
ABSTRACT
The immunogenic cell death (ICD) process represents a novel therapeutic approach to treat tumours, in which cytotoxic compounds promote both cancer cell death and the emission of damage-associated molecular patterns (DAMPs) from dying cells, to activate the immune system against the malignancy. Therefore, we explored the possibility to stimulate the key molecular players with a pivotal role in the execution of the ICD program in melanoma cells. To this aim, we used the pro-ICD agents mitoxantrone and doxorubicin and found that both agents could induce cell death and stimulate the release/exposure of the strictly required DAMPs in melanoma cells: i) calreticulin (CRT) exposure on the cell membrane; ii) ATP secretion; iii) type I IFNs gene up-regulation and iv) HMGB1 secretion, highlighting no interference by oncogenic BRAF. Importantly, although the ER stress-related PERK activation has been linked to CRT externalization, through the phosphorylation of eIF2α, we found that this stress pathway together with PERK were not involved in melanoma cells. Notably, we identified PKR and GCN2 as key mediators of eIF2α phosphorylation, facilitating the translocation of CTR on melanoma cells surface, under pro-ICD drugs stimulation. Therefore, our data indicate that pro-ICD drugs are able to stimulate the production/release of DAMPs in melanoma cells at least in vitro, indicating in this approach a potential new valuable therapeutic strategy to treat human skin melanoma malignancy.
Introduction
Human skin melanoma represents one of the most aggressive and difficult to treat form of human cancer, representing a public health issue in many countries due to its increasing incidence together with the absence of an effective therapy.Citation1 Surgical removal of primary tumours represents the main clinical intervention, particularly effective in early stages of malignancy but remains ineffective in advanced ones with consequent adverse prognosis due to the ability of tumours to invade healthy tissues. Prevention and early detection are therefore the most effective counteracting strategies that must be actively increased.Citation2 Although sun exposure represents the main cause of melanomagenesis,Citation3 several gene mutations have been associated to melanocyte transformation and tumour development, with the RAS/RAF/MEK/ERK pathway being the most affected.Citation3,Citation4 BRAFV600E is indeed associated to 40–60% of all cutaneous melanomas, responsible for uncontrolled cell proliferation, defective cell death pathways and resistance to any treatment.Citation5,Citation6 However, oncogenic BRAF alone is not sufficient to drive tumour development since this mutation is also present in some benign nevi.Citation7 Therefore, specific secondary events are required to drive melanoma development/progression and, in this contest, both ER stress and autophagy deregulation have been recently reported to take part to melanomagenesis.Citation8-Citation13
The role of adaptive ER stress in melanoma development, progression and resistance to therapy has been well established in recent years, since it has been found activated in melanoma cells responding to environmental stress,Citation13 identifying this compartment as a potential therapeutic target.Citation14,Citation15 Although autophagy and ER stress seem to act independently, these two finely regulated and evolutionary-conserved cellular processes are closely interconnected to each other with ER stress able to stimulate autophagy, and the latter able to inhibit the UPR by removing excess of ER membranes.Citation16,Citation17 More importantly, it has been recently identified a direct link existing between ER stress and autophagy in oncogenic BRAF melanoma cells, driving cell proliferation and resistance to pro-apoptotic stimuli.Citation18,Citation19 In fact, mutated BRAF results in a p38-mediated induction of chronic ER stress status resulting in downstream enhancement of basal autophagy, through the IRE1/ASK/JNK and the PERK/ATF4/TRB3 pathways, thus sustaining cell survival and inhibiting cell death.Citation18
Due to the pivotal role played by oncogenic BRAF in tumour progression and resistance to death, an effort has been made in recent years to identify specific inhibitors of the kinase, to reduce cell proliferation, tumour progression and to re-sensitize this malignancy to pro-cell death treatment. However, although specific and active BRAF kinase inhibitors, vemurafenib and dabrafenib, have been ultimately developed, these therapeutic approaches irremediably failed.Citation20 In fact, patients soon developed resistance to therapy (4÷6 months) due to intrinsic or acquired resistance, in an ERK -dependent and -independent manner, to circumvent the inhibition of BRAF pathway and to sustain cell survival.Citation20 Therefore, new effective treatments are urgently needed to cope with the complexity of cutaneous melanoma.
Recently, an alternative and interesting new therapeutic approach has been developed, consisting in combined ability of some compounds to both stimulate the apoptotic program of cancer cells and concomitant recruitment and activation of immune system resulting in a better eradication of the malignancy.Citation21,Citation22 This process known as immunogenic cell death (ICD) relies on the ability of these drugs to stimulate dying cancer cells to produce/release specific signals, namely DAMPs (damage-associated molecular pattern), to efficiently stimulate the recruitment and maturation of dendritic cells (DC), that will in turn activate T cells to kill residual cancer cells.Citation21-Citation24 This new strategy provides two potential benefits: attack the tumour on two fronts and reduce i) the concentration of cytotoxic compounds used in therapy and/or ii) the length of treatment.
To date, few DAMPs have been identifiedCitation24 showing the ability to be recognised by the immune system, but four of them are strictly required for an efficient induction of ICD: i) the exposure of ER-resident calreticulin (CRT) on cell surface, (ii) the extracellular release of ATP and (iii) the non-histone chromatin-binding protein high-mobility group box 1 (HMGB1), and (iv) the production and release of type I interferons (IFNα & β).Citation23
In the present study, we evaluated the impact of oncogenic BRAF on DAMPs production and verified the role of ER stress in CRT exposure under pro-ICD drugs treatment, elucidating part of the molecular mechanisms underlying this process. In fact, we found that PKR and GCN2 are both involved, although with different extent, in the ER stress-independent phosphorylation of eIF2α, required for CRT translocation.
Results
Pro-ICD drugs induce cell death in melanoma cells
Several compounds have cytotoxic effects in cancer cells although just few of them have the capability of causing an immunogenic cell death response, stimulating cancer cells death and the release/exposure of DAMPs at the same time.Citation25 Hence, we decided to use the two well characterized pro-ICD drugs mitoxantrone (MTX) and doxorubicin (DOXO), an anthracenedione and an anthracycline, respectively, that have been used for many years to treat different malignancies and that mainly interfere with the structure of the DNA and with the function of DNA topoisomerase II, triggering the apoptotic program.Citation26
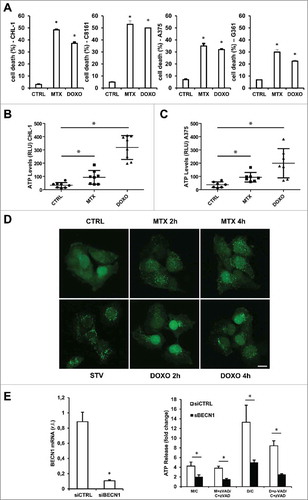
Therefore, we firstly verified the ability of these ICD-inducers to stimulate a cell death response in melanoma cells, evaluating the % of sub-G1 cells of two oncogenic (A375 & A2058) and two wild-type (CHL-1 & C8161) BRAF cell lines treated with MTX (1 µM) or DOXO (5 µM) for 24 h. Results reported in show that both drugs efficiently induced cell death in both wild-type and oncogenic BRAF cell lines although with different rates, in accordance with the known higher resistance of oncogenic BRAF cells to death stimuli.Citation18
Figure 1. Cell death induction & ATP release under MTX or DOXO treatment. Wild-type (CHL-1 & C8161) and oncogenic (A375 & G361) BRAF melanoma cell lines were treated with 1 μM MTX or 5 μM DOXO and cell death was evaluated at 24 h by flow cytometric analysis as percentage of sub-G1 population of PI-stained cells. CHL-1 (B) and A375 (C) cells were treated with 1 μM MTX or 5 μM DOXO (8 h) and ATP release into cell medium was evaluated by a luciferase-based assay. Data represent average and S.D. of 3 independent experiments. (D) Representative fluorescent micrograph from GFP-LC3 expressing CHL-1 melanoma cells treated with vehicle or 1 μM MTX or 5 μM DOXO for 2 or 4 h, and autophagic rate was evaluated by LC3-GFP puncta accumulation (in presence of 5 nM Bafilomycin A) by confocal microscopy; HBSS medium (STV) was used as positive control (bar = 10 μm). (E) Transient down-regulation of BECN1 in CHL-1 cells was evaluated by means of qRT-PCR (left panel); silenced cells were treated as in B in presence or absence of 10 μM Z-VAD-FMK and ATP was evaluated as described above (right panel, histograms reported represent the ratio of released ATP between treated and control samples in both control (siCTRL, white) and BECN1 silenced (siBECN1, black) cells (C = Control; M = MTX; D = DOXO). (n = 3; * = p < 0,05).
Analysis of DAMPs induction/release
As essential part of the ICD, the ability to induce DAMPs represents the second feature of pro-ICD drugs. In fact, once released/exposed by dying cancer cells in the extracellular space these signal molecules are recognized by the innate immune cells leading eventually to the adaptive immune response.Citation21-Citation24 Therefore, we analyzed the ability of MTX and DOXO to stimulate the production of the four required DAMPs, mentioned above.
ATP release
Stressed or dying cancer cells are able to release ATP in the extracellular space, when exposed to a few restricted molecules with chemotherapeutic activity.Citation27
In order to evaluate the release of ATP in our model, we treated wild-type (CHL-1) and mutated (A375) BRAF melanoma cell lines with MTX or DOXO (1 µM and 5 µM respectively, for 8 h) and extracellular ATP levels were measured in the cell culture medium. As shown in both drugs could stimulate the release of ATP by both melanoma cell lines, with no apparent interference by BRAF.
Although the precise molecular mechanism mediating the release of ATP is still unclear, processes such as autophagy and apoptosis might contribute.Citation28 Therefore, to evaluate the involvement of autophagy in our model, CHL-1 cells expressing a recombinant GFP-LC3 protein were treated with both drugs (MTX or DOXO) for 2 and 4 h, and cytoplasmic LC3-GFP puncta spots were analyzed by confocal microscopy.Citation29 This analysis evidenced the early appearance of LC3-GFP puncta in CHL-1 cells treated with both DOXO or MTX (), indicating the involvement of autophagy in ATP release also in melanoma cells. To further support this conclusion, we measured ATP release in CHL-1 cells in which the autophagic process was abrogated through the silencing of Beclin1 (, left panel). Data reported in (right panel) indicated that autophagy impairment strongly reduced the release of ATP stimulated by both MTX or DOXO, compared to control cells. Moreover, the presence of the caspase pan inhibitor z-VAD-FMK, in the same experimental conditions reported above, also indicated a potential involvement of active caspases in contributing to ATP release,Citation28 although with a minor impact compared to autophagy ().
HMGB1 release
In addition to its release during pathogen infection or injury,Citation30 HMGB1 is also released by dying tumor cells under anticancer treatment, contributing to the stimulation of an immune response.Citation31 Consequently, we treated our cell lines (CHL-1 and A375) with MTX or DOXO for 24 h and assessed the presence of this factor in the cell culture medium by western blotting analysis. Our data show that both agents stimulated the release of HMGB1 in tested melanoma cell lines ().
Figure 2. MTX or DOXO treatment favor HMGB1 release. (A) Wild-type (CHL-1) and oncogenic (A375) BRAF melanoma cell lines were treated with 1 μM MTX or 5 μM DOXO (24 h) and the presence of HMGB1 in growth medium was evaluated by means of western blotting analysis; cell lysate (CL) was used as positive control; (B) CHL-1 and A375 were treated 6 h as in A and IL-1β expression was evaluated by both western blot (THP-1 cells treated with LPS were used as positive control) and qRT-PCR; (C) CHL-1 (left) and A375 (right) were treated as in A for 8, 12 and 16 h and HMGB1 and PARP (full length, FL, and caspase-cleaved fragment, CL) were evaluated by western blotting; Gapdh was used as loading control in all reported experiments. (n = 3).
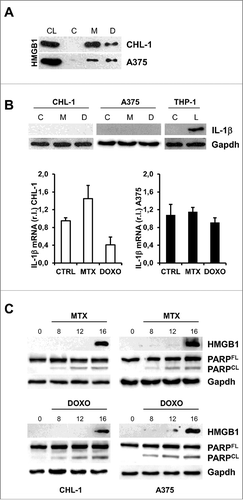
Since HMGB1 release can be a passive or active event, with the latter mediated by the inflammasome,Citation30 we assessed the expression of IL-1β, an hallmark of inflammasome activation,Citation32,Citation33 in cells treated with MTX or DOXO. Results reported in clearly indicate the absence IL-1β expression at both mRNA (bottom panels) and protein (upper panels) levels under MTX or DOXO treatment, in both A375 and CHL-1 cells (8 h). Therefore, we compared the release of HMGB1 from dying cells with the activation of caspases, in the same experimental conditions. To this aim, CHL-1 and A375 cells were treated with the indicated drugs for 8, 12 and 16 h and both HMGB1 release in the cell culture medium and PARP cleavage (active caspase marker) were evaluated by western blot analysis. As showed in , the appearance of cleaved PARP preceded the release of HMGB1, indicating that the protein is passively released by dying/dead cells. To further support this conclusion, CHL-1 and A375 cells were treated 12 or 16 h in presence or absence of the pan caspase inhibitor z-VAD and the release of HMGB1 was evaluated by western blotting analysis. As reported in Supplementary Fig. S1, the inhibition of caspase activity resulted in complete impairment of MTX- or DOXO- stimulated HMGB1 release (Suppl. S1).
Collectively these data suggest that both MTX and DOXO efficiently stimulate the release of HMGB1 in the extracellular compartment in both wild-type and mutated BRAF melanoma cells, through a passive mechanism.
CRT exposure on cell surface
CRT displays several non-endoplasmic functions in both physiological and pathological conditionsCitation34 and, importantly, the translocation of the protein on cancer cell surface is strictly correlated to the immunogenicity of dying cells during ICD execution.Citation36 As active part of UPR (unfolded protein response), CRT is also promptly up-regulated in response to both acute and chronic endoplasmic reticulum stress (ER stress), with the latter condition recently revealed to be established in BRAFV600E melanoma cells.Citation18,Citation19 Therefore, we sought whether this condition could influence both the basal and stimulated exposure of CRT on melanoma cell surface. To this aim, we first evaluated the basal expression of the two well established ER stress markers TRB3 and CHOP together with the expression of CRT, in both wild-type (CHL-1, C8161 and MeWo) and mutated (A375, A2058 and G361) BRAF melanoma cell lines. Results reported in confirm the enhanced expression of ER stress markers, comprising CRT, in oncogenic BRAF compared to wild-type cell lines but, however, the chronic ER stress status does not influence the basal exposure of CRT on melanoma cell surface, as showed by flow cytometric analysis ().
Figure 3. ER stress status and CRT exposure on cell surface after pro-ICD drugs treatment. (A) BRAF-mediated ER-stress was evaluated in both wild-type (CHL-1, C8161 and MeWo) and oncogenic (A375, A2058 and G361) BRAF melanoma cell lines by measuring the levels of the ER stress markers TRB3, CHOP and calreticulin (CRT) by qRT-PCR; (B) ecto-CRT basal level was evaluated in a panel of wild-type (CHL-1, C8161 and MeWo) and oncogenic (A375, A2058 and G361) BRAF melanoma cell lines by flow cytometry; (C) CRT exposure on cell surface was analyzed in CHL-1 and A375 cells under MTX (1 μM) or DOXO (5 μM) exposure (6 h) by means of flow cytometry; representative histograms are shown. (n = 3; * = p < 0,05).
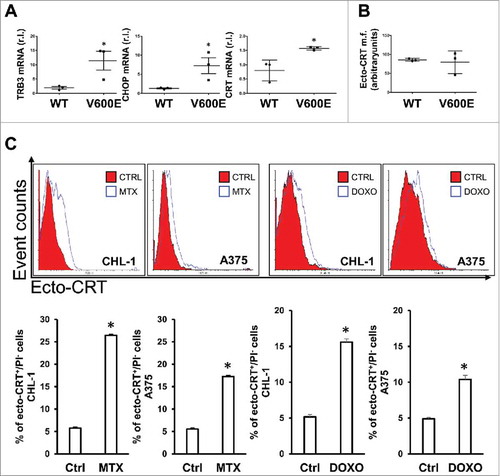
Next, in order to verify the ability of pro-ICD drugs to promote the translocation of CRT on melanoma cell surface, the early exposureCitation35 of this protein was evaluated in wild-type and oncogenic BRAF melanoma cell lines under MTX or DOXO treatment, by flow cytometric analysis. As reported in , CRT actively translocated onto cell surface under MTX or DOXO treatment, as an early event (6 h). Although the mechanism(s) underlying CRT translocation has not been fully elucidated, it has been previously reported that the process may require the presence of another ER-resident protein, ERp57.Citation36 To confirm this evidence, we down-regulated the expression of ERp57 in C8161 cells, by shRNA technology, and evaluated the exposure of CRT on cell surface after 6 h of MTX treatment. Results reported in Supplementary Fig. S2 clearly demonstrated that MTX-induced CRT translocation was completely abrogated in cells deprived of ERp57.
Collectively these data indicate that CRT is efficiently translocated onto melanoma cells under pro-ICD drug treatment, with no interference by oncogenic BRAF.
Type I IFNs expression
Although type I IFNs are commonly produced by cells in response to viral or bacterial infection to stimulate both innate and adaptive immunity,Citation37 it has been recently demonstrated that the production of these molecules is also related to ICD in vivo.Citation38 In order to verify the production of these cytokines in melanoma cells during pro-ICD drugs treatment, we treated wild-type (CHL-1 & C8161) or mutated (A375 & A2058) BRAF cell lines with MTX or DOXO and evaluated the expression of both IFN-α and IFN-β at mRNA level. shows that both drugs were able to stimulate the expression of type I IFNs in all tested cell lines, although with a different kinetic, possibly due to the sensitivity of each cell line to the treatment. This result is also supported by the observed downstream up-regulation of the type I IFNs target CXCL10 (chemokine (C-X-C motif) ligand 10),Citation38 in all tested cell lines (). However, the release of both cytokines was not detected in the cell culture media (), possibly due to their low rate of production, as evidenced in .
Figure 4. MTX- and DOXO- induced Type I IFN gene upregulation. IFN-α (A) and IFN-β (B) mRNA levels were quantified in CHL-1, C8161, A375 and G361 cell lines treated or untreated (8 h) with MTX (1 μM) or DOXO (5 μM) by qRT-PCR. (C) The downstream type I IFN target CXCL10 expression was evaluated in the same experimental conditions used in A&B, in the indicated cell lines, by qRT-PCR. (D) the release of both IFN-α and IFN-β was evaluated in the cell culture media in the indicated cells treated as in A and B. (n = 3; * = p < 0,05).
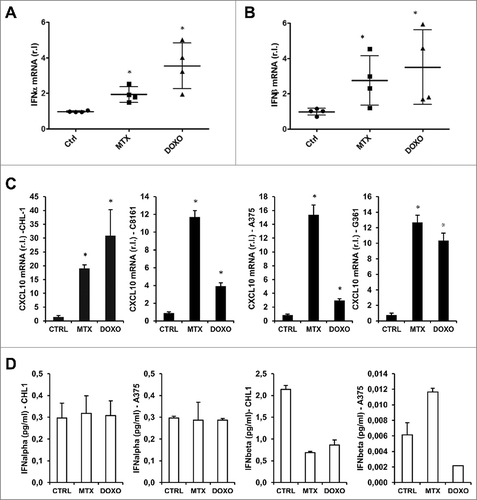
Importantly, the presence of oncogenic BRAF has no impact on IFNs expression under pro-ICD drugs treatment.
eIF2α phosphorylation occurs independently from ER stress upon pro-ICD drugs tretment in melanoma cells
Previous data indicate that few drugs, such as MTX and DOXO, induce an ER stress response during ICD execution,Citation26 with the ER stress-integrated PERK (protein kinase RNA (PKR)-like ER kinase) pathway activation playing a pivotal role in the translocation of CRT through the phosphorylation of eIF2α (eukaryotic initiation factor 2 alpha).Citation39 Interestingly, our data indicate an early enhanced phosphorylation of eIF2α in both CHL-1 and A375 cells exposed to MTX or DOXO (), thus confirming this phenomenon also in melanoma cells.Citation40 To verify the drug-mediated induction of an ER stress response, we treated our panel of wild-type and mutated BRAF melanoma cell lines with the pro-ICD drugs, in a time dependent manner. Surprisingly, our analysis indicated that none of the well-established ER stress markers TRB3, Xbp1 or CHOP was up-regulated upon ICD induction, in any of tested cell lines ( and ). Therefore, to further exclude the involvement of PERK in the eIF2α phosphorylation and CRT translocation, we directly evaluated the activation of PERK in both CHL-1 and A375 cells under MTX or DOXO treatment. As reported in , both compounds were unable to induce the phosphorylation of PERK, required for its activation, further confirming the absence of an ER stress induction. Three kinases, in addition to PERK, are known to phosphorylate eIF2α under stress conditions: HRI (Heme Regulated Initiation Factor 2 Alpha Kinase), GCN2 (general control non-repressed 2) and PKR (protein kinase RNA-activated; Suppl S3).Citation41 Therefore, we investigated their possible involvement in our model. We firstly abrogated the expression of HRI by infecting CHL-1 cells with lentiviral particles carrying an shRNA vector specific for HRI, using a scrambled sequence as control (, upper panel, and Suppl. S4 left panel). Then, cells were treated with MTX or DOXO (6 h) and eIF2α phosphorylation was evaluated. As evidenced by data reported in (bottom panel and Suppl. S4 right panel), the absence of HRI did not interfere with the MTX/DOXO-induced phosphorylation of eIF2α. Next, we silenced the expression of GCN2 by using a specific GCN2 siRNA in CHL-1 cells (, upper panel, and Suppl. S5 left panel) and evaluated the levels of P-eIF2α in cells treated as above (a scrambled siRNA was used as control). Data shown in (bottom panel and Suppl. S4 right panel) indicated that the abrogated expression of GCN2 slightly reduced the levels of P-eIF2α under MTX or DOXO treatment, compared to siCTRL cells. Finally, we down-regulated the expression of PKR by infecting CHL-1 cells with lentiviral particles carrying an shRNA vector specific for PKR, using a scrambled sequence as control (, upper panel). Cells were treated as above and P-eIF2α levels were evaluated in the same experimental condition reported above. As shown in (bottom panel), the absence of PKR strongly reduced the MTX/DOXO-mediated enhanced levels of P-eIF2α. The levels of this factor were in fact 2,0 in shCTRL treated with MTX while only 1,1 in shPKR cells, and 3,4 in shCTRL treated with DOXO while only 2,2 in shPKR cells, compared to own controls (which relative levels were set to 1). Interestingly, we noticed an enhanced basal phosphorylation of eIF2α in shPKR compared to shCTRL CHL-1 cells. This phenomenon seems to be specifically related to PKR inactivation since it was restricted to shPKR and absent in both shHRI and siGCN2 cells (compare lines 1 and 4 of WB from , and ; and in Suppl. S4-5). Moreover, we excluded that the phenomenon was related to the shPKR sequence used in the silencing experiment since the same result was obtained using two different PKR inhibitors, 2-AminopurineCitation33 and PKR inhibitor (CAS-608512-97-6)Citation42 as evidenced in Supplementary Fig. S6. To date, we can only speculate that this is the result of a still uncharacterized compensation process possibly due to unspecific PERK activation, since P-PERK appeared in shPKR cells as evidenced in Supplementary Fig. S7C & D, while no signal was evidenced in both shHRI or siGCN2 cells (Supp S7A & B). Further studies are however required to better elucidate this compensatory activity.
Figure 5. ER-stress independent eIF2α phosphorylation in response to MTX/DOXO treatment. The phosphorylation of eIF2α (eIF2α-P) and PERK (PERK-P) were evaluated in CHL-1 (A) and A375 (B) after MTX (1 μM) or DOXO (5 μM) exposure (3 and 6 h), by western blotting analysis (HSP90 was used as loading control), thapsigargin (TG, 10 μg/ml, 6 h) was used as positive control (n = 3); densitometry values of eIF2α phosphorylation ratio between treated samples and controls are reported. Relative levels (mRNA) of the ER stress markers TRB3 and Xbp-1 (spliced, mature form) were measured by qRT-PCR in cells treated as in A and B; thapsigargin (TG, 10 μg/ml, 6 h) was used as positive control (n = 3). The expression of HRI (D), GCN2 (E) or PKR (F) was down-regulated in CHL-1 cells by lentiviral infection (shHRI, or shPKR, G; scrambled shCTRL was used as control) or by siRNA transient transfection (GCN2 or CTRL siRNA oligos) and specific gene silencing was evaluated by qRT-PCR (upper panels) (n = 3); western blotting analysis was used to evaluate the role of each kinase on eIF2α phosphorylation upon MTX (1 μM) or DOXO (5 μM) treatment (6 h) (bottom panels); densitometry values of eIF2α phosphorylation ratio between treated samples and controls are reported. (G) Concomitant down-regulation of GCN2 (siGCN2) and PKR (shPKR) was performed in CHL-1 cells and qRT-PCR was performed to evaluate the efficacy of GCN2 and PKR down-regulation (upper panel; n = 3); subsequently, cells were treated with MTX (1 μM) or DOXO (5 μM) for 6 h and the phosphorylation of eIF2α was evaluated by western blotting (bottom panel); densitometry values of eIF2α phosphorylation ratio between treated samples and controls are reported. HSP90 or Tubulin was used as loading control in all experiments (n = 3; p < 0.05).
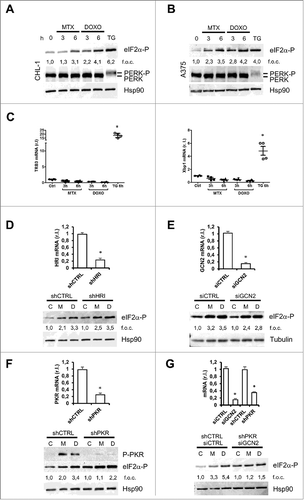
Collectively these results strongly suggest that both PKR (mostly) and GCN2 (to less extent) were responsible for eIF2α phosphorylation during MTX- or DOXO- treatment in melanoma cells. To further confirm this conclusion, we concomitantly inhibited the expression of both these kinases ( upper panel) in CHL-1 cells and evaluated the levels of P-eIF2α while exposed to MTX or DOXO, in the same experimental conditions described above. Data reported in (bottom panel) clearly indicated that the concomitant inhibition of both PKR and GCN2 completely abrogated the MTX- or DOXO- stimulated eIF2α phosphorylation in melanoma cells.
Since an ER stress -dependent and -independent ROS (reactive oxygen species) production have also been implicated in CRT translocation during ICD execution,Citation39 we evaluated their production under our experimental treatment regimen. Our results showed that ROS production was not associated neither to MTX nor DOXO exposure, thus further indicating that CRT exposure seems to be strictly associated to GCN2/PKR-mediated eIF2α phosphorylation (Suppl. S8), at least in melanoma cells.
Gadd34 is involved in the regulation of P-eIF2α/eIF2α cycle under MTX but not DOXO treatment in an ATF4/CHOP-independent manner
The ER stress-independent enhanced levels of phospho-eIF2α revealed under both MTX and DOXO treatment of melanoma cells prompted us to investigate the potential role of Gadd34 contributing to the regulation of P-eIF2α/eIF2α levels in our model. Gadd34 is the stress inducible partner of PP1, the phosphatase responsible for eIF2α dephosphorylation.Citation43 It has been previously reported that this factor might contribute to establish the ER stress mediated P-eIF2α/eIF2α ratio facilitating the CRT translocation on cell surface, upon pro-ICD drug treatment.Citation44 Therefore, we evaluated the expression levels of Gadd34 in melanoma cells under MTX or DOXO treatment. To this aim, CHL-1 and A375 cells were treated or untreated 3 and 6 h with MTX (1μM) or DOXO (5μM) and expression levels of Gadd34 were evaluated by qRT-PCR, using TG (10μg/ml) as positive control. Data reported in show no upregulation of Gadd34 in presence of DOXO, while a consistent upregulation was observed in presence of MTX, in both cell lines. Therefore, we can conclude that Gadd34 is not involved in the control of P-eIF2α/eIF2α ratio under DOXO treatment. However, the upregulation of this factor in presence of MTX prompted us to evaluate the role of the two known transcription factors responsible for Gadd34 upregulation, ATF4 and Gadd153/CHOP. To this aim, we evaluated their expression levels in both CHL-1 and A375 cells in the same experimental conditions described above. Our analysis revealed no upregulation of either ATF4 nor CHOP in presence of MTX (and DOXO) in both cell lines i) further supporting the ER stress independent phosphorylation of eIF2α in response to both MTX and DOXO treatment in our model, and ii) indicating that Gadd34 is upregulated independently from both ATF4 and CHOP as recently reported by Dalet and colleagues,Citation45 in presence of MTX (). To evaluate the real contribution of Gadd34 in the regulation of P-eIF2α/eIF2α ratio under MTX treatment, we inhibited the activity of the factor, by using SalubrinalCitation46 (4 h, 20 nM), in CHL-1 cells and evaluated the levels of P-eIF2α in presence or absence of MTX. Data reported in indicate that salubrinal increased the MTX-dependent P-eIF2α level of 1.7-fold similarly to TG, here used as positive control (). Similar results were obtained using a specific GADD34 inhibitor, Guanabenz,Citation47 as shown in Supplementary Fig. S9. Altogether, these data support the conclusion that Gadd34 has a role in regulating the levels of P-eIF2α in presence of MTX.
Figure 6. Gadd34 regulates the eIF2α/eIF2α-P ratio in cells treated with MTX but not with DOXO. CHL-1 and A375 cells were treated or untreated with MTX (1 μM) or DOXO (5 μM), as indicated, and Gadd34 (A), ATF4 (B and D) and CHOP expression levels were evaluated by qRT-PCR or western blotting analysis; TG (10 μg/ml, 6 h) was used as positive control (n = 3; * = p < 0,05). (E) CHL-1 cells treated 3 h as indicated and the expression levels of eIF2α-P were evaluate by western blotting analysis (SAL = Salubrinal, 20 αM); tubulin was used as loading control; densitometry values of eIF2α phosphorylation ratio between treated samples and controls are reported (n = 3). (F) Gadd 34 was transient transfected in CHL-1 cells to arrogate the phosphorylation of eIF2α (upper panel), and ecto-CRT was evaluated under DOXO treatment (5 μM, 6 h); (n = 3; * = p < 0,05).
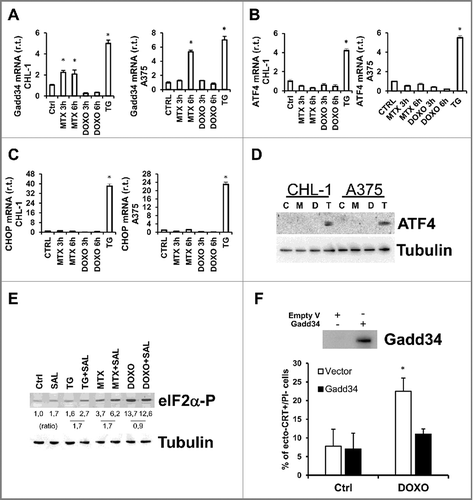
Finally, to further confirm the key role of P-eIF2α in the translocation of CTR on cell membrane surface also in melanoma cells, we inhibited the phosphorylation of eIF2α and evaluated the levels of ecto-CRT. To this aim, Gadd34 was transiently over-expressed in CHL-1 cells ( upper panel) and ecto-CRT was evaluated in cell treated or untreated with 5 μM DOXO (6 h; bottom panel). Data reported in indicate that P-eIF2α is required for drug-induced CRT exposure also in melanoma cells.
Discussion
The role of the immune system in cancer therapy has been extensively studied in the last decade to both explore the possibility to induce an anti-cancer immune response and to unveil the ability of such malignancies to evade the ‘physiological’ immune surveillance.
In this context, ICD represents a new potential therapeutic strategy to treat human tumours exploiting the ability of specific compounds to stimulate the apoptotic pathway in cancer cells and in parallel to stimulate an active immune response toward the same malignant cells, potentially resulting in a more efficient clearance of cancer.Citation48 This new therapeutic approach might be of great interest, particularly for those malignancies characterized by high resistance to conventional anti-tumour regimens, such as human skin melanoma, one of the most aggressive and difficult to treat human cancers.
Therefore, we explored the possibility of considering ICD as a new valuable therapeutic approach to treat human skin melanoma, paying particular attention to the potential impact of oncogenic BRAF.
We show here that both MTX and DOXO were efficient in stimulating a cell death program in a panel of wild type and oncogenic human melanoma cell lines with the latter group being less sensitive, in line with our and other groups previous results, further highlighting the key role of oncogenic BRAF-induced activation of pro-survival pathways.Citation18
As previously described, an efficient ICD induction is strictly linked to drug-induced production/release of specific DAMPs by dying cells, such as type I IFNs production, ATP and HMGB1 release, and CRT cell surface exposure. We verified that all the above-mentioned DAMPs were produced/released by melanoma cells, under pro-ICD drugs stimulation, at east in vitro. In particular, we observed that: i) autophagy and active caspases were required for ATP release; ii) type I IFNs genes were upregulated together with the downstream target CXCL10; iii) HMGB1 was released by dying cells through a passive and caspase-dependent process; iv) CRT was translocated onto cell surface with the cooperation of ERp57 ().
Figure 7. Schematic representation of signaling pathways stimulated by MTX or DOXO in melanoma cells. The pro-ICD drugs MTX and DOXO are able to stimulate the cell death program in melanoma cells (a); concomitantly, both drugs induce an autophagy- (mainly) and caspase- (with less extent) dependent active release of ATP (b), the upregulation of type I IFN genes (c) and a passive release of HMGB1 (d) in the extracellular compartment. The two drugs are also able to stimulate the activation of PKR and GCN2 (e) resulting in eIF2α phosphorylation and mediating the translocation of the ER resident calreticulin (CRT) on to cell surface (f). Interestingly, MTX but not DOXO up-regulates Gadd34 (g) in an ATF4/CHOP-independent manner. This factor, together with PP1, controls the P-eIF2α/eIF2α ratio.
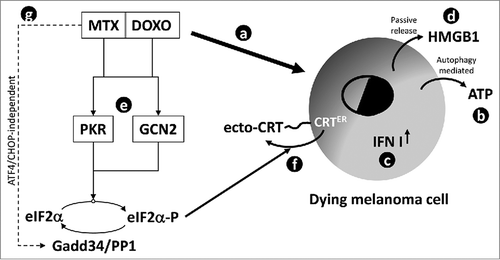
Moreover, it has been previously reported that CRT translocation is mediated by the ER stress and specifically requires the activation of the PERK/eIF2α ER stress branch,Citation26,Citation39 indicating phosphorylated eIF2α as a potential biomarker of immunogenic cell death execution.Citation40 Interestingly, our results evidenced an early phosphorylation of eIF2α during pro-ICD drugs exposure, but, surprisingly, no ER stress response was induced, since none of typical ER stress markers were up-regulated nor PERK activation was evident in the same experimental conditions. Three other kinases, besides PERK, are known to phosphorylate eIF2α under stress conditions: HRI, GCN2 and PKR. Our analysis indicates that PKR, mostly, and GCN2, with a minor extent, kinase activity is required to phosphorylate eIF2α under pro-ICD drugs treatment, in melanoma cells. Collectively these data indicate for the first time, to our knowledge, the ER stress-independent phosphorylation of eIF2α in response to pro-ICD drugs. This is of particular interest since it indicates a potential induction of ICD in vivo, also in those malignancies characterized by abrogated/compromised UPR program.
We previously showed oncogenic BRAF induces a chronic ER stress status in melanoma cells with consequent up-regulation of all UPR markers together with CRT.Citation18 Therefore, we argued if the oncogenic BRAF-induced chronic ER stress status could result in imbalanced physiological or pro-ICD drugs-stimulated translocation of CRT on cell surface. However, we found that CRT was efficiently translocated onto cell surface under pro-ICD drugs treatment and, more importantly, BRAFV600E had no effect on both basal and stimulated exposure of CRT. This result is in line with previous ones, indicating no influence played by oncogenic BRAF on production/release of the other DAMPs, such as IFNs gene up-regulation and the release of ATP and HMGB1.
In conclusion, our data indicate that pro-ICD drugs are able to stimulate the production/release of DAMPs in melanoma cells at least in vitro, indicating in this approach a potential new valuable therapeutic strategy to treat human skin melanoma malignancy.
Material and methods
Cell culture and treatments
All cell lines were cultured in DMEM (Sigma-Aldrich, St. Louis, MO, USA) except for A2058 cells that were cultured in RPMI (Sigma-Aldrich), supplemented with 10% fetal bovine serum (Sigma-Aldrich), 2 mM L-glutamine (Sigma-Aldrich), 1% penicillin/streptomycin solution (Sigma-Aldrich) at 37°C under 5% CO2. Cells were treated with thapsigargin (TG) (Sigma-Aldrich) at a final concentration of 10 μg/ml, MTX (Sigma-Aldrich) 1–5 μM, DOXO (Sigma-Aldrich) 5 μM, Fenretinide (Janssen-Cilag Ltd., Basserdorf, Switzerland) 10 μM, caspase pan-inhibitor Z-VAD-fmk (Alexis Biochemicals, San Diego, CA, USA) 10 μM; 2-Aminopurine (Sigma-Aldrich) 5 mM; PKR Inhibitor (CAS-608512-97-6; Santa Cruz) 1 nM; Bafalomycin A 5 nM (Sigma-Aldrich); Salubrinal (Sigma-Aldrich) 20 μM; Guanabenz 50 μM (Cayman Chemical). All agents were added in DMSO or methanol with an equal volume of vehicle used as control (0.1–0.5% final concentration). The Gadd34 encoding vector, pXJ40 GADD34 Wt (1-674aa), was a gift from Shirish Shenolikar (Addgene plasmid # 75478)Citation49
Retrovirus generation and infection
Co-transfection of retroviral vectors (15 μg; LC3-GFP) and vesicular stomatitis virus G protein expression plasmid (5 μg) was performed by using 293 gp/bsr cell line and calcium phosphate protocol. Supernatant with retroviral particles was harvested 48 h later and supplemented with 4 μg/ml of polybrene. These supernatants were used to infect target cells.
Lentiviral generation and infection
Co-trasfection of lentiviral vectors (shRNA-pLKO ERp57, shRNA-pLKO PKR, shRNA-pLKO HRI; Sigma) (10 μg), vesicular stomatitis virus G protein expression plasmid (2,5 μg) and psPAX2 plasmid (carrying gag, pol and rev genes) was performed using 293 T packaging cell line, by a calcium phosphate protocol. Supernatants with lentiviral particles were harvested 48 h later and supplemented with 4 μg/ml of polybrene. These supernatants were used to infect target cells.Citation50
RNA interference
hGCN2, hBECN1 and non-targeting scramble (siCTRL, used as negative control) siRNA oligoribonucleotides were obtained from Invitrogen (Carlsbad, CA, USA). 25 × 104 cells/well were seeded in six-well plates and transfected with siRNA (100 pmol) by means of RNAi Max (Invitrogen) as recommended by the supplier. 24 h later, cells were trypsinized, plated at 30 × 104 cells/well in six-well plates and treated with the indicated agents. Quantitative RT-PCR (qRT-PCR) analysis was used to assess RNA down-regulation after 48 h from transfection, as described below.
Western blotting
Protein extraction was performed by using Cell Lytic buffer (Sigma-Aldrich) supplemented with a protease inhibitors cocktail (Sigma-Aldrich) plus phosphatases inhibitors (Na3VO4 1 mM; NaF 10 mM), and resolved by electrophoresis through NuPAGE Bis-Tris gel (Invitrogen, Carlsbad, CA, USA) and electroblotted onto nitrocellulose (Protran, Sigma-Aldrich) membrane. Blots were incubated with indicated primary antibodies in 5% non-fat dry milk in PBS plus 0.1% Tween20 overnight at 4°C. Primary antibodies were: anti-eIF2α (1:1000), P-eIF2α (1:500), cleaved PARP (1:500), IL-1β (1:500), PKR (1:500) (Cell Signaling Technology, Beverly, MA, USA); anti-Gapdh (1:106; Calbiochem, Merck Millipore, Darmstadt, Germany); Tubulin-α (1:5000; Santa Cruz Biotechnology, Santa Cruz, CA, USA); anti-P-PKR (1:1000; Abcam, ab32036); anti-ATF4 (1:100; Genetex). Detection was achieved using horseradish peroxidase-conjugate secondary antibody (1:5000; Jackson ImmunoResearch, Suffolk, UK) and visualized with ECL plus (Amersham Biosciences, GE Healthcare, Buckinghamshire, UK). Images were acquired by using a ChemiDoc™ Touch Imaging System (Bio Rad) and analyzed by Image Lab software (Bio Rad).Citation51
HMGB1 evaluation
Cell culture media from treated cells was harvested and centrifuged 1 min at 12000 x g and supplemented with protease inhibitor cocktail (Sigma-Aldrich). 20 μl of media were loaded and resolved by SDS-Page, as described above. Primary antibody was: anti-HMGB1 (1:1000; Sigma-Aldrich). Total cell lysates were used as positive control. Detection was achieved as described above. Caspase inhibition was achieved by using the pan inhibitor z-VAD-FMK (Alexis Biochemicals, San Diego, CA, USA).
Quantitative RT-polymerase chain reaction
Trizol reagent (Invitrogen) was used to extract total RNA as indicated by the supplier, and a Promega reverse transcription kit (Madison, WI, USA) was used to produce cDNA following the manufacturer's recommendations. Quantitative PCR reactions were performed by using the Rotor-Gene 6000 (Corbett Research Ltd, Cambridgeshire, UK) thermocycler. Supplementary Table S1 shows the primer pair sequences for all amplicons, designed by using the online IDT PrimerQuest Tool software (IDT, Integrated DNA Technologies Inc., USA; https://eu.idtdna.com/Primerquest/Home/Index). L34 mRNA level was used as an internal control and results were expressed as previously described.Citation52
Cell death evaluation
Propidium iodide-stained cells were analyzed by flow cytometry and the percentage of cells in the sub-G1 fraction was measured, as previously described.Citation53
Autophagy assay
Autophagy was measured in GFP–LC3 transduced cells as previously described.Citation54 Briefly, cells were either starved, by using Earle's balanced salt solution medium (Sigma-Aldrich) for 2 h, or treated with MTX 1 μM or DOXO 5 μM for 2 and 4 h, in presence of 5 nM Bafilomycin A. CHL-1 GFP-LC3 cells were grown on coverslips and fixed with 4% paraformaldehyde in PBS, washed three times with PBS and examined by confocal microscopy.
Immunofluorescence assay
80 × 103 cells/well were seeded in 6-well plates and treated as indicated. Cells were harvested by trypsin, washed in PBS, fixed in 4% PFA in PBS and incubated with anti-CRT antibody (1:1000; Abcam, ab2970) and with anti-rabbit AlexaFluor 488-conjugated or AlexaFluor 647-conjugated (Life Technologies). A total of 2 × 104 fluorescent cells was acquired by using the BD FACSCalibur™ (Becton-Dickinson) flow cytometer.
ATP assay
ATP levels were measured in cell culture media by using luciferin-based ENLITEN ATP Assay (Promega) as recommended by the supplier. Bioluminescence was analyzed by Perkin Elmer Wallac 1420 Victor2 Microplate Reader.
ROS production evaluation
Briefly, 1,5 × 105 cells were treatment as indicated and cells harvested at indicate time points. Then, cells were pelleted, washed by PBS, resuspended in H2-DCFDA (10 mM in PBS), incubated at 37°C for 15 min in the dark, and 10.000 events were acquired by using a FACS Calibur cytometer (Becton-Dickinson, Mountain View, CA, USA). Data analysis was performed using FlowJo software.Citation55
Type I IFN assay
IFNα or β were measured in cell culture media by using the human IFN-alpha platinum ELISA kit or the IFN-beta Human ELISA kit (ThermoFisher Scientific) as recommended by the supplier. OD was analyzed by Perkin Elmer Wallac 1420 Victor2 Microplate Reader.
Statistical analysis
All experiments were performed at least triplicate. Data showed in this paper are representative of 3 independent experiments carried out in triplicate; western blotting images are from a representative experiment carried out in triplicate. Statistical analysis was performed using GraphPad Prism 5 and Student's t-test was used to determine statistical significance. A P-value of equal to or less than 0.05 was considered significant.
Grant support
This work was supported in part by grants from the Telethon Foundation (GEP12072), AIRC (IG2014 n. 15244 to MP), the Italian Ministry of University and Research (FIRB Accordi di Programma 2011), the Italian Ministry of Health (Ricerca Corrente and Ricerca Finalizzata), Fondazione Fibrosi Cistica (Progetto FFC#8/2015).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Suppl_material.zip
Download Zip (1.5 MB)References
- Sandru A, Voinea S, Panaitescu E, Blidaru A. Survival rates of patients with metastatic malignant melanoma. J Med Life. 2014;7(4):572. PMID:25713625.
- Shellenberger R, Nabhan M, Kakaraparthi S. Melanoma screening: A plan for improving early detection. Annals Med. 2016;48(3):142–8. doi:10.3109/07853890.2016.1145795.
- Fecher LA, Cummings SD, Keefe MJ, Alani RM. Toward a molecular classification of melanoma. J Clin Oncol. 2007;25(12):1606–20. doi:10.1200/JCO.2006.06.0442. PMID:17443002.
- Cancer Genome Atlas Network. Genomic classification of cutaneous melanoma. Cell. 2015;161(7):1681–96. doi:10.1016/j.cell.2015.05.044. PMID:26091043.
- Wan PT, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM, Jones CM, Marshall CJ, Springer CJ, Barford D, et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004;116(6):855–67. doi:10.1016/S0092-8674(04)00215-6. PMID:15035987.
- Fedorenko IV, Gibney GT, Sondak VK, Smalley KSM. Beyond BRAF: where next for melanoma therapy & quest. Br J Cancer. 2015;112(2):217–26. doi:10.1038/bjc.2014.476. PMID:25180764.
- Lopez‐Bergami P. The role of mitogen‐and stress‐activated protein kinase pathways in melanoma. Pigment Cell Melanoma Res. 2011;24(5):902–21. doi:10.1111/j.1755-148X.2011.00908.x. PMID:21914141.
- Lazova R, Klump V, Pawelek J. Autophagy in cutaneous malignant melanoma. J Cutaneous Pathol. 2010;37(2):256–68. doi:10.1111/j.1600-0560.2009.01359.x.
- Corazzari M, Fimia GM, Lovat P, Piacentini M. Why is autophagy important for melanoma? Molecular mechanisms and therapeutic implications. Semin Cancer Biol. 2013;23:337–43. Academic Press. doi:10.1016/j.semcancer.2013.07.001. PMID:23856558.
- Fimia GM, Piacentini M. Regulation of autophagy in mammals and its interplay with apoptosis. Cell Mol Life Sci. 2010;67(10):1581–8. doi:10.1007/s00018-010-0284-z. PMID:20165902.
- Croft A, Tay KH, Boyd SC, Guo ST, Jiang CC, Lai F, Tseng HY, Jin L, Rizos H, Hersey P, et al. Oncogenic activation of MEK/ERK primes melanoma cells for adaptation to endoplasmic reticulum stress. J Invest Dermatol. 2014;134(2):488–97. doi:10.1038/jid.2013.325. PMID:23921951.
- Schönthal AH. Endoplasmic reticulum stress: its role in disease and novel prospects for therapy. Scientifica. 2012;2012:85751. doi:10.6064/2012/857516.
- Sykes EK, Mactier S, Christopherson RI. Melanoma and the unfolded protein response. Cancers. 2016;8(3):30. doi:10.3390/cancers8030030.
- Corazzari M, Lovat PE, Armstrong JL, Fimia GM, Hill DS, Birch-Machin M, Redfern CPF, Piacentini M. Targeting homeostatic mechanisms of endoplasmic reticulum stress to increase susceptibility of cancer cells to fenretinide-induced apoptosis: the role of stress proteins ERdj5 and ERp57. Br J Cancer. 2007;96(7):1062–71. doi:10.1038/sj.bjc.6603672. PMID:17353921.
- Lovat PE, Corazzari M, Armstrong JL, Martin S, Pagliarini V, Hill D, Brown AM, Piacentini M, Birch-Machin M, Redfern CP. Increasing melanoma cell death using inhibitors of protein disulfide isomerases to abrogate survival responses to endoplasmic reticulum stress. Cancer Res. 2008;68(13):5363–9. doi:10.1158/0008-5472.CAN-08-0035. PMID:18593938.
- Corazzari M. ER stress & autophagy in cancer: contenders or partners in crime. Int J Mol Biol Biochem. 2013;1:23–28.
- Corazzari M, Gagliardi M, Fimia GM, Piacentini M. Endoplasmic reticulum stress, unfolded protein response, and cancer cell fate. Front Oncol. 2017;7:78. doi:10.3389/fonc.2017.00078. PMID:28491820.
- Corazzari M, Rapino F, Ciccosanti F, Giglio P, Antonioli M, Conti B, Fimia GM, Lovat PE, Piacentini M. Oncogenic BRAF induces chronic ER stress condition resulting in increased basal autophagy and apoptotic resistance of cutaneous melanoma. Cell Death Differentiation. 2015;22(6):946–58. doi:10.1038/cdd.2014.183.
- Giglio P, Fimia GM, Lovat PE, Piacentini M, Corazzari M. Fateful music from a talented orchestra with a wicked conductor: Connection between oncogenic BRAF, ER stress, and autophagy in human melanoma. Mol Cell Oncol. 2015;2(3):e995016. doi:10.4161/23723556.2014.995016. PMID:27308477.
- Sullivan RJ, Flaherty KT. Resistance to BRAF-targeted therapy in melanoma. Eur J Cancer. 2013;49(6):1297–304. doi:10.1016/j.ejca.2012.11.019. PMID:23290787.
- Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Annual Rev Immunol. 2013;31:51–72. doi:10.1146/annurev-immunol-032712-100008.
- Garg AD, Dudek-Peric AM, Romano E, Agostinis P. Immunogenic cell death. Int J Dev Biol. 2015;59(1-2-3):131–40. doi:10.1387/ijdb.150061pa. PMID:26374534.
- Bezu L, Gomes-de-Silva LC, Dewitte H, Breckpot K, Fucikova J, Spisek R, Galluzzi L, Kepp O, Kroemer G. Combinatorial strategies for the induction of immunogenic cell death. Frontiers Immunol. 2015;6:275.
- Garg AD, Galluzzi L, Apetoh L, Baert T, Birge RB, Bravo-San Pedro JM, Breckpot K, Brough D, Chaurio R, et al. Molecular and translational Classifications of DaMPs in immunogenic cell death. Frontiers Immunol. 2015;6:588. doi:10.3389/fimmu.2015.00588.
- Kepp O, Senovilla L, Kroemer G. Immunogenic cell death inducers as anticancer agents. Oncotarget. 2014;5(14):5190–1. doi:10.18632/oncotarget.2266. PMID:25114034.
- Dudek AM, Garg AD, Krysko DV, De Ruysscher D, Agostinis P. Inducers of immunogenic cancer cell death. Cytokine Growth Factor Rev. 2013;24(4):319–33. doi:10.1016/j.cytogfr.2013.01.005. PMID:23391812.
- Martins I, Tesniere A, Kepp O, Michaud M, Schlemmer F, Senovilla L, Séror C, Métivier D, Perfettini JL, Zitvogel L, et al. Chemotherapy induces ATP release from tumor cells. Cell Cycle. 2009;8(22):3723–8. doi:10.4161/cc.8.22.10026. PMID:19855167.
- Martins I, Wang Y, Michaud M, Ma Y, Sukkurwala AQ, Shen S, Kepp O, Métivier D, Galluzzi L, Perfettini JL, et al. Molecular mechanisms of ATP secretion during immunogenic cell death. Cell Death Differentiation. 2014;21(1):79–91. doi:10.1038/cdd.2013.75. PMID:23852373.
- Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD, Adeli K, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2016;12(1):1–222. doi:10.1080/15548627.2015.1100356. PMID:26799652.
- Lu B, Wang C, Wang M, Li W, Chen F, Tracey KJ, Wang H. Molecular mechanism and therapeutic modulation of high mobility group box 1 release and action: an updated review. Exp Rev Clin Immunol. 2014;10(6):713–27. doi:10.1586/1744666X.2014.909730.
- Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, Mignot G, Maiuri MC, Ullrich E, Saulnier P, et al. Toll-like receptor 4–dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. 2007;13(9):1050–9. doi:10.1038/nm1622. PMID:17704786.
- Lamkanfi M, Sarkar A, Walle LV, Vitari AC, Amer AO, Wewers MD, Tracey KJ, Kanneganti TD, Dixit VM. Inflammasome-dependent release of the alarmin HMGB1 in endotoxemia. J Immunol. 2010;185(7):4385–92. doi:10.4049/jimmunol.1000803. PMID:20802146.
- Lu B, Nakamura T, Inouye K, Li J, Tang Y, Lundbäck P, Valdes-Ferrer SI, Olofsson PS, Kalb T, Roth J, et al. Novel role of PKR in inflammasome activation and HMGB1 release. Nature. 2012;488(7413):670–4. doi:10.1038/nature11290. PMID:22801494.
- Gold LI, Eggleton P, Sweetwyne MT, Van Duyn LB, Greives MR, Naylor SM, Michalak M, Murphy-Ullrich JE. Calreticulin: non-endoplasmic reticulum functions in physiology and disease. FASEB J. 2010;24(3):665–83. doi:10.1096/fj.09-145482. PMID:19940256.
- Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, Castedo M, Mignot G, Panaretakis T, Casares N, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med. 2007;13(1):54–61. doi:10.1038/nm1523. PMID:17187072.
- Panaretakis T, Joza N, Modjtahedi N, Tesniere A, Vitale I, Durchschlag M, Fimia GM, Kepp O, Piacentini M, Froehlich KU, et al. The co-translocation of ERp57 and calreticulin determines the immunogenicity of cell death. Cell Death Differentiation. 2008;15(9):1499–509. doi:10.1038/cdd.2008.67. PMID:18464797.
- Hertzog PJ. Overview. Type I interferons as primers, activators and inhibitors of innate and adaptive immune responses. Immunol Cell Biol. 2012;90(5):471–3. doi:10.1038/icb.2012.15. PMID:22648073.
- Sistigu A, Yamazaki T, Vacchelli E, Chaba K, Enot DP, Adam J, Vitale I, Goubar A, Baracco EE, Remédios C, et al. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat Med. 2014;20(11):1301–9. doi:10.1038/nm.3708. PMID:25344738.
- Panaretakis T, Kepp O, Brockmeier U, Tesniere A, Bjorklund AC, Chapman DC, Durchschlag M, Joza N, Pierron G, van Endert P, et al. Mechanisms of pre‐apoptotic calreticulin exposure in immunogenic cell death. EMBO J. 2009;28(5):578–90. doi:10.1038/emboj.2009.1. PMID:19165151.
- Kepp O, Semeraro M, Bravo-San Pedro JM, Bloy N, Buqué A, Huang X, Zhou H, Senovilla L, Kroemer G, Galluzzi L. eIF2α phosphorylation as a biomarker of immunogenic cell death. Seminars Cancer Biol. 2015;33:86–92. Academic Press. doi:10.1016/j.semcancer.2015.02.004.
- Donnelly N, Gorman AM, Gupta S, Samali A. The eIF2α kinases: their structures and functions. Cell Mol Life Sci. 2013;70(19):3493–511. doi:10.1007/s00018-012-1252-6. PMID:23354059.
- Couturier J, Morel M, Pontcharraud R, Gontier V, Fauconneau B, Paccalin M, Page G. Interaction of double-stranded RNA-dependent protein kinase (PKR) with the death receptor signaling pathway in amyloid β (Aβ)-treated cells and in APPSLPS1 knock-in mice. J Biol Chem. 2010;285(2):1272–82. doi:10.1074/jbc.M109.041954. PMID:19889624.
- Reid DW, Tay AS, Sundaram JR, Lee IC, Chen Q, George SE, Nicchitta CV, Shenolikar S. Complementary roles of GADD34-and CReP-containing eukaryotic initiation factor 2α phosphatases during the unfolded protein response. Mol Cell Biol. 2016;36(13):1868–80. doi:10.1128/MCB.00190-16. PMID:27161320.
- Kepp O, Galluzzi L, Giordanetto F, Tesniere A, Vitale I, Martins I, Schlemmer F, Adjemian S, Zitvogel L, Kroemer G. Disruption of the PP1/GADD34 complex induces calreticulin exposure. Cell Cycle. 2009;8(23):3971–7. doi:10.4161/cc.8.23.10191. PMID:19901557.
- Dalet A, Argüello RJ, Combes A, Spinelli L, Jaeger S, Fallet M, Vu Manh TP, Mendes A, Perego J, Reverendo M, et al. Protein synthesis inhibition and GADD34 control IFN‐β heterogeneous expression in response to dsRNA. EMBO J. 2017;36(6):761–82. doi:10.15252/embj.201695000. PMID:28100675.
- Boyce M, Bryant KF, Jousse C, Long K, Harding HP, Scheuner D, Kaufman RJ, Ma D, Coen DM, Ron D, Yuan J. A selective inhibitor of eIF2α dephosphorylation protects cells from ER stress. Science. 2005;207:935–39. doi:10.1126/science.1101902.
- Tsaytler P, Harding HP, Ron D, Bertolotti A. Selective Inhibition of a Regulatory Subunit of Protein Phosphatase 1 Restores Proteostasis. Science. 2011;332:91–94. doi:10.1126/science.1201396. PMID:21385720.
- Papaioannou NE, Beniata OV, Vitsos P, Tsitsilonis O, Samara P. Harnessing the immune system to improve cancer therapy. Annals Translational Med. 2016;4(14):261. doi:10.21037/atm.2016.04.01.
- Choy MS, Yusoff P, Lee IC, Newton JC, Goh CW, Page R, Shenolikar S, Peti W. Structural and Functional Analysis of the GADD34:PP1 eIF2alpha Phosphatase. Cell Rep. 2015;11(12):1885–91. doi:10.1016/j.celrep.2015.05.043. PMID:26095357.
- Pagliarini V, Giglio P, Bernardoni P, De Zio D, Fimia GM, Piacentini M, Corazzari M. Downregulation of E2F1 during ER stress is required to induce apoptosis. J Cell Sci. 2015;128(6):1166–79. doi:10.1242/jcs.164103. PMID:25616897.
- Antunes F, Corazzari M, Pereira G, Fimia GM, Piacentini M, Smaili S. Fasting boosts sensitivity of human skin melanoma to cisplatin-induced cell death. Biochem Biophys Res Commun. 2017;485(1):16–22. doi:10.1016/j.bbrc.2016.09.149. PMID:27693581.
- Pagliarini V, Wirawan E, Romagnoli A, Ciccosanti F, Lisi G, Lippens S, Cecconi F, Fimia GM, Vandenabeele P, Corazzari M, et al. Proteolysis of Ambra1 during apoptosis has a role in the inhibition of the autophagic pro-survival response. Cell Death Differentiation. 2012;19(9):1495–504. doi:10.1038/cdd.2012.27. PMID:22441670.
- Corazzari M, Lovat PE, Armstrong JL, Fimia GM, Hill DS, Birch-Machin M, Redfern CPF, Piacentini M. Targeting homeostatic mechanisms of endoplasmic reticulum stress to increase susceptibility of cancer cells to fenretinide-induced apoptosis: the role of stress proteins ERdj5 and ERp57. Br J Cancer. 2007;96(7):1062–71. doi:10.1038/sj.bjc.6603672. PMID:17353921.
- Armstrong JL, Corazzari M, Martin S, Pagliarini V, Falasca L, Hill DS, Ellis N, Al Sabah S, Redfern CPF, Fimia GM, et al. Oncogenic B-RAF signaling in melanoma impairs the therapeutic advantage of autophagy inhibition. Clin Cancer Res. 2011;17(8):2216–26. doi:10.1158/1078-0432.CCR-10-3003. PMID:21270111.
- Ciccosanti F, Corazzari M, Soldani F, Matarrese P, Pagliarini V, Iadevaia V, Tinari A, Zaccarelli M, Perfettini JL, Malorni W, et al. Proteomic analysis identifies prohibitin down-regulation as a crucial event in the mitochondrial damage observed in HIV-infected patients. Antivir Ther. 2010;15(3):377–90. doi:10.3851/IMP1530. PMID:20516557.