
ABSTRACT
PD-L1-blocking antibodies produce significant clinical benefit in selected cancer patients by reactivating functionally-impaired antigen-experienced anticancer T cells. However, the efficacy of current PD-L1-blocking antibodies is potentially reduced by ‘on-target/off-tumor’ binding to PD-L1 widely expressed on normal cells. This lack of tumor selectivity may induce a generalized activation of all antigen-experienced T cells which may explain the frequent occurrence of autoimmune-related adverse events during and after treatment.
To address these issues, we constructed a bispecific antibody (bsAb), designated PD-L1xEGFR, to direct PD-L1-blockade to EGFR-expressing cancer cells and to more selectively reactivate anticancer T cells. Indeed, the IC50 of PD-L1xEGFR for blocking PD-L1 on EGFR+ cancer cells was ∼140 fold lower compared to that of the analogous PD-L1-blocking bsAb PD-L1xMock with irrelevant target antigen specificity. Importantly, activation status, IFN-γ production, and oncolytic activity of anti-CD3xanti-EpCAM-redirected T cells was enhanced when cocultured with EGFR-expressing carcinoma cells. Similarly, the capacity of PD-L1xEGFR to promote proliferation and IFN-γ production by CMVpp65-directed CD8+ effector T cells was enhanced when cocultured with EGFR-expressing CMVpp65-transfected cancer cells. In contrast, the clinically-used PD-L1-blocking antibody MEDI4736 (durvalumab) promoted T cell activation indiscriminate of EGFR expression on cancer cells. Additionally, in mice xenografted with EGFR-expressing cancer cells 111In-PD-L1xEGFR showed a significantly higher tumor uptake compared to 111In-PD-L1xMock. In conclusion, PD-L1xEGFR blocks the PD-1/PD-L1 immune checkpoint in an EGFR-directed manner, thereby promoting the selective reactivation of anticancer T cells. This novel targeted approach may be useful to enhance efficacy and safety of PD-1/PD-L1 checkpoint blockade in EGFR-overexpressing malignancies.
Introduction
Immune checkpoint protein programmed death-ligand 1 (PD-L1) normally serves to dampen adoptive immune responses in a timely and localized manner in order to prevent collateral damage and autoimmunity by limiting antigen-experienced PD-1+/CD8+ T cells to proliferate, produce cytokine and attack cells.Citation1 However, cancer cells misuse PD-L1 for incapacitating anticancer PD-1+/CD8+ T cells in the tumor microenvironment.Citation2 Indeed, PD-L1 expression by cancer cells was found to be associated with unfavorable prognosis in various malignancies.Citation3-Citation5 In this process, cancer cells constitutively express PD-L1 due to aberrant oncogenic signalingCitation6 or upregulate PD-L1 in response to IFN-γ locally released by activated anticancer T cells.Citation2,Citation7
Blockade of the PD-1/PD-L1 checkpoint using antagonistic antibodies has produced unprecedented antitumor responses in a subgroup of immunogenic cancer types. In particular, PD-L1-blocking antibodies, such as atezolizumab, avelumab, and durvalumab, showed prominent clinical activity in patients with advanced stage melanomaCitation8-Citation10 and non-small-cell lung carcinoma (NSCLC).Citation11,Citation12 However, the efficacy of current conventional monospecific PD-L1-blocking antibodies is potentially hampered due to on-target/off-tumor binding to numerous normal cell types that also express PD-L1. In this respect, binding to PD-L1-expressing cells in blood or other tissue may prevent antibody extravasation and accumulation at the site of the tumor.Citation13,Citation14 Moreover, lack of tumor-selectivity appears to result in generalized activation of antigen-experienced T cells, including functionally silenced auto-reactive T cells. The latter aspect is evidenced by the frequent occurrence of severe autoimmune-related adverse events during and after treatment with PD-L1-blocking antibodies.
To address these issues, we developed a bispecific antibody that aims to direct PD-L1-blockade to cancer cells and thereby reactivate anticancer T cells more selectively. Bispecific antibodies (BsAbs) are a valuable class of emerging therapeutics that combine two target functionalities harnessed into one antibody-based molecule. As target for our PD-L1-blocking bsAb we selected the epidermal growth factor receptor (EGFR), a well-established oncogenic tumor-associated surface antigen that is overexpressed and/or mutated in various epithelial malignancies, including colorectal cancer and non-small-cell lung cancer.Citation15,Citation16 Of note, FDA-approved anti-EGFR antibodies necitumumab and cetuximabCitation17 inhibit oncogenic EGFR signaling and show clinical efficacy in cancers also responsive to PD-1/PD-L1 checkpoint inhibition.Citation18
These notions prompted us to devise a targeted strategy for EGFR-directed blockade of PD-1/PD-L1 interaction. As far as we are aware, this is the first report of a bsAb that selectively directs PD-L1 blockade to EGFR-overexpressing cancer cells. This approach may represent an important step towards enhancing selectivity, efficacy and safety of PD-1/PD-L1 checkpoint inhibition approaches in EGFR-overexpressing malignancies.
Results
PD-L1xEGFR simultaneously binds to PD-L1 and EGFR
PD-L1xEGFR dose-dependently bound to CHO.PD-L1 cells and not to wt CHO cells (). Moreover, PD-L1xEGFR dose-dependent bound to A431 cells, whereas PD-L1xMock only showed minimal binding to A431 cells (). Binding levels of PD-L1xEGFR towards a panel of PD-L1+/EGFR+ cell lines closely correlated with the respective expression levels of EGFR (Suppl. Fig. 2A). In contrast, relative low binding of PD-L1xEGFR was detected towards a panel of PD-L1+/EGFR− cell lines (). Further, binding of PD-L1xEGFR to EGFRhigh A431 cells was strongly inhibited in the presence of excess amounts of EGFR-blocking mAb 425, whereas excess amounts of a PD-L1-blocking mAb only partly inhibited binding. Importantly, binding of PD-L1xEGFR to A431 cells was abrogated only in the combined presence of excess amounts of a PD-L1-blocking mAb and mAb 425 (), indicating PD-L1xEGFR selectively and simultaneously binds to PD-L1 and EGFR. Since EGFR expression levels on cancer cells typically strongly exceed those of PD-L1, we reason that binding of bsAb PD-L1xEGFR to EGFR+ cancer cells is usually dominated by its capacity to target EGFR.
Figure 1. PD-L1xEGFR selectively and simultaneously binds to PD-L1 and EGFR (A) Dose-dependent binding of PD-L1xEGFR to CHO.PD-L1 vs. parental CHO cells. (B) Dose-dependent binding of PD-L1xEGFR vs. PD-L1xMock to PD-L1+/EGFR+ A431 cells. (C) Binding of PD-L1xEGFR vs. PD-L1xMock (5 µg/ml) to a series of PD-L1+/EGFR+ and PD-L1+/EGFR− cell lines. (D) Binding of PD-L1xEGFR (1 µg/ml) to A431 cells in the presence or absence of excess PD-L1-blocking antibody and/or EGFR-blocking mAb 425. (E) Competitive binding assay in which anti-PD-L1-APC competed with increasing doses (0.01–50 µg/ml) of PD-L1xEGFR (black line) or PD-L1xMock (grey line) for binding to A431 cells. Where indicated, A431 cells were pre-treated with excess amounts of mAb 425 (50 µg/ml) (red line) or isotype control IgG2a (green line) for 15 min. All binding experiments were analyzed by flow cytometry. Statistical analysis in D was performed using One-way ANOVA followed by a Bonferroni post-hoc test (*p < 0.05, ** p < 0.01, *** p < 0.001, ns not significant).
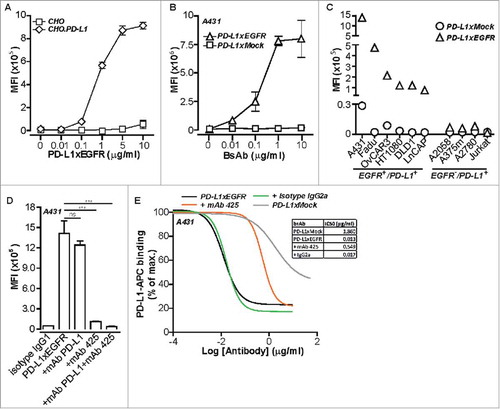
Figure 2. PD-L1xEGFR induces tumor growth inhibition and blocks the PD-1/PD-L1 interaction (A) Representative light microscopy images of PD-L1+/EGFR+ FaDu cells after 5 days treatment with 5 µg/ml PD-L1xEGFR, PD-L1xMock, mAb 425 or isotype control as indicated. (B) Cell viability of FaDu and H292 cells after treatment as in A was determined by MTS and expressed as percentage of medium control. Graphs represent mean ± SD. (C) Blockade of the PD-1/PD-L1 interaction analyzed using a commercially available PD-1/PD-L1 Blockade Bioassay (Promega). CHO.PD-L1/CD3 cells and Jurkat.PD-1-NFAT-Luc cells were treated with an increasing dose (0.01–10 µg/ml) of PD-L1xEGFR, PD-L1xMock, MEDI4736 or isotype control. NFAT-RE-mediated luciferase activity was quantified using a plate reader and expressed as fold increase compared to medium control. (D) Similar to C, mixed cultures of A431 cells and Jurkat.PD1-NFAT-luc cells were treated with increasing doses (0.01–10 µg/ml) of indicated antibodies in the presence of 75 ng/ml BIS-1. Statistical analysis in B was performed using One-way ANOVA followed by a Bonferroni post-hoc test (* p < 0.05, ** p < 0.01, *** p < 0.001, ns not significant).
PD-L1xEGFR has superior PD-L1-blocking capacity for PD-L1+/EGFR+ cancer cells
PD-L1xEGFR and PD-L1xMock were compared for their capacity to block PD-L1 on EGFR-expressing cancer cells using a competitive binding assay. In this assay, the IC50 of PD-L1xEGFR for inhibiting the binding of a competing APC-labeled PD-L1 mAb to A431 cells was calculated to be 0.013 µg/ml which was ∼140 times lower than the IC50 calculated for PD-L1xMock. Importantly, when EGFR binding to A431 cells was blocked by pre-incubation with mAb 425, the IC50 of PD-L1xEGFR increased ∼50 fold (from 0.013 to 0.549 µg/ml; ). These data indicate that, compared to PD-L1xMock, PD-L1xEGFR has superior PD-L1-blocking capacity for PD-L1+/EGFR+ cancer cells.
PD-L1xEGFR inhibits EGFR-mediated cancer cell proliferation
Treatment with PD-L1xEGFR showed similar capacity as mAb 425 ( and ) and cetuximab (data not shown) to inhibit the proliferation of FaDu and H292 cancer cells. In contrast, PD-L1xMock and isotype control antibodies did not impact the proliferation of FaDu or H292 cells.
PD-L1xEGFR blocks PD-1/PD-L1 interaction in an EGFR-directed manner
In the standard PD-1/PD-L1 Blockade Bioassay, PD-L1xEGFR and PD-L1xMock showed comparable dose-dependent blockade of PD-1/PD-L1 interaction with an IC50 value of ∼2.5 µg/ml (). Of note, in this non-targeted setting the IC50 of MEDI4736 for blocking PD-1/PD-L1 was 18 times lower than that of PD-L1xEGFR and PD-L1xMock. Next, the capacity of PD-L1xEGFR for EGFR-directed PD-1/PD-L1 blockade was assessed by replacing CHO.PD-L1/CD3 cells in the standard PD-1/PD-L1 Blockade Bioassay by A431 cells (PD-L1+/EGFR+/EpCAM+) that were pretreated with a suboptimal amount of bsAb BIS-1; an EpCAM-directed CD3-agonistic bsAb.Citation19 In the presence of BIS-1-coated A431 cells, the luciferase expression by Jurkat.PD1-NFAT-luc cells was effectively repressed. However, treatment with PD-L1xEGFR resulted in a dose-dependent increase in luciferase-mediated luminescence in Jurkat.PD1-NFAT-luc cells (). Of note, in this EGFR-directed setting, the capacity of PD-L1xEGFR to release the PD-1/PD-L1 break on luminescence in Jurkat.PD1-NFAT-luc cells became comparable to that of MEDI4736. These results indicate that the PD-L1-blocking activity of PD-L1xEGFR is markedly lower than that of MEDI4736. However, upon concurrent EGFR-binding PD-L1xEGFR regains potent PD-L1-blocking activity comparable to that of MEDI4736.
PD-L1xEGFR enhances activity of bsAb-redirected T cells
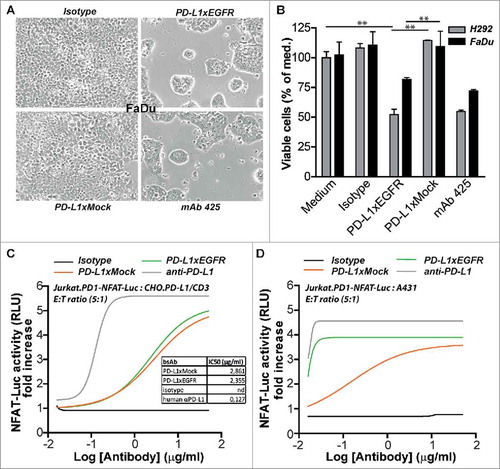
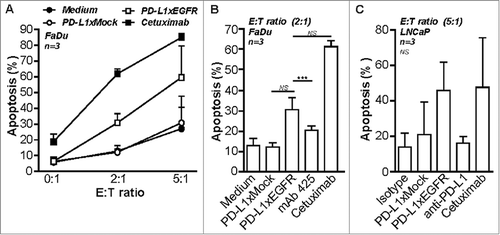
Recently, it was reported that blockade of PD-1/PD-L1 augments the killing capacity of BiTE-redirected T cells.Citation20 In our model system, we used a suboptimal amount of BIS-1 to redirect T cells towards EpCAM+ carcinoma cells that was titrated to induce low levels of apoptosis in FaDu and A431 cancer cells. In this setting, addition of PD-L1xEGFR resulted in a potent increase of apoptotic cancer cell death of ∼35% ( and ) which was accompanied by a markedly increased production of IFN-γ () and upregulation of CD25 expression (Suppl. Fig. 2B). In contrast, addition of MEDI4736 or PD-L1xMock only marginally enhanced (∼5%) the cytotoxic potential of BIS-1-redirected T cells. Of note, addition of anti-EGFR mAb 425 appeared to sensitize A431 and FaDu cancer cells to killing by BIS-1-redirected T cells (18% and 22%, respectively), which is likely attributable to its capacity to inhibit oncogenic EGFR-signaling. Addition of PD-L1xEGFR or mAb 425 to T cells that were not redirected by BIS-1 resulted in background levels of apoptosis in FaDu and A431 cells ( and ). Taken together, this indicates that the capacity of PD-L1xEGFR to enhance the anticancer activity of BIS-1-redirected T cells is attributable to its concurrent ability to block both PD-L1 signaling and oncogenic EGFR-signaling.
Figure 3. PD-L1xEGFR promotes cytotoxic activity of BIS-1-redirected T cells (A) FaDu cells were mixed with T cells at E:T ratio of 2:1 in the presence of BIS-1 (75 ng/ml) and 5 µg/ml PD-L1xEGFR or control antibodies. (B) A431 cells were treated as described in (A). Apoptosis was determined in A and B at day 3 by flow cytometry using Annexin-V staining. Apoptosis for isotype control treatments were subtracted. (C) IFN-γ levels in culture supernatant of (A) were determined by ELISA and IFN-γ levels for isotype control treatment were subtracted. (D) A431 cells were treated with the indicated antibodies, washed to remove unbound antibody and then mixed with T cells at an E:T ratio of 2:1 in the presence of BIS-1 (75 ng/ml). At day 3, T cells were carefully removed by washing, after which light microscopic images of the remaining A431 monolayer were evaluated. (E) In mixed cultures with EGFR+ FaDu and A431 or EGFR− A2058.EpCAM cells as described in D, apoptosis was determined by flow cytometry using Annexin-V staining. Apoptosis for isotype control treatments were subtracted. (F) In mixed cultures with FaDu cells as described in D, expression of T cell activation mrker CD25 was analyzed by flow cytometry. Mean fluorescence intensity (MFI) of BIS-1 treatment alone was subtracted. Three independent experiments were performed and represent mean ± SD. Statistical analysis was performed using One-way ANOVA followed by a Bonferroni post-hoc test (* p < 0.05, ** p < 0.01, *** p < 0.001, ns not significant).
Next, we evaluated whether PD-L1xEGFR enhances anticancer activity of BIS-1-redirected T cells in an EGFR-directed manner. To this end, EGFR+ and EGFR− cancer cells were briefly incubated with PD-L1xEGFR or control antibodies, after which unbound antibodies were removed by washing. In this setting, PD-L1xEGFR potently enhanced the anticancer activity of BIS-1-redirected T cells towards EGFR+ A431 and FaDu cancer cells, with essentially no enhanced activity towards EGFR− A2058.EpCAM cells (, , ). In contrast, MEDI4736 only marginally enhanced the anticancer activity of BIS-1-redirected T cells towards cancer cells indiscriminate of EGFR expression. Taken together, PD-L1xEGFR but not MEDI4736 specifically enhances the anticancer activity of BIS-1-redirected T cells in an EGFR-directed manner.
PD-L1xEGFR enhances activity of antigen-experienced T cells
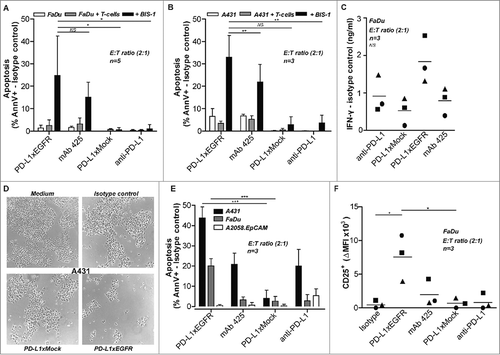
Next, we evaluated the capacity of PD-L1xEGFR to enhance the cytotoxic potential of authentic antigen-experienced T cells. To this end, CMVpp65-experienced T cells from healthy CMV-seropositive volunteers were cocultured with A431.pp65 or wt A431 cancer cells. In this setting, PD-L1xEGFR potently enhanced the cytotoxic activity of CMVpp65-experienced T cells towards A431.pp65 and not wt A431 cells. The enhanced cytotoxic potential of these T cells was corroborated by an increased cell surface expression of activation markers CD25, HLA-DR, CD137 and CD107a ( to and suppl. Fig 3), and an increased production of IFN-γ and granzyme B ( and ). Of note, expression of CD137 is reported to be restricted to T cells recently activated through TCR-mediated signaling and as such here identified the activation of pp65-experienced anti-CMV CD8+ T cells by PD-L1xEGFR. Similarly, the increased expression of degranulation marker CD107a is indicative of an increased cytotoxic activity of antigen-experienced T cells.Citation21 Taken together, these results indicated that PD-L1xEGFR enhances the activity of antigen-experienced T cells.
Figure 4. PD-L1xEGFR enhances cytotoxic potential of antigen-experienced T cells A431 or A431.pp65 cells were treated with PD-L1xEGFR or control antibodies, after which unbound antibody was washed away. Subsequently, T cells derived from CMV-seropositive donors were added at an E:T ratio of 20:1. After 8 days, expression levels of indicated activation markers were measured. (A) HLA-DR and CD25 expression, representative of 3 independent experiments. (B) CD137 expression, representative of 3 independent experiments. (C) CD137, CD107a (D) and intracellular IFN-γ (E) expression levels were analyzed within CD8+ T cell population by flow cytometry. (F) Granzyme B levels present in culture supernatants of treatment conditions as described in A were determined by ELISA.
PD-L1xEGFR induces NK cell-mediated ADCC in EGFR+ cancer cells
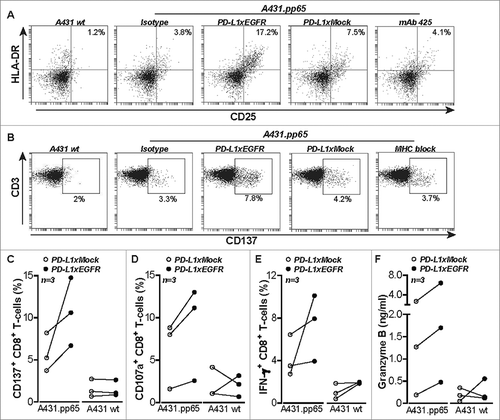
PD-L1xEGFR contains a fully functional human IgG1 domain and thus may trigger NK cell-mediated ADCC towards EGFR-expressing cancer cells. Indeed, NK cell-mediated ADCC towards FaDu and LNCaP cells was enhanced by PD-L1xEGFR or cetuximab, but not by PD-L1xMock or mAb 425 ( to ). Thus, PD-L1xEGFR combines EGFR-directed immune checkpoint blockade with ADCC towards EGFR+ cancer cells.
Figure 5. PD-L1xEGFR induces NK-cell mediated ADCC (A) FaDu cells were mixed with IL-12-pre-treated NK cells at the indicated E:T ratios and in the presence of 5 µg/ml PD-L1xEGFR or control antibodies. (B) FaDu cells were co-cultured with IL-12-pretreated NK cells at an E:T ratio of 2:1 as described in A. (C) LNCaP cells were mixed with PBMCs at an E:T ratio of 5:1 in the presence of 5 µg/ml PD-L1xEGFR or control antibodies. In all experiments apoptosis was determined by flow cytometry using Annexin-V staining procedure. All graphs represent mean ± SD. Statistical analysis in B and C were performed using One-way ANOVA followed by a Bonferroni post-hoc test (*p < 0.05, ** p < 0.01, *** p < 0.001, ns not significant).
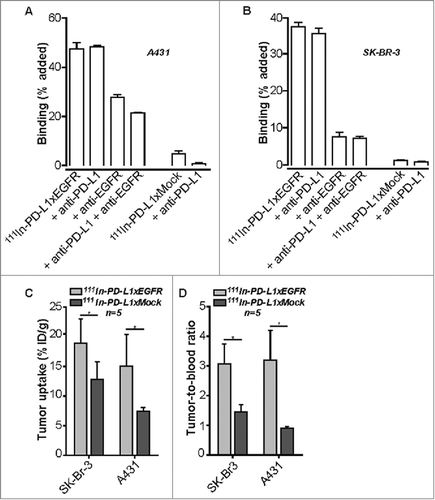
Enhanced tumor uptake of 111In-PD-L1xEGFR in EGFR+ tumor xenografted mice
Tumor uptake of PD-L1xEGFR and PD-L1xMock was compared in vivo using nude mice xenografted with EGFR+ SK-BR-3 or A431 cancer cells. PD-L1xEGFR and PD-L1xMock were labeled with 111In at a specific activity of 50 kBq/pmol with an efficiency of 95%. In vitro cell binding analysis demonstrated that radiolabeling of 111In-PD-L1xEGFR did not compromise its binding capacity for PD-L1 or EGFR ( and ). Blood clearance of 111In-PD-L1xEGFR and 111In-PD-L1xMock in non-tumor-bearing mice indicated comparable biological half-lives of ∼24 h and 30 h, respectively (data not shown). Importantly, in SK-BR-3 and A431 tumor-bearing mice, 111In-PD-L1xEGFR showed a significantly higher tumor uptake compared to 111In-PD-L1xMock (p = 0.001). Tumor uptake in A431 tumor-bearing mice for 111In-PD-L1xEGFR and 111In-PD-L1xMock were 19% and 13% ID/g, respectively. Tumor uptake in EGFR+ SK-BR-3 tumor-bearing mice for 111In-PD-L1xEGFR and 111In-PD-L1xMock were 15% and 7% ID/g, respectively ( and suppl. Fig 4). In both tumor models the tumor-to-blood-ratio for 111In-PD-L1xEGFR was notably higher compared to 111In-PD-L1xMock (p = 0.001) (). These data indicate that accumulation of PD-L1xEGFR in EGFR+ tumors is higher compared to PD-L1xMock.
Figure 6. Biodistribution of radiolabeled 111In-PD-L1xEGFR. (A) Binding of 111In-PD-L1xEGFR or 111In-PD-L1xMock to PD-L1 and EGFR in the presence or absence of excess MEDI4736 and/or EGFR blocking mAb 425 on A431 and (B) SK-BR-3 cells. (C) Tumor uptake of 111In-PD-L1xEGFR (1 µg) in mice with subcutaneous A431 (n = 5) or SK-BR-3 (n = 5) xenografts. Separate groups of mice were injected with control antibody 111In-PD-L1xMock. Tumor uptake was calculated as % injected dose per gram tissue (%ID/g) (D) Tumor-to-blood ratio was calculated, from experiment as described in C. Statistical analysis in D was performed using One-way ANOVA followed by a Bonferroni post-hoc test (* p < 0.05, ** p < 0.01, *** p < 0.001, ns not significant).
Discussion
The efficacy of current PD-L1-blocking antibodies is potentially hampered by ‘on-target/off-tumor’ binding to normal cells expressing PD-L1. This lack of tumor-selective binding may reduce tumor accretion of PD-1/PD-L1-blocking antibo-dies. Moreover, this may also induce an indiscriminate reactivation of antigen-experienced T cells, including functionally silenced yet potentially deleterious autoreactive T cells, leading to severe autoimmune-related adverse events during and after treatment.Citation8,Citation12,Citation22
Therefore, we developed a novel bsAb-based approach that aims to more selectively direct PD-L1 blockade to cancer cells. BsAbs allow to selectively target, modulate and interconnect biologic activities of otherwise separately acting cell surface receptors and ligands on the same cell or between neighboring cells in a predesigned manner.
We selected EGFR as a particularly suitable target antigen for this approach. Recently, it was reported that EGFR activation by either EGF, exon-19 deletions or L858R mutation promotes PD-L1 expression by cancer cells.Citation6,Citation23 EGFR overexpression is known to promote immune evasion of malignant cells by inhibiting the activation of signal transducer and activator of transcription 1 (STAT1) while promoting that of STAT3.Citation24,Citation25 Several EGFR-targeted agents are currently available that inhibit oncogenic EGFR signaling, including small-molecule EGFR kinase inhibitors (gefitinib, erlotinib and osimertinib) and antagonistic antibodies (necitumumab, nimotuzumab and cetuximab). Indeed, our results show that gefitinib and erlotinib sensitize cancer cells to apoptosis induction by BIS-1-redirected T cell (Suppl. Fig. 2C and D). Moreover, our results suggest that the capacity of PD-L1xEGFR to enhance the anticancer activity of BIS-1-redirected T cells is attributable to its concurrent ability to block both PD-L1 signaling and oncogenic EGFR-signaling.
PD-L1xEGFR is constructed in a so-called bispecific taFv-Fc format,Citation26 a class of symmetric tetravalent bispecific molecules that can be equipped with an Fc domain of choice. Our data demonstrate that the capacity of PD-L1xEGFR to simultaneously bind to PD-L1 and EGFR results in an significantly enhanced overall binding strength (avidity) for PD-L1+/EGFR+ cancer cells. Of note, EGFR expression by carcinoma cells usually exceeds expression of PD-L1. Indeed, in mice with EGFR+ xenografts the tumor accumulation of 111In-PD-L1xEGFR was significantly higher than that of control 111In-PD-L1xMock. Obviously, more detailed fit-for-purpose evaluation of our approach needs to be performed e.g. in patient-derived xenograft (PDX) models using EGFR-transgenic and humanized NSG mice, but such is clearly beyond the scope of the current report.
Interestingly, in a bioassay for PD-1/PD-L1 blockade PD-L1xEGFR showed reduced capacity to block PD-1/PD-L1 compared to PD-L1-blocking antibody MEDI4736. However, upon binding to EGFR-overexpressing target cells the PD-L1-blocking capacity of PD-L1xEGFR was restored to that of MEDI4736. Compared to indiscriminate high-affinity PD-L1-binding activity by MEDI4736, the EGFR-dependent PD-L1-blocking capacity of PD-L1xEGFR may be beneficial to reduce ‘on-target/off-tumor’ effects.
Our results indicate that PD-L1xEGFR enhances activation of antigen-experienced T cells in an EGFR-directed manner. In particular, treatment of CMV-pp65 transfected EGFR+ cancer cells with PD-L1xEGFR, followed by removal of unbound antibody, promoted the activity of HLA-matched CMV-specific CD8+ T cells which was corroborated by increased expression of CD137, CD107a, granzyme B, and IFN-γ production. Previously, it was reported that PD-L1-blocking antibody avelumab has similar in vitro capacity to enhance activation of antigen-experienced T cells directed against CMV, EBV, Flu or tetanus,Citation27 albeit obviously not in a tumor-directed manner.
Typically, PD-1/PD-L1-blocking antibodies are engineered as human IgG4 or to have reduced or silenced ADCC activity to avoid elimination of PD-1/PD-L1-expressing immune cells.Citation28,Citation29 However, human IgG1 containing avelumab was shown to have a toxicity profile comparable to ADCC-null PD-L1-blocking antibodiesCitation30-Citation32 with low levels of lysis of PBMCs in vitro.Citation27,Citation33 Moreover, NK cell-mediated ADCC by avelumab was shown to enhance its therapeutic functionality.Citation33,Citation34 Similarly, the human IgG1 domain present in PD-L1xEGFR may enhance its therapeutic activity as it promotes NK cell-mediated ADCC towards EGFR-expressing cancer cells.
Collectively, our data demonstrates that PD-L1xEGFR has multiple mutually reinforcing anticancer activities not available in any of the current conventional PD-L1-blocking antibodies. In particular, PD-L1xEGFR: 1. simultaneously binds to both PD-L1 and EGFR resulting in enhanced avidity towards PD-L1+/EGFR+ cancer cells; 2. blocks oncogenic EGFR-signaling which sensitizes cancer cells to therapy; 3. blocks PD-1/PD-L1 interaction in an EGFR-directed manner; 4. promotes anticancer activity of both anti-CD3xanti-EpCAM-redirected and antigen-experienced T cells in an EGFR-directed manner; 5. induces elimination of EGFR+ tumor cells by NK cell-mediated ADCC; 6. accumulates more selectively in EGFR+ xenografts.
Taken together, our PD-L1xEGFR-based approach may represent a next step towards enhancing selectivity, efficacy and safety of PD-1/PD-L1 checkpoint inhibition approaches in EGFR-overexpressing malignancies and as such warrants further development.
Materials and methods
Antibodies and reagents
Goat anti-human Ig-PE (Southern Biotech), anti-PD-L1-APC (clone 29E.2A3, BioLegend), anti-EGFR-FITC (clone 528, Santa Cruz Biotechnology), anti-CD107a-APC (clone H4A3, BD Pharmingen), anti-CD137-PE (clone 4B4), anti-IFN-γ-PerCP-Cyanine5.5 (clone 4S.B3,), anti-CD3-PerCP-Cyanine5.5 (clone OKT-3, all from eBioscience), and anti-CD3-FITC (clone Ucht1), anti-CD8-FITC, APC (clone HIT8a), anti-CD56-PE (clone B-A19), anti-CD14-FITC, PE (clone MEM-15), anti-CD25-FITC, APC (clone MEM-181), anti-HLA-DR-FITC, PE (clone MEM-12), mouse IgG1-FITC, PE, Mouse IgG2b-APC, Annexin-V-FITC (all from Immunotools). Recombinant human IFN-γ, TNF-α, PGE2, GM-CSF, IL-1β, IL-4, IL-6, IL12 and anti-CD3 mAb UCHT-1 were from Immunotools. PD-L1-blocking mAb was from BPS Bioscience. Anti-EGFR mAb 425 was from Merck. Cetuximab was obtained from the Department of Hospital Pharmacy, UMCG, The Netherlands. Secretion of cytokines by T cells was measured using appropriate ELISA kits (IFN-γ from eBioscience and granzyme B from Mabtech).
Cell lines and transfectants
Cell lines A431, FaDu, H292, OVCAR3, HT1080, DLD-1, SK-BR-3, LNCaP, A2058, A375 m, A2780, CHO-K1 and Jurkat cells were obtained from the American Type Culture Collection (ATCC). Cells were cultured in RPMI-1640 or DMEM (Lonza), supplemented with 10% fetal calf serum (FCS, Thermo Scientific), CHO-K1 cells were cultured in GMEM (First Link), supplemented with 5% dialyzed FBS (Sigma Aldrich) at 37°C in a humidified 5% CO2 atmosphere. Cell line authentication was carried out by short tandem repeat (STR) analysis. CHO.PD-L1 cells stably expressing human PD-L1 were generated by lipofection (Fugene-HD, Promega) with plasmid pCMV6-PD-L1 (Origene). A431.pp65 cells stably expressing cytomegalovirus (CMV) protein pp65 were generated by lipofection with plasmid pCMV6-pp65 (Origene). A2058.EpCAM cells stably expressing EpCAM-YFP were generated by lipofection with plasmid pEpCAM-YFP-N1 (kindly provided by Dr. Olivier Gires, Munich, Germany). Clones with stable expression of the indicated transgenes were selected using culture media supplemented with the appropriate antibiotic. PD-L1, EGFR and EpCAM expression was analyzed for all cell lines by flow cytometry using anti-PD-L1-APC, anti-EGFR-FITC and anti-EpCAM-FITC antibodies and appropriate isotype controls. The relative expression levels of PD-L1, EGFR and EpCAM are listed in Supplementary Table 1.
Construction of PD-L1xEGFR
DNA fragments encoding scFvPD-L1 and scFv425 were generated by commercial gene synthesis service (Genscript) based on published VH and VL sequence data of PD-L1-blocking antibody 3G10 and EGFR-directed antibody mAb 425, respectively. For construction and production of PD-L1xEGFR we used eukaryotic expression plasmid pEE14-bsAb,Citation35 which contains 3 consecutive multiple cloning sites (MCS). MCS#1 and MCS#2 are interspersed by a 22 amino acid flexible linker derived from a CH1 IgG domain.Citation36 MCS#1, MCS#2 and MCS#3 were used for directional and in-frame insertion of DNA fragments encoding scFvPD-L1, scFv425, and human IgG1 Fc domain, respectively, yielding plasmid pEE14-PD-L1xEGFR (Suppl. Fig. 1A and B). Of note, pEE14 is equipped with a strong CMV promotor and a murine kappa light-chain leader peptide for driving expression of PD-L1xEGFR through the ER and Golgi complex ensuring excretion of correctly folded and biological active recombinant protein with authentic port-translational modifications. A human IgG1 hinge region and G4S spacers between antigen recognizing domains were applied to promote flexibility of bsAb PD-L1xEGFR (Suppl. Fig. 1C). Analogously, pEE14-PD-L1xMock encoding PD-L1xMock was constructed by replacing scFv425 in pEE14-PD-L1xEGFR by scFv4-4-20 directed against fluorescein.
Eukaryotic production of bsAbs
PD-L1xEGFR and PD-L1xMock were produced using the Expi293 expression system (ThermoFisher). Briefly, Expi293 cells were transfected with plasmid pEE14-PD-L1xEGFR or pEE14-PD-L1xMock and cultured for 7 days on a shaker platform (125 rpm) at 37°C, 8% CO2. Next, culture supernatant was harvested and cleared by centrifugation (3000 x g, 30 min), after which PD-L1xEGFR and PD-L1xMock were purified using an HiTrap protein A HP column connected to an ÄKTA Start chromatography system (GE Healthcare Life Sciences).
SDS-PAGE analysis of PD-L1xEGFR
Protein A-purified PD-L1xEGFR or MEDI4736 (2,5 μg) were separated by SDS-PAGE (10% acrylamide) under reducing (with β-mercaptoethanol) or nonreducing conditions (without β-mercaptoethanol), followed by staining of the gel with Coomassie Brilliant Blue. Under nonreducing conditions PD-L1xEGFR migrated as a single protein band with an apparent molecular weight of 170 kDa, which dropped to 80 kDa when separated under reducing conditions. This is in good agreement with the calculated molecular weight of 84 kDa for PD-L1xEGFR monomer and the proposed disulfide-stabilized dimeric single chain composition of the native protein. MEDI4736 showed the expected heterodimeric composition of heavy and light chain characteristic for conventional antibodies (Suppl. Fig. 1D).
Binding activity of PD-L1xEGFR for PD-L1 and EGFR
Flow cytometry was used to evaluate the binding activity of PD-L1xEGFR for PD-L1 and EGFR using a broad panel of cell lines (CHO, CHO.PD-L1, EGFR+ cancer cell lines A431, FaDu, OVCAR3, HT1080, DLD1, LNCaP and EGFR− cancer cell lines A2058, A375 m, A2780, Jurkat). In short, cells were incubated with increasing amounts of PD-L1xEGFR (0.01–10 µg/ml, 45 min at 4°C), washed 3 times with PBS, and then incubated with anti-human-Ig-PE mAb (45 min at 4°C) and evaluated by flow cytometry. Binding for PD-L1xEGFR (1 µg/ml) to A431 cells was blocked by either PD-L1-blocking mAb (10 µg/ml), mAb 425 (10 µg/ml), or a combination of PD-L1-blocking mAb and mAb 425.
Competitive binding assay
Overall binding strength (avidity) of PD-L1xEGFR and PD-L1xMock, for PD-L1+/EGFR+ cancer cells was compared in a competitive binding assay, essentially as described previously.Citation37 In short, A431 cells were pre-incubated (or not) with excess amounts of mAb 425 (50 µg/ml) for 15 min at 4°C, after which PD-L1xEGFR or PD-L1xMock was added in a concentration range from 0.01 to 50 µg/ml, in the presence of an APC-labeled PD-L1-blocking mAb (8 µg/ml). After 45 min the tumor cell-bound APC levels were quantified by flow cytometry. Of note, in this assay, the binding capacity of the tested bispecific antibody is inversely proportional to its capacity to reduce binding of the APC-labeled PD-L1-blocking antibody.
Bioassay for PD-1/PD-L1 blockade
Blockade of PD-1/PD-L1 interaction was assessed using a commercially available PD-1/PD-L1 Blockade Bioassay (Promega). This assay exploits Jurkat.PD1-NFAT-luc T cells (expressing PD-1 and NFAT-inducible luciferase) and CHO-PD-L1-CD3 cells (expressing PD-L1 and a membrane-linked agonistic anti-CD3 antibody). When co-cultured, PD-1/PD-L1 interaction between both cell types inhibits TCR signaling and NFAT-mediated luciferase activity in Jurkat.PD1-NFAT-luc T cells. Addition of a PD-1/PD-L1 blocking agent, such as PD-LxEGFR, PD-L1xMock and MEDI4736, results in releasing PD-1/PD-L1 block on NFAT-mediated luciferase activity in Jurkat.PD1-NFAT-luc T cells.
Bioassay for EGFR-directed PD-1/PD-L1 blockade by PD-L1xEGFR
The capacity of PD-L1xEGFR for EGFR-directed PD-1/PD-L1 blockade was assessed using a modified version of the above PD-1/PD-L1 Blockade Bioassay by replacing CHO.PD-L1/CD3 cells by A431 cells that were pretreated with a suboptimal amount of BIS-1. BIS is an EpCAM-directed CD3-agonistic bsAb.Citation38 This modification allows for redirecting the Jurkat.PD1-NFAT-luc T cells towards EpCAM+ A431 carcinoma cells. In short, Jurkat.PD1-NFAT-luc T cells were mixed BIS-1-pretreated A431 cells at a cell ratio of 5 to 1 and then cultured for 18 h in the presence of PD-L1xEGFR or appropriate control antibodies. Subsequently, Bio-Glo reagent was added after which bioluminescence was quantified using a Victor V3 multilabel plate reader (Perkin Elmer).
Inhibition of EGFR-mediated cancer cell proliferation by PD-L1xEGFR
FaDu and H292 cancer cells were pre-cultured in 48-wells plates for 6 h at 8.000 cells/well, followed by addition of PD-L1xEGFR, PD-L1xMock or appropriate control antibodies (each 5 µg/ml). After 5 days, cancer cell proliferation was determined in an MTS-based colorimetric assay (CellTiter 96, Promega) using a Victor V3 multi-label plate counter at 490 nm. Absorbance data for maximum cell death were obtained by control treatment with 70% ethanol for 15 min.
Activation BIS-1-redirected T cells by PD-L1xEGFR
T cells were sorted from PBMCs by MACS (human Pan T cell isolation kit, Miltenyi Biotec). T cells were incubated with a suboptimal amount of bsAb BIS-1 (75 ng/ml) and then added to A431, FaDu or A2058.EpCAM target cells in an E:T ratio of 2 to 1, in the presence or absence of PD-L1xEGFR, appropriate control antibodies (each 5 µg/ml), gefitinib, erlotinib (10 nM/ml) or DMSO control. At day 3, apoptosis induction in cancer cells (Annexin-V) and CD25 expression on T cells were evaluated by flow cytometry.
Activation of CMV-specific T cells by PD-L1xEGFR
A431.CMVpp65 or wild-type (wt) A431 cells were incubated with PD-L1xEGFR or appropriate control antibodies (5 µg/ml), washed to remove unbound antibody and then cultured in 48-wells plates (3 × 104 cells/well). Freshly isolated PBMCs from a healthy CMV+ donor were added to cancer cells at an E:T ratio of 20 to 1. After 4 days, the various experimental conditions were restimulated by adding fresh A431.pp65 or wt A431 cells. At day 7, ∼50% of PBMCs of the various conditions was used for restimulation with A431.pp65 or wt A431 cancer cells overnight. At day 8, the cytotoxic potential of CMV-specific CD8+ T cells was assessed by measurement of degranulation marker CD107a (FastImmune CD107a APC reagent, BD Pharmingen) and intracellular IFN-γ (FIX&PERM, Nordic-MUbio and anti-IFN-γ-PerCP-Cyanine5.5). The remaining 50% of the CD3+/CD8+ T cells was used for evaluation of cell surface expression of HLA-DR, CD25 and CD137 by flow cytometry. Supernatant was used for granzyme B ELISA.
NK cell-mediated ADCC by PD-L1xEGFR
Natural killer (NK) cells were sorted from PBMCs using the Magnisort™ Negative Selection kit (eBioscience). Sorted NK cells (1 × 106 cells/ml) were stimulated using in RPMI-1640/FBS medium supplemented with IL-12 (10 ng/ml) for 24 h as described previously.Citation33 NK cells were washed with PBS and then mixed with cancer cells at the indicated E to T ratios and treated with PD-L1xEGFR, PD-L1xMock or appropriate control antibodies (each 5 µg/ml). After 18 h of treatment, apoptosis was assessed by flow cytometry using Annexin-V staining.
Alternatively, LNCaP cells were co-cultured with PBMCs at an E to T cell ratio of 5 to 1 and then treated with PD-L1xEGFR or indicated control antibodies (each 5 µg/ml). After 48 h, ADCC-mediated apoptosis induction in cancer cells was assessed by flow cytometry using Annexin-V staining procedure.
Radiolabeling and in vitro binding of 111In-PD-L1xEGFR and 111In-PD-L1xMock
BsAbs PD-L1xEGFR and PD-L1xMock were conjugated with isothiocyanatobenzyl- diethylenetriaminepentaacetic acid (ITC-DTPA, Macro Cyclics) and radiolabeled with 111In as described previously.Citation39 To assess binding of radiolabeled bsAbs to PD-L1 and EGFR, A431 and SK-Br-3 cells were incubated for 4 h at 37°C with 21.6 pM 111In-PD-L1xEGFR or 111In-PD-L1xmock. To show binding specificity for PD-L1 and EGFR, separate wells were incubated with 66 nM non-labeled mAb 425 or MEDI4736. After incubation, cells were lysed using 0.1 M NaOH and cell-associated activity was measured in a shielded well-type gamma counter (Perkin-Elmer, Boston, MA, USA).
Biodistribution in tumor-bearing nude mice
Animal studies were performed using 6–8 weeks old female BALB/c nude mice (Janvier, France) in accordance with the Dutch Act on Animal Experimentation and approved by the institutional Animal Welfare Committee at Radboud University Nijmegen. To determine blood kinetics, two groups of 5 non-tumor bearing mice were injected with 5 µg (0.2 MBq) 111In-PD-L1xEGFR or 111In-PD-L1xmock. Blood samples were obtained at 5 min, 1 h, 4 h, 6 h, 24 h, 48 h and 72 h, and 168 post injection. To compare the tumor targeting properties of the bsAbs, two groups of 10 mice were injected subcutaneously on the right flank with either 5 × 106 SK-BR-3 cells, representing an EGFRlow low expression pattern (n = 10) in 33% Matrigel matrix (Corning) RPMI 1640 or 3 × 106 A431 cells, representing an EGFRhigh expression pattern (n = 10) in RPMI 1640. After ten days, mice were injected in the tail vein with 1 µg 111In-PD-L1xEGFR or 111In-PD-L1xMock (0.2 MBq, n = 5 per group). Three days post injection, mice were euthanized using CO2/O2-asphyxation. The biodistrubution of radiolabel was determined ex vivo. Tumor, blood, muscle, lung, spleen, pancreas, intestine, kidney, liver, bone, bone marrow and lymph nodes were excised and weighed and evaluated in a gamma counter. To determine the uptake of radiolabeled antibodies in each sample as a fraction of the injected dose, aliquots of the injected dose were counted simultaneously. Results were decay corrected and expressed as percentage injected dose per gram tissue (%ID/g).
Statistical analysis
IC50 values were determined by nonlinear regression analysis of concentration response curves using GraphPad Prism. Unless otherwise noted, values are mean ± SD. Statistical analysis was done by one-way ANOVA followed by Bonferroni post-hoc test, as indicated using Prism software. P<0.05 was defined as a statistically significant difference. Where indicated * = P<0.05; ** = P<0.01; *** = P<0.001.
Conflicts of interest
No potential conflicts of interest were disclosed.
Author contributions
IK, DH, DFS performed experiments, analyzed data, and wrote the manuscript. RJG, SH and JLW analyzed data. E.B and W.H conceived and designed the study and wrote the manuscript.
2017ONCOIMM1032R-f07-z-bw.docx
Download MS Word (1.8 MB)Additional information
Funding
References
- Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26:677–704. doi:10.1146/annurev.immunol.26.021607.090331. PMID:18173375.
- Taube JM, Anders RA, Young GD, Xu H, Sharma R, McMiller TL, Chen S, Klein AP, Pardoll DM, Topalian SL, et al. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci Transl Med. 2012 Mar 28;4(127):127ra37. doi:10.1126/scitranslmed.3003689.
- Massi D, Brusa D, Merelli B, Ciano M, Audrito V, Serra S, Buonincontri R, Baroni G, Nassini R, Minocci D, et al. PD-L1 marks a subset of melanomas with a shorter overall survival and distinct genetic and morphological characteristics. Ann Oncol. 2014 Dec;25(12):2433–42. doi:10.1093/annonc/mdu452.
- Shimoji M, Shimizu S, Sato K, Suda K, Kobayashi Y, Tomizawa K, Takemoto T, Mitsudomi T. Clinical and pathologic features of lung cancer expressing programmed cell death ligand 1 (PD-L1). Lung Cancer. 2016 Aug;98:69–75. doi:10.1016/j.lungcan.2016.04.021.
- Thompson RH, Gillett MD, Cheville JC, Lohse CM, Dong H, Webster WS, Krejci KG, Lobo JR, Sengupta S, Chen L, et al. Costimulatory B7-H1 in renal cell carcinoma patients: Indicator of tumor aggressiveness and potential therapeutic target. Proc Natl Acad Sci U S A. 2004 Dec 7;101(49):17174–9. doi:10.1073/pnas.0406351101.
- Chen N, Fang W, Zhan J, Hong S, Tang Y, Kang S, Zhang Y, He X, Zhou T, Qin T, et al. Upregulation of PD-L1 by EGFR activation mediates the immune escape in EGFR-driven NSCLC: Implication for optional immune targeted therapy for NSCLC patients with EGFR mutation. J Thorac Oncol. 2015 Jun;10(6):910–23. doi:10.1097/JTO.0000000000000500.
- Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, Roche PC, Lu J, Zhu G, Tamada K, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat Med. 2002 Aug;8(8):793–800. doi:10.1038/nm730.
- Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012 Jun 28;366(26):2443–54. doi:10.1056/NEJMoa1200690.
- Weber JS, D'Angelo SP, Minor D, Hodi FS, Gutzmer R, Neyns B, Hoeller C, Khushalani NI, Miller WH, Jr, Lao CD, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): A randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015 Apr;16(4):375–84. doi:10.1016/S1470-2045(15)70076-8.
- Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, Daud A, Carlino MS, McNeil C, Lotem M, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med. 2015 Jun 25;372(26):2521–32. doi:10.1056/NEJMoa1503093.
- Brahmer J, Reckamp KL, Baas P, Crino L, Eberhardt WE, Poddubskaya E, Antonia S, Pluzanski A, Vokes EE, Holgado E, et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N Engl J Med. 2015 Jul 9;373(2):123–35. doi:10.1056/NEJMoa1504627.
- Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, Patnaik A, Aggarwal C, Gubens M, Horn L, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. 2015 May 21;372(21):2018–28. doi:10.1056/NEJMoa1501824.
- Nedrow JR, Josefsson A, Park S, Ranka S, Roy S, Sgouros G. Imaging of programmed death ligand-1 (PD-L1): Impact of protein concentration on distribution of anti-PD-L1 SPECT agent in an immunocompetent melanoma murine model. J Nucl Med. 2017 Oct;58(10):1560–66. doi:10.2967/jnumed.117.193268.
- Josefsson A, Nedrow JR, Park S, Banerjee SR, Rittenbach A, Jammes F, Tsui B, Sgouros G. Imaging, biodistribution, and dosimetry of radionuclide-labeled PD-L1 antibody in an immunocompetent mouse model of breast cancer. Cancer Res. 2016 January 15;76(2):472–9. doi:10.1158/0008-5472.CAN-15-2141.
- Rokita M, Stec R, Bodnar L, Charkiewicz R, Korniluk J, Smoter M, Cichowicz M, Chyczewski L, Niklinski J, Kozlowski W, et al. Overexpression of epidermal growth factor receptor as a prognostic factor in colorectal cancer on the basis of the allred scoring system. Onco Targets Ther. 2013 July 24;6:967–76. doi:10.2147/OTT.S42446.
- Selvaggi G, Novello S, Torri V, Leonardo E, De Giuli P, Borasio P, Mossetti C, Ardissone F, Lausi P, Scagliotti GV. Epidermal growth factor receptor overexpression correlates with a poor prognosis in completely resected non-small-cell lung cancer. Ann Oncol. 2004 January 01;15(1):28–32. doi:10.1093/annonc/mdh011.
- Ciardiello F, Tortora G. EGFR antagonists in cancer treatment. N Engl J Med. 2008 Mar 13;358(11):1160–74. doi:10.1056/NEJMra0707704.
- Thakur MK, Wozniak AJ. Spotlight on necitumumab in the treatment of non-small-cell lung carcinoma. Lung Cancer (Auckl). 2017 February 13;8:13–9.
- Kroesen BJ, Nieken J, Sleijfer DT, Molema G, de Vries EG, Groen HJ, Helfrich W, The TH, Mulder NH, de Leij L. Approaches to lung cancer treatment using the CD3 x EGP-2-directed bispecific monoclonal antibody BIS-1. Cancer Immunol Immunother. 1997;45(3–4):203–6. doi:10.1007/s002620050433. PMID:9435874.
- Krupka C, Kufer P, Kischel R, Zugmaier G, Lichtenegger FS, Kohnke T, Vick B, Jeremias I, Metzeler KH, Altmann T, et al. Blockade of the PD-1/PD-L1 axis augments lysis of AML cells by the CD33/CD3 BiTE antibody construct AMG 330: Reversing a T-cell-induced immune escape mechanism. Leukemia. 2016 February 01;30(2):484–91. doi:10.1038/leu.2015.214.
- Aktas E, Kucuksezer UC, Bilgic S, Erten G, Deniz G. Relationship between CD107a expression and cytotoxic activity. Cell Immunol. 2009;254(2):149–54. doi:10.1016/j.cellimm.2008.08.007. PMID:18835598.
- Zimmer L, Goldinger SM, Hofmann L, Loquai C, Ugurel S, Thomas I, Schmidgen MI, Gutzmer R, Utikal JS, Goppner D, et al. Neurological, respiratory, musculoskeletal, cardiac and ocular side-effects of anti-PD-1 therapy. Eur J Cancer. 2016 Jun;60:210–25. doi:10.1016/j.ejca.2016.02.024.
- Akbay EA, Koyama S, Carretero J, Altabef A, Tchaicha JH, Christensen CL, Mikse OR, Cherniack AD, Beauchamp EM, Pugh TJ, et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov. 2013 December 01;3(12):1355–63. doi:10.1158/2159-8290.CD-13-0310.
- Concha-Benavente F, Srivastava RM, Ferrone S, Ferris RL. EGFR-mediated tumor immunoescape: The imbalance between phosphorylated STAT1 and phosphorylated STAT3. Oncoimmunology. 2013 December 01;2(12):e27215. doi:10.4161/onci.27215.
- Leibowitz MS, Srivastava RM, Andrade Filho PA, Egloff AM, Wang L, Seethala RR, Ferrone S, Ferris RL. SHP2 is overexpressed and inhibits pSTAT1-mediated APM component expression, T-cell attracting chemokine secretion, and CTL recognition in head and neck cancer cells. Clin Cancer Res. 2013 February 15;19(4):798–808. doi:10.1158/1078-0432.CCR-12-1517.
- Brinkmann U, Kontermann RE. The making of bispecific antibodies. MAbs. 2017 March 01;9(2):182–212. doi:10.1080/19420862.2016.1268307.
- Grenga I, Donahue RN, Lepone LM, Richards J, Schlom J. A fully human IgG1 anti-PD-L1 MAb in an in vitro assay enhances antigen-specific T-cell responses. Clin Transl Immunology. 2016 May 20;5(5):e83. doi:10.1038/cti.2016.27.
- Stewart R, Morrow M, Hammond SA, Mulgrew K, Marcus D, Poon E, Watkins A, Mullins S, Chodorge M, Andrews J, et al. Identification and pre-clinical characterization of MEDI4736, an antagonistic anti-PD-L1 monoclonal antibody. Cancer Immunol Res. 2015 Sep 3;(9):1052–62. doi:10.1158/2326-6066.CIR-14-0191.
- Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, Sosman JA, McDermott DF, Powderly JD, Gettinger SN, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014 Nov 27;515(7528):563–7. doi:10.1038/nature14011.
- Heery CR, O'Sullivan Coyne GH, Madan RA, Schlom J, von Heydebreck A, Cuillerot J-M, Sabzevari H, Gulley J L. Phase I open-label, multiple ascending dose trial of MSB0010718C, an anti-PD-L1 monoclonal antibody, in advanced solid malignancies. J Clin Oncol. 2014; 32(Suppl 15):3064.
- Kelly K, Patel MR, Infante JR, Iannotti N, Nikolinakos P, Leach J, Wang D, Chandler JC, Jerusalem GHC, Gurtler JS, et al. Avelumab (MSB0010718C), an anti-PD-L1 antibody, in patients with metastatic or locally advanced solid tumors: Assessment of safety and tolerability in a phase I, openlabel expansion study. J Clin Oncol. 2015; 33(suppl; abstr 3044).
- Kaufman HL, Russell J, Hamid O, Bhatia S, Terheyden P, D'Angelo SP, Shih KC, Lebbe C, Linette GP, Milella M, et al. Avelumab in patients with chemotherapy-refractory metastatic merkel cell carcinoma: A multicentre, single-group, open-label, phase 2 trial. Lancet Oncol. 2016 October 01;17(10):1374–85. doi:10.1016/S1470-2045(16)30364-3.
- Boyerinas B, Jochems C, Fantini M, Heery CR, Gulley JL, Tsang KY, Schlom J. Antibody-dependent cellular cytotoxicity activity of a novel anti-PD-L1 antibody avelumab (MSB0010718C) on human tumor cells. Cancer Immunology Research. 2015 Oct;3(10):1148–57. doi:10.1158/2326-6066.CIR-15-0059.
- Fujii R, Friedman ER, Richards J, Tsang KY, Heery CR, Schlom J, Hodge JW. Enhanced killing of chordoma cells by antibody-dependent cell-mediated cytotoxicity employing the novel anti-PD-L1 antibody avelumab. Oncotarget. 2016 June 07;7(23):33498–511. doi:10.18632/oncotarget.9256.
- He Y, Hendriks D, van Ginkel R, Samplonius D, Bremer E, Helfrich W. Melanoma-directed activation of apoptosis using a bispecific antibody directed at MCSP and TRAIL receptor-2/death receptor-5. J Invest Dermatol. 2016 Feb;136(2):541–4. doi:10.1016/j.jid.2015.11.009.
- Helfrich W, Haisma HJ, Magdolen V, Luther T, Bom VJ, Westra J, van der Hoeven R, Kroesen BJ, Molema G, de Leij L. A rapid and versatile method for harnessing scFv antibody fragments with various biological effector functions. J Immunol Methods. 2000 April 03;237(1–2):131–45. doi:10.1016/S0022-1759(99)00220-3.
- Piccione EC, Juarez S, Liu J, Tseng S, Ryan CE, Narayanan C, Wang L, Weiskopf K, Majeti R. A bispecific antibody targeting CD47 and CD20 selectively binds and eliminates dual antigen expressing lymphoma cells. MAbs. 2015;7(5):946–56. doi:10.1080/19420862.2015.1062192. PMID:26083076.
- Kroesen BJ, Ter Haar A, Spakman H, Willemse P, Sleijfer DT, de Vries EG, Mulder NH, Berendsen HH, Limburg PC, The TH. Local antitumour treatment in carcinoma patients with bispecific-monoclonal-antibody-redirected T cells. Cancer Immunol Immunother. 1993 Nov;37(6):400–7. doi:10.1007/BF01526797.
- Heskamp S, Hobo W, Molkenboer-Kuenen JD, Olive D, Oyen WJ, Dolstra H, Boerman OC. Noninvasive imaging of tumor PD-L1 expression using radiolabeled anti-PD-L1 antibodies. Cancer Res. 2015 July 15;75(14):2928–36. doi:10.1158/0008-5472.CAN-14-3477.