
ABSTRACT
Checkpoint inhibitors targeting the PD-1/PD-L1 axis are promising immunotherapies shown to elicit objective responses against multiple tumor types, yet these agents fail to benefit most patients with carcinomas. This highlights the need to develop effective therapeutic strategies to increase responses to PD-1/PD-L1 blockade. Histone deacetylase (HDAC) inhibitors in combination with immunotherapies have provided preliminary evidence of anti-tumor effects. We investigated here whether exposure of either natural killer (NK) cells and/or tumor cells to two different classes of HDAC inhibitors would augment (a) NK cell‒mediated direct tumor cell killing and/or (b) antibody-dependent cellular cytotoxicity (ADCC) using avelumab, a fully human IgG1 monoclonal antibody targeting PD-L1. Treatment of a diverse array of human carcinoma cells with a clinically relevant dose of either the pan-HDAC inhibitor vorinostat or the class I HDAC inhibitor entinostat significantly enhanced the expression of multiple NK ligands and death receptors resulting in enhanced NK cell‒mediated lysis. Moreover, HDAC inhibition enhanced tumor cell PD-L1 expression both in vitro and in carcinoma xenografts. These data demonstrate that treatment of a diverse array of carcinoma cells with two different classes of HDAC inhibitors results in enhanced NK cell tumor cell lysis and avelumab-mediated ADCC. Furthermore, entinostat treatment of NK cells from healthy donors and PBMCs from cancer patients induced an activated NK cell phenotype, and heightened direct and ADCC-mediated healthy donor NK lysis of multiple carcinoma types. This study thus extends the mechanism and provides a rationale for combining HDAC inhibitors with PD-1/PD-L1 checkpoint blockade to increase patient responses to anti-PD-1/PD-L1 therapies.
Introduction
Immune checkpoint blockade constitutes a major paradigm shift in cancer therapy, aimed at interrupting a major tumor immune evasion axis. Checkpoint inhibitors targeting programmed death-1/programmed death ligand-1 (PD-1/PD-L1) inhibit the interaction of PD-1 on immune cells with its ligand PD-L1 on tumor and myeloid cells present in the tumor microenvironment (TME), resulting in reduction of immunosuppressive signals and enhancement of immune-cell activation and tumor lysis.Citation1–Citation5 Despite eliciting unprecedented clinical benefits for 20–50% of patients with diverse cancers, these agents still fail to benefit most patients with solid malignancies, including those with lung, prostate, ovarian, and triple-negative breast cancer.Citation6,Citation7 Thus, there is an unmet clinical need to develop therapeutic strategies that can augment clinical responses to this class of immune checkpoint inhibitors across multiple solid malignancies.
Despite notable exceptions, mounting evidence supports the notion that poor response to therapies targeting PD-1/PD-L1 is most prevalent in tumors with limited lymphocyte infiltration and poor PD-L1 expression in the TME, such as prostate cancer, where these therapies have failed thus far to elicit significant objective responses.Citation1,Citation6 In addition, previous research, mostly in melanoma and non-small cell lung (NSCL) cancer, strongly suggests that resistance to checkpoint blockade, including targeting the PD-1/PD-L1 axis, is highly associated with innate or acquired tumor defects in interferon (IFN)‒γ signaling and antigen processing and presentation pathways.Citation7–Citation11
Along the same lines, epigenetic silencing of immune-associated genes, including those involved in antigen processing and immune recognition, has been identified as a determining factor in tumor evasion of host immune surveillance.Citation10,Citation12,Citation13 One type of epigenetic silencing occurs through histone deacetylase enzymes (HDAC) that induce histone hypoacetylation, which is associated with gene silencing. Thus, HDAC inhibitors have emerged as novel immunomodulatory drugs (reviewed inCitation14,Citation15). Although HDAC inhibitors have demonstrated limited clinical benefit as a monotherapy against solid tumors, mounting preclinical and clinical evidence suggests there is potential to treat solid tumors by combining HDAC blockade with other treatment modalities, including radiation, chemotherapy, hormonal therapy, and, most importantly for the focus of this paper, immunotherapy such as PD-1 checkpoint inhibitors.Citation16–Citation20
Natural killer (NK) cells are effector lymphocytes of the innate immune system that have been shown to control tumor growth.Citation21 HDAC inhibitors have been shown to modulate the expression of NK ligands on the surface of neuroblastoma, melanoma, osteosarcoma, colon and Merkel cell carcinomas.Citation22–Citation26 Although few studies have investigated the role of epigenetic modulation on NK cell activation and cytotoxicity, reports indicate that histone acetylation is involved in the regulation of NK cell activation and effector function.Citation25,Citation27–Citation29 NK-mediated lysis of malignant cells is dictated by the direct engagement of a combination of inhibitory and activating NK cell surface receptors with ligands on the tumor, such as major histocompatibility complex (MHC) class I polypeptide-related chain A and B (MIC-A/B), UL16 binding proteins (ULBPs), Nectin-2 (CD112), poliovirus receptor (PVR, CD155), and intracellular adhesion molecule 1 (ICAM-1).Citation30–Citation32 A shift in the balance between activating and inhibitory signals dictates NK cell activation.Citation33 In addition, tumor cells can be eliminated by NK cells through antibody-dependent cellular cytotoxicity (ADCC) if the proper IgG1 monoclonal antibody (mAb) target is present on the surface of tumor cells.Citation34–Citation39 We have previously reported the ability of avelumab to mediate the lysis of a range of human carcinomas in vitro by ADCC in the presence of peripheral blood mononuclear cells (PBMCs) or NK cell effectors.Citation34,Citation39
Data from our laboratory have previously shown that clinically relevant exposure of breast and prostate carcinoma cells to HDAC inhibitors increases their expression of human leukocyte antigen (HLA) and antigen processing and presentation proteins, reversing tumor resistance to T cell‒mediated lysis.Citation40 Here, we used two distinct classes of HDAC inhibitors, vorinostat and entinostat, to examine the potential of epigenetic priming of multiple human carcinoma cell types and NK cell effectors to modulate the expression of NK ligands and receptors, and PD-L1. Vorinostat, a pan-HDAC inhibitor that suppresses the activity of class I and IIb HDACs, is currently approved by the Food and Drug Administration for the treatment of cutaneous T-cell lymphoma.Citation41,Citation42 Entinostat is a class I HDAC inhibitor under clinical investigation for the treatment of multiple malignancies.Citation42 We also investigated the effect of entinostat on NK effector function and carcinoma sensitivity to lysis in the presence or absence of the PD-L1 targeting mAb avelumab. To the best of our knowledge, our data demonstrate for the first time that HDAC inhibition of NK and/or tumor cells enhanced avelumab-mediated ADCC. Of note, entinostat treatment promoted a more active phenotype on NK cells from healthy donor and heavily pretreated cancer patient PBMCs. Data presented here offer a rationale for combining HDAC inhibitors with mAbs targeting the PD-1/PD-L1 axis, including for patients who are refractory or expected to not respond to these therapies alone due to absent or low PD-L1 tumor expression.
Results
Clinically relevant exposure of prostate and NSCL carcinoma cells to HDAC inhibitors modulates MIC-A/B and PD-L1 expression
Throughout this study, clinically relevant exposures of both HDAC inhibitors were used and were performed as follows. Carcinoma cells were exposed to DMSO or entinostat (500 nM) for 72 hours, which is the range of entinostat exposure (Cmax, AUC) attained in cancer patients dosed orally once weekly at 4 mg/m2.Citation43 Alternatively, tumor cells were exposed daily for 5 hours to DMSO or vorinostat (3 µM) for 4 consecutive days, mimicking the range of vorinostat exposure (Cmax, AUC) attained in cancer patients after a once-daily oral dose of 400 mg.Citation44
Tumor cell lysis by NK cells is partially dictated by direct NK cell engagement with stimulatory ligands, such as MHC class I-related chain molecules A and B (MIC-A/B).Citation25,Citation27 Therefore, we began by assessing the effect that vorinostat and entinostat had on the extracellular expression of MIC-A/B on prostate (DU145 and PC-3) and NSCL (NCI-H44 and NCI-H460) carcinoma cells. The data in are represented as fold increases of percent positive or geometric mean fluorescence intensity (gMFI) of MIC-A/B or PD-L1 induced by HDAC inhibitor treatment over DMSO-treated cells. The raw data of percent positive and gMFI for this table are in Supplemental Table 1. Exposure to vorinostat induced a substantial fold increase in the percentage of cells expressing MIC-A/B and on a per cell basis (gMFI) on 3/4 cell lines, namely PC-3, DU145, and H460 (). Exposure to entinostat also substantially increased the gMFI and/or the percentage of cells with MIC-A/B expression on 3/4 cell lines examined (DU145, H460, and H441). In only 2/4 cells lines, however, did both vorinostat and entinostat enhance MIC-A/B. No significant effects of either HDAC inhibitor were observed on the viability of tumor cells (Supplemental Table 1).
Table 1. Effect of tumor cell exposure to HDAC inhibitors on cell-surface expression of MIC-A/B and PD-L1
In multiple tumor types, cell-surface expression of PD-L1 has been shown to determine tumor-cell sensitivity to ADCC mediated by the anti-PD-L1 mAb avelumab in vitro.Citation34,Citation39 We investigated if cell surface expression of PD-L1 in prostate and NSCL cancer cell lines was altered by HDAC inhibitor treatment. In two out of four cancer cell lines, H460 and DU145, exposure to vorinostat increased the fold change of the gMFI and percentage of cells with PD-L1 expression compared to DMSO-treated cells (). A minor increase in PD-L1 expression was also observed on PC-3 cells. Vorinostat had no significant effect on PD-L1 expression on H441 carcinoma cells (). In contrast, entinostat exposure increased cell-surface expression of PD-L1 in all four prostate and NSCL carcinoma cell lines examined. H460, H441, and PC-3 cells exposed to entinostat showed a substantial fold increase in PD-L1 expression both in percentage and on a per cell basis relative to DMSO-treated cells, while a slight increase in PD-L1 expression was observed on DU145 cells. Overall, these data indicate that different HDAC inhibitors can differentially increase MIC-A/B and PD-L1 expression on carcinoma cells in vitro.
HDAC inhibitors modulate PD-L1 cell-surface expression on human NSCL and prostate carcinoma cells in vivo
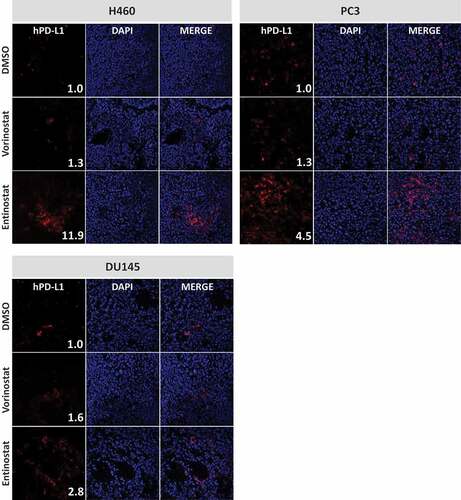
Next, we examined the effect of both HDAC inhibitors on PD-L1 expression in vivo. Female nu/nu mice (n = 3) were implanted with NCI-H460 (lung), DU145 (prostate), or PC-3 (prostate) carcinoma cells. When tumors reached 0.5–1 cm3, animals received four daily doses of DMSO or vorinostat (150 mg/kg, p.o.). Alternatively, animals received a single dose of entinostat (20 mg/kg, p.o.) or DMSO 72 hours prior to tumor excision. Frozen specimens were examined for cell-surface expression of human PD-L1 by immunofluorescence. Quantification of PD-L1 expression levels was determined by analysis of fluorescent image intensity (ImageJ). As shown in , all control tumors had very low cell-surface expression of PD-L1 in vivo. However, treatment of animals with HDAC inhibitors promoted upregulation of PD-L1 in the TME. In H460 tumors, PD-L1 levels (MFI) were increased 11.91-fold after entinostat administration relative to DMSO-treated controls. Vorinostat treatment also augmented expression of PD-L1 in these tumors, albeit to a lower degree (1.32-fold). Similar effects were attained in prostate tumor xenografts where PD-L1 expression in the TME of PC-3 tumors was increased by 1.31-fold and 4.49-fold after treatment with vorinostat and entinostat, respectively. Similarly, vorinostat and entinostat induced 1.58-fold and 2.83-fold increase in mean PD-L1 expression in DU145 tumors, respectively. These data support our in vitro observations () that the two classes of HDAC inhibitors can differentially upregulate PD-L1 in distinct tumor types. Interestingly, entinostat increased PD-L1 levels in vivo and in vitro to a greater extent than vorinostat at clinically relevant concentrations tested here (, ).
Figure 1. Vorinostat and entinostat modulate PD-L1 expression in human lung and prostate tumor xenografts. Female nu/nu mice implanted with NCI-H460 (lung), PC-3 (prostate), or DU145 (prostate) carcinoma cells received four daily doses of DMSO or vorinostat (150 mg/kg, p.o.). Tumors were excised 24 h after the last dose. Alternatively, animals received a single dose of entinostat (20 mg/kg, p.o.) or DMSO 72 h prior to tumor excision. Frozen sections were examined for cell-surface expression of human PD-L1 by immunofluorescence, using anti-human PD-L1 (clone SP142) and goat anti-rabbit AF594, and counterstained with DAPI-containing mounting medium, as described in Materials and Methods. Confocal images are shown at 20X magnification and are representative of 3 animals/treatment. Inset numbers: relative PD-L1 expression levels were calculated using ImageJ software by normalizing the intensity values to their respective DMSO-treated controls
Prostate and NSCL carcinoma cells exposed to HDAC inhibitors are significantly more sensitive to avelumab-mediated ADCC and direct NK lysis
To determine whether the augmented PD-L1 and MIC-A/B expression observed in carcinoma cells after exposure to HDAC inhibitors affected the sensitivity of prostate and NSCL tumor cells to NK cell‒mediated attack, DU145, PC-3, H460, and H441 cells were exposed to vorinostat, entinostat, or DMSO, and then washed prior to being used as targets for avelumab-mediated and direct NK cell lysis in vitro. NK cells purified from two healthy donor PBMCs were used as effectors. For both direct NK lysis and ADCC-mediated lysis, the fold increase induced by the HDAC inhibitor over DMSO was calculated (). Focusing first on prostate cancer cells, tumor cell exposure to vorinostat significantly enhanced avelumab-mediated NK lysis of DU145 cells () and PC-3 cells () compared to DMSO-treated cells with an average fold increase from the two healthy donors of 1.7 and 1.3, respectively (). The same cell lines exposed to entinostat, however, had a heightened sensitivity to avelumab-mediated NK lysis ( and ) with the average fold increase for DU145 and PC-3 of 2.9 and 4.0, respectively (). In addition, entinostat rendered DU145 cells (fold increase 3.9) and PC-3 cells (fold increase 40.5) significantly more sensitive to direct NK cell lysis relative to DMSO-treated cells, whereas vorinostat showed no effect on either cell line (-, gray bars; ).
Table 2. Fold changes in direct NK lysis or avelumab-mediated ADCC lysis with HDAC inhibitor exposure of prostate and NSCLC cell lines
Figure 2. Prostate and NSCL human carcinoma cells exposed to vorinostat or entinostat are more sensitive to avelumab-mediated ADCC. DU145 (A, C) and PC-3 (B, D) prostate carcinoma cells and H460 (E, G) and H441 (F, H) NSCL carcinoma cells were exposed to vorinostat (A, B, E, F), entinostat (C, D, G, H), or DMSO, as described in Materials and Methods, prior to being used as targets for NK cell lysis (4 h), in the presence or absence of avelumab or isotype control (2 ng/mL). Purified NK cells from 2 healthy donors were used as effectors at an effector:target ratio of 30:1. Results are presented as mean ± S.E.M. from 3 replicate wells, and are representative of 2–4 independent experiments. Asterisks denote statistical significance relative to controls (2-way ANOVA). * p < 0.05; ** p < 0.01; *** p < 0.001
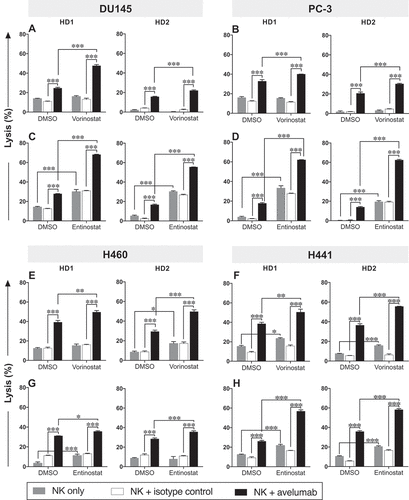
Similar results were observed with NSCLC cell lines. Vorinostat exposure significantly augmented avelumab-mediated NK lysis of H460 () and H441 cells () relative to DMSO-treated cells with the average fold increase of 1.5 and 1.4, respectively (). Increased direct NK lysis of H460 (1/2 healthy donors) and H441 cells (2/2 healthy donors) was also observed with exposure to either HDAC inhibitor (-, gray bars; ). In addition, entinostat exposure significantly enhanced avelumab-mediated NK lysis of H460 cells () and H441 cells () compared to DMSO-treated cells with the average fold increase of 1.2 and 1.9, respectively (). These data indicate that exposure to either HDAC inhibitor significantly enhances both avelumab-mediated lysis and direct NK cell‒mediated lysis of prostate and/or NSCL carcinoma cell lines.
Avelumab-mediated NK lysis of carcinoma cells exposed to HDAC inhibition is dependent on CD16 engagement
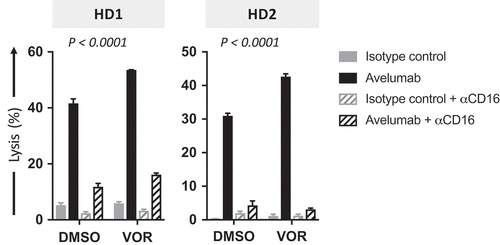
ADCC lysis mediated by human NK cells occurs when CD16 (FcγRIII) on NK effectors interacts with the Fc portion of IgG1 antibodies bound to target cells.Citation31,Citation35 CD16 engagement has been shown to be a critical component of ADCC lysis mediated by avelumab.Citation34 To confirm that increased NK cell lysis of carcinoma cells exposed to HDAC inhibitors in the presence of avelumab is specifically mediated by ADCC, NK effectors isolated from PBMCs of two healthy donors were exposed to a CD16-blocking antibody prior to being used in an in vitro ADCC assay. As shown in , the presence of CD16-blocking antibody significantly inhibited the lysis of both DMSO- and vorinostat-treated H460 cells in the presence of avelumab, confirming that the augmented NK cell lysis of carcinoma cells exposed to HDAC inhibitors in the presence of avelumab is mediated by ADCC. Similar results were observed with DU145 carcinoma cells exposed to entinostat (Supplemental Figure 1).
Figure 3. Avelumab-mediated lysis of carcinoma cells is decreased by CD16 neutralization. NCI-H460 lung carcinoma cells were exposed to vorinostat or DMSO, as described in Materials and Methods, prior to being used as targets for NK cell lysis (4 h) in the presence or absence of avelumab or isotype control (2 ng/mL). Where applicable, NK cells were pretreated for 2 h with anti-CD16 blocking antibody (12 µg/mL) prior to being used as effectors. Purified NK cells from 2 healthy donors were used as effectors at an effector:target ratio of 30:1. Results are presented as mean ± S.E.M. from 3 replicate wells, and are representative of 2–4 independent experiments. P values denote statistical significance relative to DMSO controls (2-way ANOVA). HD, healthy donor
Exposure of multiple carcinoma types to entinostat modulates various ligands and immune-relevant proteins involved in NK cell recognition and tumor cell lysis
Because entinostat is a more specific HDAC inhibitor targeting only class I enzymes and having a lower frequency of adverse events compared to vorinostat,Citation45 from this point forward we performed a more comprehensive analysis focusing on the effect of entinostat on additional carcinoma types and on NK cell function. Previously, we showed that entinostat and vorinostat augmented the cell surface expression of MIC-A/B and PD-L1 on prostate and NSCL carcinomas (), but NK-mediated recognition and elimination of tumor cells is dictated by direct NK cell engagement with multiple other inhibitory and activating ligands. Therefore, we expanded our examination of cell surface proteins on tumors cells to include the NK activating ligands for NKG2D (MIC-A/B and ULBP proteins), DNAM-1 (CD112 (nectin-2) and CD155 (PVR)), LFA-1 (ICAM-1), and NKp30 (B7-H6), as well as the inhibitor of NK cell function, MHC class I/HLA-ABC.Citation27,Citation31,Citation35 Additionally, we examined the death receptors TRAIL-R2 (DR5) and Fas, both known to be involved in NK-mediated tumor lysis.Citation46,Citation47 The carcinoma types we examined were also expanded to include not only prostate (DU145 and PC-3) and non-small cell lung (H441 and H460), but also small-cell lung (H1882), triple-negative breast (MDA-MB-231 and SUM159PT), and ovarian (HEY-T30) cancers. The effect of entinostat was assessed by exposing these carcinoma cells in vitro to DMSO or entinostat as described before analyzing by flow cytometry the surface expression of the aforementioned proteins. Consistent with results in prostate and NSCL cancer cell lines ( and Supplemental Table 1), entinostat exposure substantially upregulated the NKG2D ligand MIC-A/B in 2/4 additional cell lines, namely SUM159PT (triple-negative breast; TNB) and HEY-T30 (ovarian) ( and Supplemental Table 1). Entinostat exposure also significantly increased the cell-surface expression of ULBP proteins across diverse tumor types (). This included ULBP-1 upregulation in 3/8 tumor cell lines (prostate and SCL), ULBP-3 in 3/8 tumor cell lines (NSCL, SCL, and TNB), and ULBP-2/5/6 in 4/8 cell lines (NSCL, SCL, TNB, and ovarian) examined (). Entinostat had lesser effects on ULBP-4 and B7-H6 expression (Supplemental Table 2). On a per cell basis (MFI), exposure to entinostat substantially augmented the expression of the DNAM-1 ligands CD112 and CD155 in half of the cell lines examined, and expression of ICAM-1 in 6/8 cell lines examined. Similar results were observed with augmented expression of death receptors Fas (4/8 cell lines) and DR5 (5/8 cell lines). While all cell lines examined had HLA-ABC expression levels greater than 90%, entinostat increased the NK inhibitor HLA-ABC on a per cell basis (MFI) in 6/9 cell lines. These data will be discussed below in light of other data. Consistent with our observations in prostate and NSCL cancer cell lines (), entinostat significantly increased the expression of PD-L1 in all four cell lines examined, H1882 (SCLC), MDA-MB-231 (TNBC), SUM159PT (TNBC), and HEY-T30 (ovarian) ( and Supplemental Table 1). These data indicate that entinostat exposure increases the expression of a variety of immune relevant proteins on multiple carcinoma cell lines, suggesting that a more productive immunological synapse could form between NK and tumor cells.
Table 3. Effect of tumor cell exposure to entinostat on cell-surface expression of immune-relevant proteins
Diverse carcinoma cell types exposed to entinostat are more sensitive to NK-mediated attack through direct lysis and anti-PD-L1‒mediated ADCC
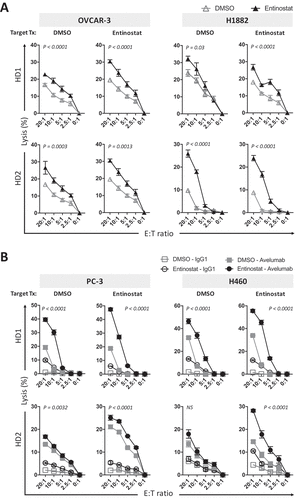
Extending what was shown in to additional cancer types, we investigated if entinostat exposure of cells from ovarian and small cell lung cancers also enhanced NK-mediated direct lysis. Ovarian (OVCAR-3, SK-OV-3, HEY-T30) and SCL (H1882) carcinoma cells were exposed to entinostat or DMSO as described before, and then washed prior to being used in vitro as targets for direct NK cell lysis. NK cells purified from PBMCs from two healthy donors were used as effectors. As shown in , OVCAR-3 cells exposed to entinostat were significantly more sensitive to NK-mediated lysis at various effector:target (E:T) ratios than control DMSO-exposed cells. Similar results were attained with ovarian cancer cell lines SK-OV-3 and HEY-T30, as well as the SCL carcinoma cell line H1882 with NK cells from both healthy donors. Ovarian (OVCAR-3, SK-OV-3, HEY-T30) and TNB (MDA-MB-231) carcinoma cells were also significantly more sensitive to PD-L1‒specific NK targeting through avelumab-mediated ADCC (). Interestingly, entinostat treatment of OVCAR-3 and SK-OV-3 cells elevated NK-mediated direct lysis to a level that was above or equal to the level of lysis with avelumab-mediated ADCC of DMSO treated cells (). These data demonstrate that carcinomas of diverse origins exposed to entinostat are more sensitive to human NK cell lysis, either directly or through avelumab-mediated ADCC.
Figure 4. Entinostat exposure of carcinoma cells increases their sensitivity to NK-mediated attack through direct lysis and anti-PD-L1‒mediated ADCC. (A) Ovarian (OVCAR-3, SK-OV-3, HEY-T30) and SCL (H1882) human carcinoma cells were exposed to entinostat or DMSO, as described in Materials and Methods, prior to being used as targets for NK cell lysis (15 h). (B) Ovarian (OVCAR-3, SK-OV-3, HEY-T30) and TNB (MDA-MB-231) carcinoma cells were exposed to entinostat, or DMSO as above, prior to being used as targets for NK cell lysis (15 h) in the presence of avelumab or isotype control (2 ng/ml). Purified NK cells from 2 healthy donors were used as effectors at the designated effector:target (E:T) ratios. (C) DU145 cells were treated as in (B) with the addition M7824 (2.46ng/mL) and at a E:T ratio of 20:1. Results are presented as mean ± S.E.M. from 3 replicate wells. Results on direct NK lysis and PD-L1‒mediated ADCC are representative of 2–4 independent experiments. Results on M7824-mediated ADCC are representative of 1 independent experiment. P values denote statistical significance relative to DMSO controls (2-way ANOVA). HD, healthy donor; TNBC, triple negative breast cancer
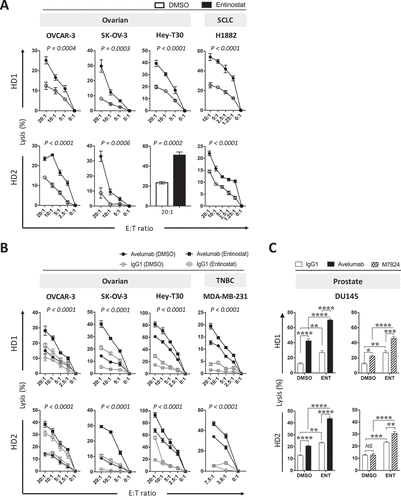
We also investigated if the PD-L1 targeting antibody M7824 (MSB0011359C) could elicit similar increases in ADCC of entinostat-treated targets. M7824 is a first-in-class fully human bifunctional molecule composed of the extracellular domain of human TGFβRII (TGFβ Trap) linked to the C-terminus of anti-PD-L1 heavy chain. Our laboratory has previously shown M7824 to be capable of mediating ADCC of a wide range of human carcinoma cells in vitro, albeit to a lesser extent than avelumab.Citation48 Using an equimolar concentration of M7824 to avelumab, entinostat treatment of DU145 significantly enhanced the M7824-mediated ADCC (). However, M7824-mediated ADCC of DU145 cells was reduced compared to avelumab-mediated ADCC ().
Treatment of cancer patient PBMCs with HDAC inhibitors reduced Tregs and had minimal impact on other immune cell types
Next, we investigated if these drugs had any impact in vitro on NK cells and other immune subsets in PBMCs of cancer patients. Via flow cytometry, we identified nine classic immune cell types and 114 refined subsets relating to their maturation and function in PBMCs from seven heavily pretreated metastatic breast cancer patients after entinostat, vorinostat, or DMSO exposure (Supplemental Table 3). Neither HDAC inhibitor altered the viability of the PBMCs (Supplemental Tables 4 and 5). Furthermore, there was minimal to no change in the frequency of CD4+ T cells, CD8+ T cells, NKT, B cells, plasmacytoid dendritic cells (DCs), and myeloid derived suppressor cells (MDSCs). No change was observed in the frequency of NK cells with either HDAC inhibitor, but there was a decrease in the double negative non-lytic, non-cytokine producing NK cells after exposure to entinostat (Supplemental Table 4). Of note, entinostat exposure significantly increased the frequency of conventional DCs in 7/7 patient samples (p = 0.0156) and markedly decreased the frequency of regulatory T cells (Tregs) and MFI of FoxP3 in 5/7 patient samples (p = 0.0156) (Supplemental Table 4 and Supplemental Figure 2). Vorinostat exposure also decreased Treg frequency and FoxP3 expression, albeit to a lesser degree as compared to entinostat (Supplemental Figure 2 and Supplemental Table 5). Thus, treatment of cancer patient PBMCs with HDAC inhibitors had no adverse effects on classic immune subsets. However, both HDAC inhibitors reduced Tregs, which are known immune suppressors.
Entinostat increases effector function of human NK cells
Mounting evidence suggests that NK effector function is epigenetically regulated, including through control of histone acetylation.Citation27,Citation28 Thus, we investigated if NK cell function could be modulated by exposure to entinostat. Human NK cells isolated from healthy donors were exposed to DMSO or entinostat for 24 hours and then washed prior to being used as effectors for NK-mediated lysis of DMSO or entinostat-treated carcinoma cells. As shown in , treating NK cells with entinostat resulted in significantly enhanced lysis of OVCAR-3 exposed to either DMSO or entinostat. Similar results were observed on the lysis of the SLC carcinoma line H1882. In addition, NK cells exposed to entinostat were able to lyse prostate (PC-3) and NSCL (H460) carcinoma cells exposed to either DMSO or entinostat more efficiently through avelumab-mediated ADCC (). These data indicate that HDAC inhibition with entinostat in human healthy donor NK cells augments their cytotoxicity against diverse tumor types.
Figure 5. Entinostat modulates the cytotoxicity of NK cells from healthy donors to augment the lysis of multiple human carcinoma cells. Human NK cells isolated from 2 healthy donors were exposed to DMSO or entinostat for 24 h prior to being used as effectors at the designated E:T ratios for NK-mediated lysis (15 h) in the presence of avelumab or IgG1 isotype control, as described in Materials and Methods. Targets were (A) DMSO or entinostat-treated OVCAR-3 or H1882 carcinoma cells, or (B) DMSO or entinostat-treated PC-3 and H460 carcinoma cells. Results are presented as mean ± S.E.M. from 3–5 replicate wells, and are representative of 2 independent experiments. P values denote statistical significance relative to DMSO controls. NS, not statistically significant. HD, healthy donor
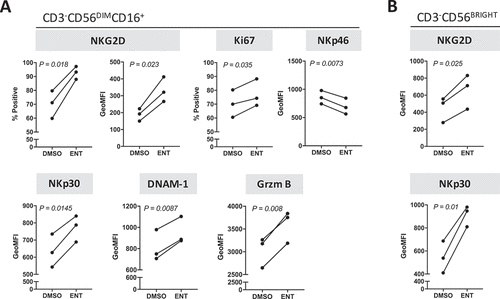
Entinostat modulates the phenotype of human healthy donor NK cells towards a more active and cytotoxic signature
To understand the mechanism associated with entinostat-induced heightened cytotoxicity of NK cells, purified NK cells from three healthy donors treated for 48 hours with entinostat or DMSO were examined for the expression of multiple activation markers by flow cytometry. As shown in , entinostat induced a significant increase in protein expression in 3/4 activating receptors, namely NKG2D (p = 0.018), NKp30 (p = 0.0145), and DNAM-1 (p = 0.0087) in mature CD3−CD56DIMCD16+ NK cells from all healthy donors examined, albeit decreasing NKp46 expression (p = 0.0073). Entinostat exposure also significantly increased the proliferative capacity of mature NK cells as denoted by Ki67 expression (p = 0.035). For all healthy donors, granzyme B expression was markedly increased (p = 0.008) upon NK exposure to entinostat, suggesting augmented NK cytolytic function. Entinostat also increased the expression of the activating receptors NKG2D (p = 0.025) and NKp30 (p = 0.01) on a per cell basis on 3/3 healthy donors’ CD56BRIGHT immature NK cells (). Altogether these data indicate that entinostat promotes activation of both mature and immature human NK cells through multiple mechanisms.
Figure 6. Entinostat modulates the phenotype of human NK cells towards a more active and cytotoxic signature. Purified NK cells from 3 healthy donors exposed to DMSO or entinostat for 48 h as described in Materials and Methods were examined for the expression of multiple activation markers by flow cytometry. (A) Mature CD3−CD56DIMCD16+ NK cells. (B) Immature CD3−CD56BRIGHT NK cells. P values denote statistical significance relative to DMSO controls
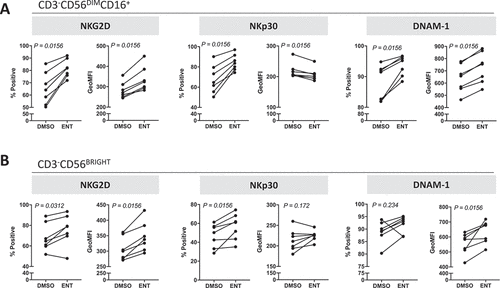
Entinostat modulates the phenotype of NK cells from cancer patients towards a more active signature
It has been shown that NK cells from cancer patients’ PBMCs have decreased expression of activation receptors and functional defects;Citation49,Citation50 we thus investigated if entinostat treatment could increase the activation phenotype of these NK cells. PBMCs from seven heavily pretreated metastatic breast cancer patients were treated for 48 hours with a clinically relevant exposure of entinostat or DMSO. As stated before, entinostat had no effect on the viability of PBMCs and did not alter NK cell frequency (Supplemental Table 4). However, entinostat exposure markedly increased the population of CD3−CD56DIMCD16+ mature NK cells expressing NKG2D, NKp30, and DNAM-1 (p = 0.0156) (), indicating a more active phenotype. This effect was also observed on a per cell basis, as reflected by an increase in NKG2D and DNAM-1 MFI in NK cells from all seven patients. Similar results were observed in CD3−CD56BRIGHT immature NK cells when PBMCs were exposed to entinostat relative to DMSO control (). Overall, these results suggest entinostat promotes an NK cell phenotype associated with activation in metastatic cancer patients.
Figure 7. Entinostat modulates the phenotype of NK cells from cancer patients towards a more active signature. PBMCs from 7 heavily pretreated breast cancer patients exposed to entinostat or DMSO for 48 h as described in Materials and Methods were examined for the expression of multiple activation markers by flow cytometry. (A) Mature CD3−CD56DIMCD16+ NK cells. (B) Immature CD3−CD56BRIGHT NK cells. P values denote statistical significance relative to DMSO controls
Discussion
In this study, we demonstrated that clinically relevant exposure of tumor cells to two different classes of HDAC inhibitors resulted in altered tumor cell phenotype and enhanced NK cell direct killing and ADCC-mediated killing of a variety of carcinoma cells ( and , and ). Further, entinostat treatment of NK cells enhanced their ability to lyse tumors cells () and promoted an activated phenotype in NK cells from both healthy donors and heavily pretreated breast cancer patients ( and ). The fact that two different types of HDAC inhibitors both enhanced NK cell lysis of tumor cells indicates this was most likely not due to off target effects. Furthermore, these results are in agreement with what other reports have shown for entinostat, vorinostat, and other HDAC inhibitors (sodium butyrate and sodium valproate) in other tumor types.Citation22,Citation51–Citation54 One study found that hepatocellular carcinoma cells treated with sodium valproate had increased recognition by cytotoxic lymphocytes in an NKG2D-dependent manner, and increased the expression of MIC-A/B on these cells.Citation54 Another study reported that treatment of DAOY (medulloblastoma) and PC-3 cells with multiple HDAC inhibitors resulted in elevated expression of MIC-A/B, ULBP1-3, CD112, and CD155, and enhanced lysis of these cells by IL-2‒activated PBMCs.Citation53 A third study demonstrated that exposure of Ewing sarcoma cells to HDAC inhibitors elevated the expression of MIC-A/B, ULBP1-3, CD112, and CD155 and enhanced their lysis by IL-15 activated NK cells.Citation52 Importantly, in our studies, both inhibitors were used with clinically relevant exposures in an attempt to closely approximate effects that would be seen in patients. Additionally, NK cells in our study were freshly isolated from PBMCs and not activated with IL-2 or IL-15 prior to being used in killing assays.
Previous studies that investigated HDAC inhibitors enhancing NK killing focused on either a few cells lines from melanoma, osteosarcoma, hepatocellular carcinoma, prostate cancer, or medulloblastoma.Citation22,Citation51–Citation54 Our study simultaneously investigated tumor cell lines from NSCL, SCL, prostate, TNB, and ovarian origins, extending the effect of HDAC inhibition enhancing NK killing to novel tumor types. Additionally, our study provided a more comprehensive analysis of 12 different NK lysis relevant proteins on the surface of nine different cell lines encompassing five different tumor types ( and , Supplemental Tables 1 and 2). These data highlight the generality of our findings by extending them to multiple different tumor types.
It has been reported that MHC class I/HLA-ABC presence on tumor cells can inhibit NK cell‒mediated lysis.Citation27,Citation31,Citation35 In our tumor cell phenotype, we observed upregulation of HLA-ABC with HDAC inhibition (). However, despite HLA-ABC upregulation, HDAC inhibition of tumor cells augmented their recognition and direct lysis by NK cells, as well as through avelumab-mediated ADCC (–). One possible explanation for this would be that the immunological synapse between a tumor cell and NK cell is a sum of both inhibitory and activating interactions. Since there was upregulation of many activating ligands and receptors on both tumor and NK cells with HDAC inhibition (, ), we may be tipping the balance towards a more NK cell‒activating interaction. This suggests HDAC inhibition of tumor cells and/or NK cells promotes the formation of a more productive immunological synapse.
To investigate the impact HDAC inhibitors would have on the immune subsets in the blood, we treated seven cancer patients' PBMCs with vorinostat or entinostat. Entinostat has recently been shown to decrease the frequency of circulating Tregs in cancer patients.Citation55 To our knowledge, our findings are the first comprehensive analysis on the effects of these agents spanning 123 immune subsets in PBMCs from heavily pretreated cancer patients (Supplemental Table 3). We observed that vorinostat treatment of PBMCs elevated the frequency of NKT cells (Supplemental Table 5). Entinostat treatment had very different effects, significantly increasing the frequency of conventional DCs and reducing the frequency of Tregs (Supplemental Table 4 and Supplemental Figure 2). No deleterious effects to the other immune subsets were observed. These data further support the use of HDAC inhibitors in combination with immunotherapies.
Our findings also indicate that epigenetic priming of CD56DIM mature NK cells from healthy donors and metastatic cancer patients with entinostat induces a phenotypic signature associated with increased activation and effector function ( and ). However, entinostat decreased the expression of the activating receptor NKp46 in NK cells from healthy donors. NKp46 is an important receptor in NK lytic function in infection and cancer, known to recognize several virus-specific ligands in virus-infected cells.Citation56–Citation58 Currently, its ligand(s) in cancer cells remains to be identified.Citation58 Thus it is possible that its decreased expression in NK cells upon entinostat exposure does not impact NK lysis of the tumor cell lines used in our study or is overcome by the overall observed increase in other activating ligands and effector molecules. Entinostat also induced activation of CD56BRIGHT immature NK cells from healthy donors and cancer patients ( and ). Interestingly, it was recently demonstrated that CD56BRIGHT NK cells are not only involved with immunomodulation, but are also capable of lysing tumor cells especially after priming with IL-15.Citation59 Therefore, by enhancing the activation of CD56BRIGHT NK cells with entinostat, we may also increase the cytolytic function of these cells. Moreover, the fact that NK cells from heavily pretreated breast cancer patients had phenotypic changes after exposure to entinostat similar to healthy donor NK cells was a key novel finding of our study since it has been shown that cancer patients often have dysfunctional NK cells that are lacking an active phenotype.Citation49,Citation50
We have previously shown that ALT-803, the IL-15 superagonist/IL-15RαSushi-Fc fusion complex, expands and enhances the function of NK cells.Citation48,Citation60 Additionally, it has previously been shown by our laboratory that ALT-803 enhances NK lysis and ADCC induced by M7824.Citation48 One could speculate that the combination of ALT-803 and entinostat may further increase NK cell direct killing and anti-PD-L1‒mediated ADCC.
To our knowledge, our data show for the first time that HDAC inhibition elevated NK anti-PD-L1-mediated ADCC of tumor cells. A previous study demonstrated PD-L1 elevation in melanoma cell lines and patient melanoma biopsies after entinostat treatment.Citation24 Here we demonstrate that HDAC inhibition elevates PD-L1 levels across diverse carcinoma cell types in vitro and in human prostate and NSCL cancer xenografts (, ). Normally, this would be considered a disadvantageous effect of HDAC inhibition because PD-L1 is involved in promoting tolerance and decreasing immune surveillance.Citation61,Citation62 However, the data from our study suggest the enhanced PD-L1 levels on tumor cells could increase the accumulation of anti-PD-L1 antibody in the TME and subsequently enhance ADCC tumor cell lysis (, , and ). This concept could be applied to other antibody therapies that target PD-L1 and mediate ADCC, such as M7824, which combines an anti-PD-L1 antibody with a TGFβ Trap, increasing the amount of the bifunctional antibody conjugate in the TME ().Citation48,Citation63
Also, it is important to note that the effectiveness of this combination of entinostat and avelumab may, in part, be due to the substantial elevation in PD-L1 on the tumor cell surface. For instance, entinostat exposure did not consistently elevate the percent of cells expressing epidermal growth factor receptor (EGFR) on their cell surface (2/9 cell lines) or its expression on a per cell basis (4/9 cell lines) (Supplemental Table 2) as it did for PD-L1. Accordingly, targeting these cells with the anti-EGFR antibody cetuximab showed that entinostat exposure only modestly and inconsistently enhanced cetuximab-mediated ADCC of four out of 13 different cell lines (Supplemental Figure 3).
From a therapeutic perspective, HDAC inhibitors may sensitize patients to therapies targeting PD-1/PD-L1, such as avelumab or M7824. Innate or acquired resistance to therapies targeting PD-1/PD-L1 has been shown to be most prevalent in patients with poor PD-L1 expression in the TME as well as those harboring defects in IFN-γ signaling and antigen processing and presentation pathways. Citation8–Citation11 HDAC inhibitors enhance these factors. First, we show in this study that HDAC inhibition enhances PD-L1 expression on human carcinoma cells in vitro and in vivo (, ). Additionally, we and others have previously shown that HDAC inhibitors can restore protein expression of MHC class I and antigen processing and presentation molecules.Citation23,Citation40,Citation64 This sensitization to PD-1/PD-L1 therapies by HDAC inhibition may be translated into tumor types where this class of agents has not yet generated significant clinical outcome, such as prostate, SCL, ovarian, and TNB carcinomas. One could speculate that this sensitization could transform a cold/non-inflamed tumor into a hot/inflamed tumor that is supported by preclinical studies demonstrating that romidepsin, a class I HDAC inhibitor, enhances CD8 T cell infiltration.Citation65 Moreover, an ongoing clinical trial with pembrolizumab (anti-PD-1) and entinostat in melanoma patients previously treated with anti-PD-1 therapy has shown promising signs of clinical efficacy in this patient population. Of note, a patient who previously progressed on therapy with nivolumab (anti-PD-1) and ipilimumab (anti-CTLA-4) had a confirmed partial response. This patient converted from a PD-L1 negative non-inflamed gene signature (“cold”) to a PD-L1 positive inflamed gene signature (“hot”) after 2 weeks of treatment.Citation66 Currently, entinostat is being examined clinically in combination with the PD-L1 targeting mAbs avelumab and atezolizumab in patients with ovarian (NCT02915523) and TNB (NCT02708680) cancer, respectively. Studies combining entinostat with other immune checkpoints are also planned.
These findings underscore the potential clinical benefit of HDAC inhibition to reprogram solid tumors and human NK cells for subsequent response to immunotherapy, including immune checkpoint blockade. Overall, this provides a rationale for combining HDAC inhibitors with therapies relying on activation of the innate immune system, such as mAbs targeting PD-L1. In particular, entinostat immunotherapy combinations may show promise for patients who have failed monotherapy regimens with epigenetic therapies and/or whose tumors respond poorly to anti-PD-1/PD-L1 checkpoint inhibitors.
Materials and methods
Tumor-cell lines
Human carcinoma cells of the ovary (OV) [SK-OV-3 (ATCC® HTB-77), HEY-T30 (ATCC® CRL-3252), OVCAR-3 (ATCC® HTB-161)], small cell lung (SCL) [NCI-H1882 (ATCC® CRL-5903)], NSCL [NCI-H441 (ATCC® HTB-174™), NCI-H460 [H460] (ATCC® HTB-177™)], prostate [DU145 (ATCC® HTB-81™), PC-3 (ATCC® CRL-1435™)], and TNB [MDA-MB-231 (ATCC® HTB-26)] were obtained from American Type Culture Collection (Manassas, VA, USA). The triple-negative breast carcinoma cell line SUM-159PT [CS-08] was obtained from Asterand Bioscience (Detroit, MI, USA). All cell lines were used at low passage number, free of Mycoplasma and cultured at 37°C/5% CO2 in medium designated by the provider for propagation and maintenance.
Human healthy donor NK cells
PBMCs from healthy donors obtained from the National Institutes of Health Clinical Center Blood Bank (NCT00001846) were separated by Ficoll-Hypaque density gradient separation and cryopreserved (in Thermo Scientific Mr. FrostyTM containers at −1°C/minute) in 90% heat-inactivated fetal bovine serum and 10% DMSO at a concentration of 1-5 × 107 cells/mL until assayed. PBMCs were counted and assessed for cell viability with trypan blue exclusion; median cell viability of healthy donor PBMCs was 96% (85% min–99% max). NK effector cells were isolated from PBMCs using the Human NK Cell Isolation (negative selection) Kit 130–092-657 (Miltenyi Biotech, San Diego, CA, USA), according to the manufacturer’s protocol. Isolated NK cells were > 90% pure. Purified NK cells were incubated overnight in RPMI-1640 medium (Mediatech, Manassas, VA, USA) containing 10% fetal bovine serum (Gemini Bio-Products, West Sacramento, CA, USA), glutamine, and antibiotics (Mediatech) prior to use.
Analysis of immune cell subsets in PBMCs from cancer patients
PBMCs were isolated and cryopreserved as described above from the blood of seven patients with metastatic breast cancer enrolled in a Phase II study at the National Cancer Institute (NCI; NCT00179309Citation67) prior to treatment. PBMCs were defrosted and rested overnight at 37 °C/5% CO2 in Iscove’s DMEM medium (Mediatech) supplemented with 10% heat-inactivated human AB serum (Omega Scientific, Terzana, CA, USA, virus negative, sterile filtered), 2 mM glutamine (Mediatech), 100 units per ml penicillin and 100 μg ml−1 streptomycin (Mediatech). PBMCs were counted and assessed for cell viability with trypan blue exclusion; median cell viability of cancer patient PBMCs was 89.5% (84.3% min–94.6% max). The following day, 5 × 106 PBMCs were exposed to DMSO or entinostat (0.5 μM) for 48 hours. Alternatively, 5 × 106 PBMCs were exposed to vorinostat (3 μM) or DMSO daily for 5 hours for 2 consecutive days; at the end of the first treatment, cells were washed in fresh medium and returned to incubation at 37°C with 5% CO2. Following full treatment, PBMCs were harvested, assessed for number and viability by trypan blue exclusion, and examined by multicolor flow cytometry to identify 123 immune cell subsets, as previously described.Citation68 Immune subsets with a potentially biologically relevant change following treatment with HDAC inhibitors were defined as those with a p value of < 0.05, the majority of patients having a > 20% change, difference in medians > 0.05% of PBMCs, and a frequency >0.01% of total PBMCs.
Antibodies
Avelumab, matching IgG1 isotype control, and bintrafusp alpha (M7824) (MSB0011359C), a fully human bifunctional molecule composed of the extracellular domain of human TGFβRII (TGFβ Trap) linked to the C-terminus of anti-PD-L1 heavy chain, were obtained from EMD Serono (Rockland, MA) as part of a Cooperative Research and Development Agreement with the NCI, NIH. Anti-human CD16-neutralizing mAb was obtained from eBioscience (San Diego, CA, USA).
Chemicals and drug exposure
Vorinostat was obtained from Selleck Chemicals (Houston, TX, USA). Entinostat was obtained from Syndax Pharmaceuticals, as part of a Cooperative Research and Development Agreement with the NCI, NIH. Adherent tumor cells in log-growth phase were exposed daily for 5 hours to vehicle control (DMSO) or vorinostat (3 µM) for 4 consecutive days. At the end of each treatment, cells were washed in fresh medium and returned to incubation at 37°C/5% CO2. Alternatively, cells were continuously exposed for 72 hours to DMSO or entinostat (500 nM). Matrigel was obtained from Invitrogen (Carlsbad, CA, USA). For selected experiments, purified NK cells from healthy donors or PBMCs from metastatic breast cancer patients were exposed to DMSO or entinostat (500 nM) for 24 or 48 hours and then washed prior to being used as effectors or examined by flow cytometry, respectively.
Analysis of cell viability
After drug exposure as described above, cells were harvested and viable cells were counted by trypan blue exclusion using a Cellometer Auto T4 automated cell counter (Nexcelom Bioscience, Lawrence, MA, USA). Cell viability was confirmed by flow cytometry using LIVE/DEAD® Fixable Dead Cell Stain Kits (Life Technologies, Carlsbad, CA, USA).
NK lysis in vitro assay
The lysis of tumor targets in the absence or presence of avelumab was examined using a standard 4 hours or overnight (15 hours) 111In-release assay, as previously described.Citation34 Human tumor cells incubated with DMSO, vorinostat or entinostat as previously described were washed in fresh media before being used as targets. Purified human NK cells that were either untreated or treated with DMSO or entinostat and then washed were used as effectors at various effector to target ratios depicted in the figures and figure legends. Viability of NK cells with entinostat treatment was decreased by 5% relative to DMSO. Avelumab and isotype control antibodies were used at a concentration of 2 ng/mL. M7824 was used at the equimolar concentration of 2.46 ng/mL. Where applicable, purified NK cells were incubated at 37°C for 2 hours with CD16-blocking antibody (12 µg/mL) prior to being used as effectors.
Flow cytometry
Analysis of cell-surface proteins in 3 × 104 tumor cells was performed on a FACSVerse™ flow cytometer (BD Biosciences, San Jose, CA, USA) as previously described,Citation34 using the primary labeled mAbs HLA-ABC [G46-2.6], CD274 (PD-L1) [MIH1], MICA/B [6D(24)4], and the appropriate isotype-matched controls (BD Biosciences). Analysis of purified healthy donor NK cells was performed using LIVE/DEAD Fixable Blue Dead Cell Stain Kit (ThermoFisher, Grand Island, NY, USA) prior to labeling with the primary mAbs CD3-FITC [HIT3a], NKp30-PE [P30-15], CD226-PE-Cy7 [11A8], CD56-BV605 [HCD56], CD16-AF700 [3G8], NKG2D-APC-Cy7 [1D11], Ki67-PerCP-Cy5.5 [Ki-67], CD158-PE-Cy7 [HP-MA4], Granzyme B-BV421 [gb11], NKp44-PE [P44-8], NKp46-PE-Cy7 [9E2], Perforin-APC [dG9], CD107a-APC-Cy7 [H4A3], and PD-1-PE [EH12.2H7]. Data were collected from 1 × 105 cells on a BD LSRFortessa Flow Cytometer and analyzed using FlowJo software (V9.7.6 or V9.9.5 for MAC; Treestar Inc., Ashland, OR, USA).
In vivo studies
Female nu/nu mice were implanted with NCI-H460, DU145, or PC-3 carcinoma cells (5 × 106, s.c., 1:1 in Matrigel). When tumors reached 0.5–1 cm3, animals received four daily doses of DMSO or vorinostat (150 mg/kg, p.o.) prior to sacrifice. Alternatively, animals received a single dose of entinostat (20 mg/kg, p.o.) or DMSO 72 hours prior to sacrifice. Tumors were harvested, embedded in OCT, and frozen immediately in a liquid nitrogen vapor phase. Six- to 10-week old female nu/nu mice were obtained from the National Cancer Institute’s Frederick Cancer Research Facility, Frederick, MD, and were maintained in microisolator cages under specific pathogen-free conditions in accordance with the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) guidelines. All experimental studies were carried out under approval of the NIH Intramural Animal Care and Use Committee.
Immunofluorescence
Frozen tumor sections (10 μm) were incubated for 10 min at–20°C with ice-cold methanol (EMD Millipore, Billerica, MA, USA) and washed three times with PBS. Sections were incubated at room temperature for 1 hour with PBS containing 3% BSA/0.1% Tween 20, followed by a 1-hour incubation with 10% normal goat serum in PBS. Sections were then incubated with antihuman PD-L1 mAb (SP142, Spring Bioscience, Pleasanton, CA, USA) diluted at 1:100 in PBS containing 1% BSA/0.1% Tween 20 overnight at 4°C. After extensive washing in PBS, sections were incubated with goat anti-rabbit AF594 (Molecular Probes, Eugene, OR, USA; 1:750 dilution) for 45 minutes at room temperature. Sections were extensively washed with PBS and mounted with DAPI-containing Vectashield mounting medium (Vector Laboratories, Inc., Burlingame, CA, USA) in the dark. Images were acquired on an LSM 780 confocal microscope (Carl Zeiss Microimaging, Thornwood, NY, USA), and confocal data were analyzed using Zen 2012 SP1 software (black edition). Relative PD-L1 expression levels were calculated using ImageJ software by normalizing the intensity values to their respective DMSO-treated controls.
Statistical analysis
Significant differences in the distribution of cell populations by flow cytometry analyses were examined using the Kolmogorov-Smirnov test and considered biologically significant if they differed by ≥ 25% relative to respective controls. Significant differences between two treatment groups were determined by a 2-tailed t test, and differences between multiple treatment groups were examined by 2-way ANOVA, using GraphPad Prism 7.0 software. Differences were considered significant when the p value was < 0.05. For the analysis of immune subsets in peripheral blood, p values were determined using the predefined Wilcoxon matched-pairs signed rank test, and given the exploratory nature of the study, are presented without adjustment for multiple comparisons.
Authors’ Contributions
Conception and design: K. Hicks, M. Fantini, S. Gameiro, K. Tsang, J. Hodge, J. Schlom
Development of methodology: K. Hicks, M. Fantini, S. Gameiro, K. Tsang, R. Donahue, C. Allen, P. Cavijo, J. Hodge
Acquisition of data: K. Hicks, S. Gameiro, M. Fantini, K. Knudson, A. Schwab, S. Tritsch, P. Cavijo, C. Allen, J. Hodge
Analysis and interpretation of data: K. Hicks, M. Fantini, S. Gameiro, K. Knudson, P. Cavijo, A. Schwab, C. Jochems, R. Donahue, J. Hodge, J. Schlom
Writing/review of manuscript: K. Hicks, M. Fantini, S. Gameiro, K. Knudson, C. Jochems, R. Donahue, P. Cavijo, K. Tsang, J. Hodge, J. Schlom
Administrative, technical, or material support: J. Schlom
Study supervision: J. Hodge, J. Schlom
Disclosure of Potential Conflicts of Interest
The authors have no potential conflicts of interest to disclose.
2018ONCOIMM0227R-s02.pdf
Download PDF (604.1 KB)Acknowledgments
The authors thank Curtis Randolph for his excellent technical assistance, and Debra Weingarten for her excellent editorial assistance in the preparation of this manuscript.
Supplemental material
Supplemental data for this article can be accessed online at http://dx.doi.org/10.1080/2162402X.2018.1466018.
Additional information
Funding
References
- Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366(26):2455–2465. doi:10.1056/NEJMoa1200694.
- Ansell SM, Lesokhin AM, Borrello I, Halwani A, Scott EC, Gutierrez M, Schuster SJ, Millenson MM, Cattry D, Freeman GJ, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N Engl J Med. 2015;372(4):311–319. doi:10.1056/NEJMoa1411087.
- Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, Sosman JA, McDermott DF, Powderly JD, Gettinger SN, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515(7528):563–567. doi:10.1038/nature14011.
- Powles T, Eder JP, Fine GD, Braiteh FS, Loriot Y, Cruz C, Bellmunt J, Burris HA, Petrylak DP, Teng SL, et al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature. 2014;515(7528):558–562. doi:10.1038/nature13904.
- Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, Segal NH, Ariyan CE, Gordon RA, Reed K, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369(2):122–133. doi:10.1056/NEJMoa1302369.
- Martin AM, Nirschl TR, Nirschl CJ, Francica BJ, Kochel CM, van Bokhoven A, Meeker AK, Lucia MS, Anders RA, DeMarzo AM, et al. Paucity of PD-L1 expression in prostate cancer: innate and adaptive immune resistance. Prostate Cancer Prostatic Dis. 2015;18(4):325–332. doi:10.1038/pcan.2015.39.
- Zou W, Wolchok JD, Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: mechanisms, response biomarkers, and combinations. Sci Transl Med. 2016;8(328):328rv4–328rv4. doi:10.1126/scitranslmed.aad7118.
- Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, Torrejon DY, Abril-Rodriguez G, Sandoval S, Barthly L, et al. Mutations associated with acquired resistance to PD-1 blockade in Melanoma. N Engl J Med. 2016;375(9):819–829. doi:10.1056/NEJMoa1604958.
- Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, Chen T, Roszik J, Bernatchez C, Woodman SE, et al. Loss of IFN-gamma pathway genes in tumor cells as a mechanism of resistance to anti-CTLA-4 therapy. Cell. 2016;167(2):397–404 e399. doi:10.1016/j.cell.2016.08.069.
- Gettinger S, Choi J, Hastings K, Truini A, Datar I, Sowell R, Wurtz A, Dong W, Cai G, Melnick MA, et al. Impaired HLA class I antigen processing and presentation as a mechanism of acquired resistance to immune checkpoint inhibitors in lung cancer. Cancer Discov. 2017;7(12):1420–1435. doi:10.1158/2159-8290.CD-17-0593.
- Patel SJ, Sanjana NE, Kishton RJ, Eidizadeh A, Vodnala SK, Cam M, Gartner JJ, Jia L, Steinberg SM, Yamamoto TN, et al. Identification of essential genes for cancer immunotherapy. Nature. 2017;548(7669):537–542. doi:10.1038/nature23477.
- Hellebrekers DM, Castermans K, Vire E, Dings RP, Hoebers NT, Mayo KH, Oude Egbrink MG, Molema G, Fuks F, Van Engeland M, et al. Epigenetic regulation of tumor endothelial cell anergy: silencing of intercellular adhesion molecule-1 by histone modifications. Cancer Res. 2006;66(22):10770–10777. doi:10.1158/0008-5472.CAN-06-1609.
- Topper MJ, Vaz M, Chiappinelli KB, DeStefano Shields CE, Niknafs N, Yen RC, Wenzel A, Hicks J, Ballew M, Stone M, et al. Epigenetic therapy ties MYC depletion to reversing immune evasion and treating lung cancer. Cell. 2017;171(6):1284–1300 e1221. doi:10.1016/j.cell.2017.10.022.
- Licciardi PV, Karagiannis TC. Regulation of immune responses by histone deacetylase inhibitors. ISRN Hematol. 2012;2012:690901. doi:10.5402/2012/690901.
- Villagra A, Sotomayor EM, Seto E. Histone deacetylases and the immunological network: implications in cancer and inflammation. Oncogene. 2010;29(2):157–173. doi:10.1038/onc.2009.334.
- Azad N, Zahnow CA, Rudin CM, Baylin SB. The future of epigenetic therapy in solid tumours–lessons from the past. Nat Rev Clin Oncol. 2013;10(5):256–266. doi:10.1038/nrclinonc.2013.42.
- Juo YY, Gong XJ, Mishra A, Cui X, Baylin SB, Azad NS, Ahuja N. Epigenetic therapy for solid tumors: from bench science to clinical trials. Epigenomics. 2015;7(2):215–235. doi:10.2217/epi.14.73.
- Thurn KT, Thomas S, Moore A, Munster PN. Rational therapeutic combinations with histone deacetylase inhibitors for the treatment of cancer. Future Oncol. 2011;7(2):263–283. doi:10.2217/fon.11.2.
- West AC, Christiansen AJ, Smyth MJ, Johnstone RW. The combination of histone deacetylase inhibitors with immune-stimulating antibodies has potent anti-cancer effects. Oncoimmunology. 2012;1(3):377–379. doi:10.4161/onci.18804.
- Kim K, Skora AD, Li Z, Liu Q, Tam AJ, Blosser RL, Diaz LA Jr., Papadopoulos N, Kinzler KW, Vogelstein B, et al. Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. Proc Natl Acad Sci U S A. 2014;111(32):11774–11779. doi:10.1073/pnas.1410626111.
- Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol. 2008;9(5):503–510. doi:10.1038/ni1582.
- Kiany S, Huang G, Kleinerman ES. Effect of entinostat on NK cell-mediated cytotoxicity against osteosarcoma cells and osteosarcoma lung metastasis. Oncoimmunology. 2017;6(8):e1333214. doi:10.1080/2162402X.2017.1333214.
- Ritter C, Fan K, Paschen A, Reker Hardrup S, Ferrone S, Nghiem P, Ugurel S, Schrama D, Becker JC. Epigenetic priming restores the HLA class-I antigen processing machinery expression in Merkel cell carcinoma. Sci Rep. 2017;7(1):2290. doi:10.1038/s41598-017-02608-0.
- Woods DM, Sodre AL, Villagra A, Sarnaik A, Sotomayor EM, Weber J. HDAC inhibition upregulates PD-1 ligands in melanoma and augments immunotherapy with PD-1 blockade. Cancer Immunol Res. 2015;3(12):1375–1385. doi:10.1158/2326-6066.CIR-15-0077-T.
- Raulet DH, Marcus A, Coscoy L. Dysregulated cellular functions and cell stress pathways provide critical cues for activating and targeting natural killer cells to transformed and infected cells. Immunol Rev. 2017;280(1):93–101. doi:10.1111/imr.2017.280.issue-1.
- Zhu S, Denman CJ, Cobanoglu ZS, Kiany S, Lau CC, Gottschalk SM, Hughes DP, Kleinerman ES, Lee DA. The narrow-spectrum HDAC inhibitor entinostat enhances NKG2D expression without NK cell toxicity, leading to enhanced recognition of cancer cells. Pharm Res. 2015;32(3):779–792. doi:10.1007/s11095-013-1231-0.
- Schenk A, Bloch W, Zimmer P. Natural killer cells–an epigenetic perspective of development and regulation. Int J Mol Sci. 2016;17(3):326. doi:10.3390/ijms17030326.
- Ogbomo H, Michaelis M, Kreuter J, Doerr HW, Cinatl J Jr. Histone deacetylase inhibitors suppress natural killer cell cytolytic activity. FEBS Lett. 2007;581(7):1317–1322. doi:10.1016/j.febslet.2007.02.045.
- Ni L, Wang L, Yao C, Ni Z, Liu F, Gong C, Zhu X, Yan X, Watowich SS, Lee DA, et al. The histone deacetylase inhibitor valproic acid inhibits NKG2D expression in natural killer cells through suppression of STAT3 and HDAC3. Sci Rep. 2017;7:45266. doi:10.1038/srep45266.
- Kim JS, Shin BR, Lee HK, Lee JH, Kim KH, Choi JE, Ji AY, Hong JT, Kim Y, Han SB. Cd226(-/-) natural killer cells fail to establish stable contacts with cancer cells and show impaired control of tumor metastasis in vivo. Oncoimmunology. 2017;6(8):e1338994. doi:10.1080/2162402X.2017.1338994.
- Iannello A, Thompson TW, Ardolino M, Marcus A, Raulet DH. Immunosurveillance and immunotherapy of tumors by innate immune cells. Curr Opin Immunol. 2016;38:52–58. doi:10.1016/j.coi.2015.11.001.
- Urlaub D, Hofer K, Muller ML, Watzl C. LFA-1 activation in NK cells and their subsets: influence of receptors, maturation, and cytokine stimulation. J Immunol. 2017;198(5):1944–1951. doi:10.4049/jimmunol.1601004.
- Long EO, Kim HS, Liu D, Peterson ME, Rajagopalan S. Controlling natural killer cell responses: integration of signals for activation and inhibition. Annu Rev Immunol. 2013;31:227–258. doi:10.1146/annurev-immunol-020711-075005.
- Boyerinas B, Jochems C, Fantini M, Heery CR, Gulley JL, Tsang KY, Schlom J. Antibody-dependent cellular cytotoxicity activity of a novel anti-PD-L1 antibody avelumab (MSB0010718C) on human tumor cells. Cancer Immunol Res. 2015;3(10):1148–1157. doi:10.1158/2326-6066.CIR-15-0059.
- Guillerey C, Huntington ND, Smyth MJ. Targeting natural killer cells in cancer immunotherapy. Nat Immunol. 2016;17(9):1025–1036. doi:10.1038/ni.3518.
- Junttila TT, Parsons K, Olsson C, Lu Y, Xin Y, Theriault J, Crocker L, Pabonan O, Baginski T, Meng G, et al. Superior in vivo efficacy of afucosylated trastuzumab in the treatment of HER2-amplified breast cancer. Cancer Res. 2010;70(11):4481–4489. doi:10.1158/0008-5472.CAN-09-3704.
- Wild J, Schmiedel BJ, Maurer A, Raab S, Prokop L, Stevanovic S, Dorfel D, Schneider P, Salih HR. Neutralization of (NK-cell-derived) B-cell activating factor by Belimumab restores sensitivity of chronic lymphoid leukemia cells to direct and Rituximab-induced NK lysis. Leukemia. 2015;29(8):1676–1683. doi:10.1038/leu.2015.50.
- Lo Nigro C, Ricci V, Vivenza D, Monteverde M, Strola G, Lucio F, Tonissi F, Miraglio E, Granetto C, Fortunato M, et al. Evaluation of antibody-dependent cell-mediated cytotoxicity activity and cetuximab response in KRAS wild-type metastatic colorectal cancer patients. World J Gastrointest Oncol. 2016;8(2):222–230. doi:10.4251/wjgo.v8.i2.222.
- Fujii R, Friedman ER, Richards J, Tsang KY, Heery CR, Schlom J, Hodge JW. Enhanced killing of chordoma cells by antibody-dependent cell-mediated cytotoxicity employing the novel anti-PD-L1 antibody avelumab. Oncotarget. 2016;7(23):33498–33511. doi:10.18632/oncotarget.v7i23.
- Gameiro SR, Malamas AS, Tsang KY, Ferrone S, Hodge JW. Inhibitors of histone deacetylase 1 reverse the immune evasion phenotype to enhance T-cell mediated lysis of prostate and breast carcinoma cells. Oncotarget. 2016;7(7):7390–7402. doi:10.18632/oncotarget.v7i7.
- West AC, Johnstone RW. New and emerging HDAC inhibitors for cancer treatment. J Clin Invest. 2014;124(1):30–39. doi:10.1172/JCI69738.
- Knipstein J, Gore L. Entinostat for treatment of solid tumors and hematologic malignancies. Expert Opin Investig Drugs. 2011;20(10):1455–1467. doi:10.1517/13543784.2011.613822.
- Pili R, Salumbides B, Zhao M, Altiok S, Qian D, Zwiebel J, Carducci MA, Rudek MA. Phase I study of the histone deacetylase inhibitor entinostat in combination with 13-cis retinoic acid in patients with solid tumours. Br J Cancer. 2012;106(1):77–84. doi:10.1038/bjc.2011.527.
- Iwamoto M, Friedman EJ, Sandhu P, Agrawal NG, Rubin EH, Wagner JA. Clinical pharmacology profile of vorinostat, a histone deacetylase inhibitor. Cancer Chemother Pharmacol. 2013;72(3):493–508. doi:10.1007/s00280-013-2220-z.
- Subramanian S, Bates SE, Wright JJ, Espinoza-Delgado I, Piekarz RL. Clinical toxicities of histone deacetylase inhibitors. Pharmaceuticals (Basel). 2010;3(9):2751–2767. doi:10.3390/ph3092751.
- Smyth MJ, Cretney E, Kelly JM, Westwood JA, Street SE, Yagita H, Takeda K, van Dommelen SL, Degli-Esposti MA, Hayakawa Y. Activation of NK cell cytotoxicity. Mol Immunol. 2005;42(4):501–510. doi:10.1016/j.molimm.2004.07.034.
- Zamai L, Ahmad M, Bennett IM, Azzoni L, Alnemri ES, Perussia B. Natural killer (NK) cell-mediated cytotoxicity: differential use of TRAIL and Fas ligand by immature and mature primary human NK cells. J Exp Med. 1998;188(12):2375–2380. doi:10.1084/jem.188.12.2375.
- Jochems C, Tritsch SR, Pellom ST, Su Z, Soon-Shiong P, Wong HC, Gulley JL, Schlom J. Analyses of functions of an anti-PD-L1/TGFbetaR2 bispecific fusion protein (M7824). Oncotarget. 2017;8(43):75217–75231. doi:10.18632/oncotarget.20680.
- Mamessier E, Sylvain A, Thibult ML, Houvenaeghel G, Jacquemier J, Castellano R, Goncalves A, Andre P, Romagne F, Thibault G, et al. Human breast cancer cells enhance self tolerance by promoting evasion from NK cell antitumor immunity. J Clin Invest. 2011;121(9):3609–3622. doi:10.1172/JCI45816.
- Garcia-Iglesias T, Del Toro-Arreola A, Albarran-Somoza B, Del Toro-Arreola S, Sanchez-Hernandez PE, Ramirez-Duenas MG, Balderas-Pena LM, Bravo-Cuellar A, Ortiz-Lazareno PC, Daneri-Navarro A. Low NKp30, NKp46 and NKG2D expression and reduced cytotoxic activity on NK cells in cervical cancer and precursor lesions. BMC Cancer. 2009;9:186. doi:10.1186/1471-2407-9-186.
- Lopez-Cobo S, Pieper N, Campos-Silva C, Garcia-Cuesta EM, Reyburn HT, Paschen A, Vales-Gomez M. Impaired NK cell recognition of vemurafenib-treated melanoma cells is overcome by simultaneous application of histone deacetylase inhibitors. Oncoimmunology. 2018;7(2):e1392426. doi:10.1080/2162402X.2017.1392426.
- Berghuis D, Schilham MW, Vos HI, Santos SJ, Kloess S, Buddingh EP, Egeler RM, Hogendoorn PC, Lankester AC. Histone deacetylase inhibitors enhance expression of NKG2D ligands in Ewing sarcoma and sensitize for natural killer cell-mediated cytolysis. Clin Sarcoma Res. 2012;2(1):8. doi:10.1186/2045-3329-2-8.
- Schmudde M, Braun A, Pende D, Sonnemann J, Klier U, Beck JF, Moretta L, Broker BM. Histone deacetylase inhibitors sensitize tumour cells for cytotoxic effects of natural killer cells. Cancer Lett. 2008;272(1):110–121. doi:10.1016/j.canlet.2008.06.027.
- Armeanu S, Bitzer M, Lauer UM, Venturelli S, Pathil A, Krusch M, Kaiser S, Jobst J, Smirnow I, Wagner A, et al. Natural killer cell-mediated lysis of hepatoma cells via specific induction of NKG2D ligands by the histone deacetylase inhibitor sodium valproate. Cancer Res. 2005;65(14):6321–6329. doi:10.1158/0008-5472.CAN-04-4252.
- Pili R, Quinn DI, Hammers HJ, Monk P, George S, Dorff TB, Olencki T, Shen L, Orillion A, Lamonica D, et al. Immunomodulation by entinostat in renal cell carcinoma patients receiving high-dose Interleukin 2: a multicenter, single-arm, phase I/II trial (NCI-CTEP#7870). Clin Cancer Res. 2017;23(23):7199–7208. doi:10.1158/1078-0432.CCR-17-1178.
- Hadad U, Thauland TJ, Martinez OM, Butte MJ, Porgador A, Krams SM. NKp46 clusters at the immune synapse and regulates NK cell polarization. Front Immunol. 2015;6:495. doi:10.3389/fimmu.2015.00495.
- Mandelboim O, Lieberman N, Lev M, Paul L, Arnon TI, Bushkin Y, Davis DM, Strominger JL, Yewdell JW, Porgador A. Recognition of haemagglutinins on virus-infected cells by NKp46 activates lysis by human NK cells. Nature. 2001;409(6823):1055–1060. doi:10.1038/35059110.
- Koch J, Steinle A, Watzl C, Mandelboim O. Activating natural cytotoxicity receptors of natural killer cells in cancer and infection. Trends Immunol. 2013;34(4):182–191. doi:10.1016/j.it.2013.01.003.
- Wagner JA, Rosario M, Romee R, Berrien-Elliott MM, Schneider SE, Leong JW, Sullivan RP, Jewell BA, Becker-Hapak M, Schappe T, et al. CD56bright NK cells exhibit potent antitumor responses following IL-15 priming. J Clin Invest. 2017;127(11):4042–4058. doi:10.1172/JCI90387.
- Kim PS, Kwilas AR, Xu W, Alter S, Jeng EK, Wong HC, Schlom J, Hodge JW. IL-15 superagonist/IL-15RalphaSushi-Fc fusion complex (IL-15SA/IL-15RalphaSu-Fc; ALT-803) markedly enhances specific subpopulations of NK and memory CD8+ T cells, and mediates potent anti-tumor activity against murine breast and colon carcinomas. Oncotarget. 2016;7(13):16130–16145.
- Fife BT, Pauken KE, Eagar TN, Obu T, Wu J, Tang Q, Azuma M, Krummel MF, Bluestone JA. Interactions between PD-1 and PD-L1 promote tolerance by blocking the TCR-induced stop signal. Nat Immunol. 2009;10(11):1185–1192. doi:10.1038/ni.1790.
- Topalian SL, Drake CG, Pardoll DM. Targeting the PD-1/B7-H1(PD-L1) pathway to activate anti-tumor immunity. Curr Opin Immunol. 2012;24(2):207–212. doi:10.1016/j.coi.2011.12.009.
- Knudson KM, Hicks KC, Luo X, Chen J-Q, Schlom J, Gameiro SR. M7824, a novel bifunctional anti-PD-L1/TGFβ Trap fusion protein, promotes anti-tumor efficacy as monotherapy and in combination with vaccine. OncoImmunology. 2018;7(5):e1426519. doi:10.1080/2162402X.2018.1426519.
- Kortenhorst MS, Wissing MD, Rodriguez R, Kachhap SK, Jans JJ, Van der Groep P, Verheul HM, Gupta A, Aiyetan PO, van der Wall E, et al. Analysis of the genomic response of human prostate cancer cells to histone deacetylase inhibitors. Epigenetics. 2013;8(9):907–920. doi:10.4161/epi.25574.
- Zheng H, Zhao W, Yan C, Watson CC, Massengill M, Xie M, Massengill C, Noyes DR, Martinez GV, Afzal R, et al. HDAC inhibitors enhance T-cell chemokine expression and augment response to PD-1 immunotherapy in lung adenocarcinoma. Clin Cancer Res. 2016;22(16):4119–4132. doi:10.1158/1078-0432.CCR-15-2584.
- Johnson ML, Gonzalez R, Opyrchal M, Gabrilovich D, Ordentlich P, Brouwer S, Sankoh S, Schmidt EV, Meyers ML, Agarwala SS. ENCORE 601: A phase II study of entinostat (ENT) in combination with pembrolizumab (PEMBRO) in patients with melanoma. J Clin Oncol. 2017;35(15_suppl):9529.
- Heery CR, Ibrahim NK, Arlen PM, Mohebtash M, Murray JL, Koenig K, Madan RA, McMahon S, Marte JL, Steinberg SM, et al. Docetaxel alone or in combination with a therapeutic cancer vaccine (PANVAC) in patients with metastatic breast cancer: a randomized clinical trial. JAMA Oncol. 2015;1(8):1087–1095. doi:10.1001/jamaoncol.2015.2736.
- Donahue RN, Lepone LM, Grenga I, Jochems C, Fantini M, Madan RA, Heery CR, Gulley JL, Schlom J. Analyses of the peripheral immunome following multiple administrations of avelumab, a human IgG1 anti-PD-L1 monoclonal antibody. J Immunother Cancer. 2017;5:20. doi:10.1186/s40425-017-0220-y.