Giurisato E, Shaw AS, Colonna M. Cutting edge: CD96 (tactile) promotes NK cell-target cell adhesion by interacting with the poliovirus receptor (CD155). J Immunol. 2004;172:3994–8. doi:10.4049/jimmunol.172.7.3994. PMID:15034010.
- Shibuya A, Campbell D, Hannum C, Yssel H, Franz-Bacon K, McClanahan T, Kitamura T, Nicholl J, Sutherland GR, Lanier LL, et al. DNAM-1, a novel adhesion molecule involved in the cytolytic function of T lymphocytes. Immunity. 1996;4:573–81. doi:10.1016/S1074-7613(00)70060-4. PMID:8673704.
- Bottino C, Castriconi R, Pende D, Rivera P, Nanni M, Carnemolla B, Cantoni C, Grassi J, Marcenaro S, Reymond N, et al. Identification of PVR (CD155) and Nectin-2 (CD112) as cell surface ligands for the human DNAM-1 (CD226) activating molecule. J Exp Med. 2003;198:557–67. doi:10.1084/jem.20030788. PMID:12913096.
- Zhu Y, Paniccia A, Schulick AC, Chen W, Koenig MR, Byers JT, Yao S, Bevers S, Edil BH. Identification of CD112R as a novel checkpoint for human T cells. J Exp Med. 2016;213:167–76. doi:10.1084/jem.20150785. PMID:26755705.
- Chan CJ, Martinet L, Gilfillan S, Souza-Fonseca-Guimaraes F, Chow MT, Town L, Ritchie DS, Colonna M, Andrews DM, Smyth MJ. The receptors CD96 and CD226 oppose each other in the regulation of natural killer cell functions. Nat Immunol. 2014;15:431–8. doi:10.1038/ni.2850. PMID:24658051.
- Blake SJ, Stannard K, Liu J, Allen S, Yong MC, Mittal D, Aguilera AR, Miles JJ, Lutzky VP, de Andrade LF, et al. Suppression of metastases using a new lymphocyte checkpoint target for cancer immunotherapy. Cancer Discov. 2016;6:446/59. doi:10.1158/2159-8290.CD-15-0944. PMID:26787820.
- Chauvin JM, Pagliano O, Fourcade J, Sun Z, Wang H, Sander C, Kirkwood JM, Chen TH, Maurer M, Korman AJ, et al. TIGIT and PD-1 impair tumor antigen-specific CD8(+) T cells in melanoma patients. J Clin Invest. 2015;125:2046–58. doi:10.1172/JCI80445. PMID:25866972.
- Johnston Robert J, Comps-Agrar L, Hackney J, Yu X, Huseni M, Yang Y, Park S, Javinal V, Chiu H, Irving B, et al. The Immunoreceptor TIGIT Regulates Antitumor and Antiviral CD8+ T Cell Effector Function. Cancer Cell. 2014;26:923–37. doi:10.1016/j.ccell.2014.10.018. PMID:25465800.
- Lozano E, Dominguez-Villar M, Kuchroo V, Hafler DA. The TIGIT/CD226 axis regulates human T cell function. J Immunol. 2012;188:3869–75. doi:10.4049/jimmunol.1103627. PMID:22427644.
- Kurtulus S, Sakuishi K, Ngiow SF, Joller N, Tan DJ, Teng MW, Smyth MJ, Kuchroo VK, Anderson AC. TIGIT predominantly regulates the immune response via regulatory T cells. J Clin Invest. 2015;125:4053–62. doi:10.1172/JCI81187. PMID:26413872.
- Joller N, Lozano E, Burkett Patrick R, Patel B, Xiao S, Zhu C, Xia J, Tan TG, Sefik E, Yajnik V, et al. Treg Cells Expressing the Coinhibitory Molecule TIGIT Selectively Inhibit Proinflammatory Th1 and Th17 Cell Responses. Immunity. 2014;40:569–81. doi:10.1016/j.immuni.2014.02.012. PMID:24745333.
- Ellestad KK, Thangavelu G, Ewen CL, Boon L, Anderson CC. PD-1 is not required for natural or peripherally induced regulatory T cells: Severe autoimmunity despite normal production of regulatory T cells. Eur J Immunol. 2014;44:3560–72. doi:10.1002/eji.201444688. PMID:25236923.
- Chen X, Fosco D, Kline DE, Meng L, Nishi S, Savage PA, Kline J. PD-1 regulates extrathymic regulatory T-cell differentiation. Eur J Immunol. 2014;44:2603–16. doi:10.1002/eji.201344423. PMID:24975127.
- Ngiow SF, Young A, Jacquelot N, Yamazaki T, Enot D, Zitvogel L, Smyth MJ. A Threshold Level of Intratumor CD8+ T-cell PD1 Expression Dictates Therapeutic Response to Anti-PD1. Cancer Research. 2015;75:3800–11. doi:10.1158/0008-5472.CAN-15-1082. PMID:26208901.
- Knight DA, Ngiow SF, Li M, Parmenter T, Mok S, Cass A, Haynes NM, Kinross K, Yagita H, Koya RC, et al. Host immunity contributes to the anti-melanoma activity of BRAF inhibitors. J Clin Invest. 2013;123:1371–81. doi:10.1172/JCI66236. PMID:23454771.
- Haynes NM, Hawkins ED, Li M, McLaughlin NM, Hammerling GJ, Schwendener R, Winoto A, Wensky A, Yagita H, Takeda K, et al. CD11 c+ dendritic cells and B cells contribute to the tumoricidal activity of anti-DR5 antibody therapy in established tumors. J Immunol. 2010;185:532–41. doi:10.4049/jimmunol.0903624. PMID:20505139.
- Gao Y, Souza-Fonseca-Guimaraes F, Bald T, Ng SS, Young A, Ngiow SF, Rautela J, Straube J, Waddell N, Blake SJ, et al. Tumor immunoevasion by the conversion of effector NK cells into type 1 innate lymphoid cells. Nat Immunol. 2017;18:1004–15. PMID:28759001.
- Young A, Ngiow SF, Gao Y, Patch AM, Barkauskas DS, Messaoudene M, Lin G, Coudert JD, Stannard KA, Zitvogel L, et al. A2AR adenosine signaling suppresses natural killer cell maturation in the tumor microenvironment. Cancer Res. 2018;78:1003–16. doi:10.1158/0008-5472.can-17-2826. PMID:29229601.
- Bettini M, Szymczak-Workman AL, Forbes K, Castellaw AH, Selby M, Pan X, Drake CG, Korman AJ, Vignali DA. Cutting edge: accelerated autoimmune diabetes in the absence of LAG-3. J Immunol. 2011;187:3493–8. doi:10.4049/jimmunol.1100714. PMID:21873518.
- Okazaki T, Okazaki IM, Wang J, Sugiura D, Nakaki F, Yoshida T, Kato Y, Fagarasan S, Muramatsu M, Eto T, et al. PD-1 and LAG-3 inhibitory co-receptors act synergistically to prevent autoimmunity in mice. J Exp Med. 2011;208:395–407. doi:10.1084/jem.20100466. PMID:21300912.
- Wang J, Yoshida T, Nakaki F, Hiai H, Okazaki T, Honjo T. Establishment of NOD-Pdcd1-/- mice as an efficient animal model of type I diabetes. Proc Natl Acad Sci U S A. 2005;102:11823–8. doi:10.1073/pnas.0505497102. PMID:16087865.
- Hughes J, Vudattu N, Sznol M, Gettinger S, Kluger H, Lupsa B, Herold KC. Precipitation of autoimmune diabetes with anti-PD-1 immunotherapy. Diabetes Care. 2015;38:e55–7. PMID:25805871.
- Byun DJ, Wolchok JD, Rosenberg LM, Girotra M. Cancer immunotherapy – immune checkpoint blockade and associated endocrinopathies. Nat Rev Endocrinol. 2017;13:195–207. doi:10.1038/nrendo.2016.205. PMID:28106152.
- Juneja VR, McGuire KA, Manguso RT, LaFleur MW, Collins N, Haining WN, Freeman GJ, Sharpe AH. PD-L1 on tumor cells is sufficient for immune evasion in immunogenic tumors and inhibits CD8 T cell cytotoxicity. J Exp Med. 2017;214:895–904. doi:10.1084/jem.20160801. PMID:28302645.
- Nishimura H, Minato N, Nakano T, Honjo T. Immunological studies on PD-1 deficient mice: implication of PD-1 as a negative regulator for B cell responses. Int Immunol. 1998;10:1563–72. doi:10.1093/intimm/10.10.1563. PMID:9796923.
- Stagg J, Divisekera U, Duret H, Sparwasser T, Teng MW, Darcy PK, Smyth MJ. CD73-deficient mice have increased antitumor immunity and are resistant to experimental metastasis. Cancer Res. 2011;71:2892–900. doi:10.1158/0008-5472.CAN-10-4246. PMID:21292811.
- Ferrari de Andrade L, Ngiow SF, Stannard K, Rusakiewicz S, Kalimutho M, Khanna KK, Tey SK, Takeda K, Zitvogel L, Martinet L, et al. Natural killer cells are essential for the ability of BRAF inhibitors to control BRAFV600E-mutant metastatic melanoma. Cancer Res. 2014;74:7298–308. doi:10.1158/0008-5472.CAN-14-1339. PMID:25351955.
- Groom JR, Fletcher CA, Walters SN, Grey ST, Watt SV, Sweet MJ, Smyth MJ, Mackay CR, Mackay F. BAFF and MyD88 signals promote a lupuslike disease independent of T cells. J Exp Med. 2007;204:1959–71. doi:10.1084/jem.20062567. PMID:17664289.
- Mayer CT, Tian L, Hesse C, Kuhl AA, Swallow M, Kruse F, Thiele M, Gershwin ME, Liston A, Sparwasser T. Anti-CD4 treatment inhibits autoimmunity in scurfy mice through the attenuation of co-stimulatory signals. J Autoimmun. 2014;50:23–32. doi:10.1016/j.jaut.2013.08.010. PMID:24075450.
- Teng MW, Ngiow SF, von Scheidt B, McLaughlin N, Sparwasser T, Smyth MJ. Conditional regulatory T-cell depletion releases adaptive immunity preventing carcinogenesis and suppressing established tumor growth. Cancer Res. 2010;70:7800–9. doi:10.1158/0008-5472.CAN-10-1681. PMID:20924111.