
ABSTRACT
The Wiskott-Aldrich syndrome protein (WASp) is a key regulator of the actin cytoskeleton in hematopoietic cells and mutated in two severe immunodeficiency diseases with high incidence of cancer. Wiskott-Aldrich syndrome (WAS) is caused by loss-of-function mutations in WASp and most frequently associated with lymphoreticular tumors of poor prognosis. X-linked neuropenia (XLN) is caused by gain-of-function mutations in WASp and associated with acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS). To understand the role of WASp in tumorigenesis, we bred WASp+, WASp−, and WASp-XLN mice onto tumor susceptible p53+/- background and sub-lethally irradiated them to enhance tumor development. We followed the cohorts for 24 weeks and tumors were characterized by histology and flow cytometry to define the tumor incidence, onset, and cell origin. We found that p53+/-WASp+ mice developed malignancies, including solid tumors and T cell lymphomas with 71.4% of survival 24 weeks after irradiation. p53+/-WASp− mice showed lower survival rate and developed various early onset malignancies. Surprisingly, the p53+/-WASp-XLN mice developed malignancy mostly with late onset, which caused delayed mortality in this colony. This study provides evidence for that loss-of-function and gain-of-function mutations in WASp influence tumor incidence and onset.
Introduction
Patients that suffer from primary immunodeficiency are at risk to develop haematological malignancies often associated with Epstein-Barr virus infection and of poor prognosis.Citation1 The reason for increased cancer risk could be failure of the immune system to eradicate tumors or due to intrinsic failure during myelopoiesis and lymphopoiesis. Mutations in the WAS gene that encodes for the cytoskeletal regulator WASp are associated with two immunodeficiency syndromes, Wiskott-Aldrich syndrome (WAS) and X-linked neutropenia (XLN). WAS is caused by loss-of-function mutations in WASp leading to severe immunodeficiency. The tumor incidence in WAS is estimated to be 13–22% with a median age of onset of 9.5 years and with poor prognosis.Citation2,Citation3 WAS patient tumors include non-Hodgkin lymphoma, EBV positive and EBV negative lymphoma, Hodgkin lymphoma, Burkitt lymphoma, and less frequently myelodysplasia, acute lymphoblastic leukemia, myelomonocytic leukaemia, and nonhematopoietic malignancies.Citation3-Citation10 XLN is caused by mutations that destroy the auto-inhibitory folding of WASp thereby rendering WASp constitutively active.Citation11-Citation15 XLN patients show bone marrow arrest at the promyelocyte stage associated with development of myelodysplastic syndrome and acute myeloid leukemia. This is associated with aberrant segregation of chromosomes to the daughter cells and impaired cytokinesis during mitosis, leading to increased cell death.Citation14-Citation16 Moreover, somatic XLN mutations in WASp correlate with poor prognosis in patients with juvenile myelomonocytic leukemia and the XLN-WASp expressing leukemic clone becomes dominant in these patients.Citation17
Table 1. Malignancies in the p53+/- irradiation model.
We herein sought to identify the tumor incidence, onset, and cell origin in WAS and XLN. Since no spontaneous tumor development has been observed in WASp− knockout and WASp-XLN knockin mouse colonies, we established a tumor model where WASp mutant strains were bred onto tumor prone Trp53 (p53) deficient mice. As the p53 gene is responsible for maintaining genomic integrity during genotoxic stress, p53 deficient mice develop spontaneous tumors with a high incidence.Citation18 In p53±/- heterozygous mice, which shows only moderate tumor susceptibility, gamma radiation dramatically reduces the latency of tumor development.Citation19-Citation21 Due to irradiation induced mutations, the intact wild type p53 allele is mutated in p53+/- tumors, resulting in loss of heterozygosity (LOH), no functional p53 expression, and tumor growth.Citation19 Importantly, synergy between p53 inactivation and other mutations lead to the emergence of specific tumor types such as p53 loss in KrasG12D knock-in leads to AML Citation22 or Eμ-myc transgene to B cell lymphomas.Citation23
Here we used the p53+/-WASp− and p53+/-WASp-XLN mice to test if WASp mutations synergize with p53 in carcinogenesis. Using this model, we provide evidence for that WASp deficiency is associated with earlier onset of tumors. In contrast, gain-of-function XLN mutations in WASp led to later onset of tumors.
Materials and methods
Mice
Mice were bred and housed at animal facility of the Department of Microbiology Tumor and Cell biology at Karolinska Institutet under defined pathogen-free conditions. The WASp− and the WASp-XLN (WASp-I296T and WASp-L272P) mice were backcrossed to the C57BL/6 (B6) background for at least 8 generations. Trp53tm1Brd (p53−/-) mice Citation18 were bred with the WASp mutant mice to generate p53+/-WASp−, WASp-XLN, and p53+/-WASp+ mice as littermate controls. Twenty eight male p53+/-WASp+, 14 p53+/-WASp−, 16 p53+/-WASp-XLN (10 p53+/-WASp-I296T+6 p53+/-WASp-L272P), and 9 p53+/+WASp+ mice were sub-lethally irradiated with a single exposure of 4 Gy at a median age of 20 weeks (14–32 weeks) of age. All animal experiments were performed after approval from the local ethical committee (the north Stockholm district court, Dnr 272/13).
Histopathological examination of mice
Mice were sacrificed when they developed visible tumors, showed a severe weight loss, and/or showed signs of severe discomfort. A small number of mice died spontaneously with no early indication of deterioration of their health. Spleen, liver, lymph nodes, thymus, or sternum were collected during autopsy and tissues fixed in 4% neutral buffered formaldehyde solution (Sigma-Aldrich). Tissues were routinely processed, paraffin embedded, and 4µm sections were mounted on glass slides, deparaffinized and hematoxylin-eosin stained. For immunohistochemistry, sections were mounted on charged glass slides and deparaffinized. Following heat-induced epitope retrieval (HIER) at pH9 sections were incubated with monoclonal rabbit anti F4/80 (macrophage marker; dilution 1:100, clone SP115, Abcam), or monoclonal rabbit anti CD3 (T-cell marker; dilution 1:200, clone SP7, Thermo Scientific) and visualized by Leica Bond polymer refine detection on a Leica Bond RXm staining platform. For detection of B-cells, sections were heated for 40 min in citrate buffer (pH 6), manually incubated overnight with polyclonal goat anti Pax5 (dilution 1:2500; C-20, Santa Cruz), detected using rabbit-anti-goat (1:300, Dako) and visualized using Vectastain ABC and ImmPACT DAB kits (Vector Laboratories) according to the manufacturer’s instructions, and countersained with hematoxylin. Stained sections were reviewed by a veterinary pathologist (R.K.).
Flow cytometry
To determine early onset changes of the cellularity in peripheral blood, blood samples were collected at regular intervals up to 15 weeks after irradiation and during autopsy. To quantify the amount of hematopoietic cells, AccuCheck counting beads (Invitrogen by Thermo Fisher Scientific) were added to heparin treated blood samples before staining. Red blood cells were lysed with hypotonic salt. For other tissues, single-cell suspensions of spleen, liver, thymus, and tumor were prepared, red blood cells were lysed, and cells were labeled with fluorescently conjugated anti-mouse antibodies: CD45 (APC-Cy-7; hematopoietic cells), CD3 (PE; T lymphocytes), B220 (FITC; B lymphocytes), CD11b (Pacific Blue; myeloid cells) and Ly6G (PE; neutrophils). Spleen suspensions and peripheral blood samples of mice were stained with CD3e (PE; T lymphocytes), CD19 (APC-Cy-7; B lymphocytes), CD11b and CD34 (FITC; hematopoietic progenitor cells). To block Fc receptor binding, purified anti-CD16/32 was added to the staining solutions. All antibodies were purchased from BioLegend. Dead cells were detected by using 4ʹ,6-Diamidino-2-phenylindol (DAPI) and excluded from the gating. Flow cytometric analyses were performed on an LSRFortessa X-20 using FACSDiva software (Becton Dickinson). Data were analyzed using the FlowJo software (Tree Star, Inc.).
Tumor killing assay
The YAC-1 T cell lymphoma cell line was stable transfected with Luciferase-pcDNA3.Citation24 Luciferase-pcDNA3 was a gift from William Kaelin (Addgene plasmid #18,964). Natural killer (NK) cells were purified from spleens of poly I:C injected mice with negative selection (Miltenyi Biotec). YAC-1-luciferase cells were co-incubated with NK cells in various ratios for 6h in 96 well plates. Luciferase activity was measured in lysed cells with luciferin substrate (Sigma-Aldrich). NK cell killing activity was calculated as percentage of luciferase signal of the total signal of YAC-1 cells in the absence of NK cells.
In vitro neutrophil differentiation
Neutrophil differentiation in liquid culture was performed as described by Gupta et al. Citation25. Hematopoietic progenitors and stem cells were purified from bone marrow suspension with negative selection (Stemcell Technologies). Equal cell density of each genotype was cultured for 3 days in the presence of SCF and IL-3. 50,000 cells were plated in each well of 96 well plates, and supplemented with SCF, IL-2, and G-CSF to induce promyelocyte differentiation for 2 days. Where indicated, samples were irradiated at day 3 with 1 Gy using X-ray. At day 5, cell numbers were determined with AccuCheck counting beads using flow cytometry.
Statistical analysis
Data were analyzed and processed using Prism 7.0 software (GraphPad Software). Survival curves were generated by using the Kaplan-Meier method and compared by Gehan-Breslow-Wilcoxon test. Laplace regression Citation26 was used to estimate the differences in the 10th and 20th percentiles of survival time between p53±/-WASp± and p53±/-WASp− or p53±/-WASp-XLN groups.
Results
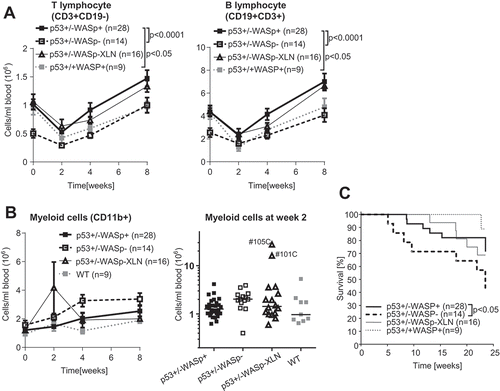
To evaluate tumor incidence, onset, and cell origin, we bred WASp mutant mice with p53−/- mice to generate p53+/-WASp+ (28 mice), p53+/-WASp− (14 mice), and p53+/-WASp-XLN mice (16 mice). To induce genotoxic stress, these mice were sublethally irradiated and followed for 24 weeks. Two weeks after irradiation, the blood B and T lymphocyte count dropped in all genotypes and then recovered at week 4 (). p53+/-WASp+ rebounded to a higher lymphocyte count than p53+/+WASp+ (WT). p53+/-WASp− mice had significantly lower number of blood T and B lymphocytes than the p53+/-WASp+ control mice during the whole period of monitoring, including the time before irradiation (). Myeloid cell number in blood did not drop ( left panel), however 2 weeks after irradiation 2/16 p53+/-WASp-XLN mice (12.5%) showed a sharp temporary expansion of the myeloid compartment ( right panel). The expanded cells in these two p53±/-WASp-XLN mice represented a 7–9 fold increase in monocytes (CD11b±SSClo cells) and 11–20 fold expansion of neutrophils (CD11b±SSChi cells). Although this cell expansion occurred stochastic in vivo, in vitro bone marrow myeloid cultures of WASp-XLN (WASp-I296T and WASp-L272P) consistently showed higher rate of proliferation than WASp± control cultures, regardless of p53 genotype or irradiation (Fig. S1A). Deaths in the cohort started at 6 weeks after irradiation and through the whole 24 week period, the survival curve shows that p53+/-WASp− mice had lower survival rate when compared to p53+/-WASp+ and p53+/-WASp-XLN mice ().
Figure 1. Blood leucocyte counts and survival rate in the p53+/-WASp mutant tumor model. Blood lymphocyte (A) and (B) myeloid cell counts were determined with flow cytometry for 8 weeks after irradiation. Two-way ANOVA and Bonferroni’s multiple comparison test. Mean±SEM. (B right panel: median myeloid count) (C) Kaplan-Maier survival curve of p53+/-WASp+, p53+/-WASp−, p53+/-WASp – XLN, and p53+/+WASp+ mice. (p value: Gehan-Breslow-Wilcoxon test).
Mice were sacrificed upon detection of a palpable tumor, upon severe weight loss, signs of severe suffering, or when reaching the end point set at 24 weeks. Terminal events during the experiment were initially categorized by macroscopic features and light microscopy on H&E sections. The presence of a visible (sub) cutaneous tumor () with spindle shape cells showing frequent mitoses ( left panel) was classified as sarcoma (). Severely enlarged thymus ( middle panel) or splenomegaly with extensive lymphoid infiltration of liver ( right panel) and lymphoid organs was classified as lymphoma (). Six other cases of deaths showed signs of both lymphoid and myeloid proliferation () or there was no information to establish cause of death ().
Figure 2. Phenotypical features of malignancies in the p53+/- irradiation model. (A) Sarcomas (upper panel) were initially identified as visible tumors on exteriors. Lymphomas (lower panels) were recognized in animals showing severe weight loss, enlarged thymus or a combination of extreme splenomegaly and other microscopic features. T: tumor; Th: thymus; Sp: spleen; Liv: liver. (B) Hematoxylin-eosin stained tissue sections from sarcoma (#135A left panel) and lymphoma (#65B right panel). (C) Body weight before sacrificing in control (survived until endpoint) mice vs. mice with various pathology and (D) spleen weight at endpoint in control (survived until endpoint) mice vs. mice with various pathology. ANOVA and Bonferroni’s multiple comparison test. Mean±SD. (E) Cumulative % of tumor deaths was calculated by binning the frequencies of deaths into 4 weeks periods.
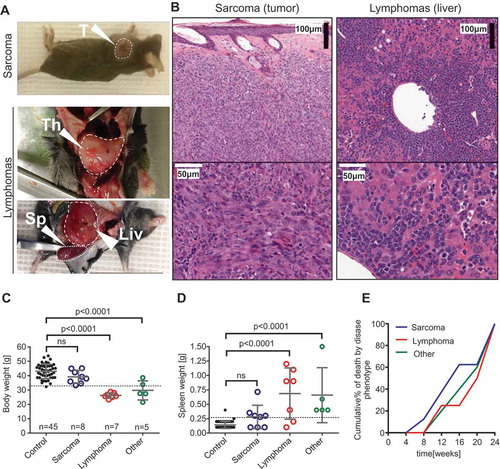
Examination of body weight revealed that mice with lymphoma and the uncharacterized cohort of mice showed marked weight loss shortly before death (). Mice with sarcomas maintained their normal body weights (). Similarly, mice with lymphoma and the uncharacterized cohort had splenomegaly while sarcoma mice had relatively small spleens at the endpoint (). Plotting deaths over time showed that sarcomas were rising earlier while lymphomas appeared later ().
Immune phenotyping showed that a large proportion (more than 50%) of spindle cells in sarcomas were positive for F4/80 (macrophage, myeloid lineage) (). This phenotype is consistent with histiocytic sarcoma, a frequent tumor in C57BL/6 mice that can be triggered with chronic low-dose irradiation.Citation27 FACS analysis of the tumor cell suspension indicated a large proportion of hematopoietic (CD45+) cells in the tumor mass, which mostly came from the myeloid lineage and contained neutrophils when the tumor was ulcerating ( and ).
Figure 3. Immuno phenotypical features of maligancies in p53+/- irradiation model. (A) F4/80 immunohistochemistry of subcutaneous tumor. (#135A) (B) FACS analysis of large ulcerating tumor. (#87A) (C) Immunohistochemistry of liver in lymphoma with the indicated antibodies. (#124A). (D) FACS analysis of thymus, liver, and spleen in mouse with thymic lymphoma. (#78A) (E) Immunohistochemistry of liver of mouse with severe mixed hematopoietic proliferation. (#44C). (F) FACS Liver (#114A).
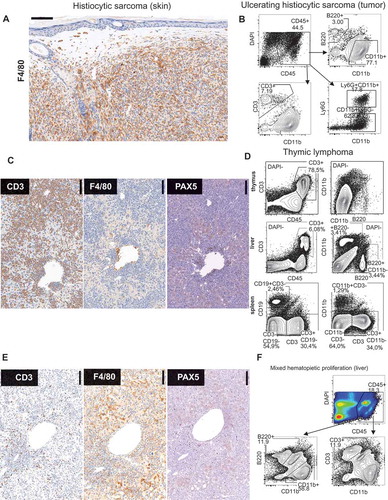
In the cases diagnosed as lymphomas, tumor cells dominating in lymphoid tissues and liver stained positive for the pan T cell marker CD3 (, ). Interestingly, spleens frequently contained lineage negative (CD3-CD19-CD11b-) cells with lymphocyte morphology possibly due to downregulation of specific surface markers on the neoplastic cells. The third group of mice showed mild (#117C and #111A) and severe (#44C, 114A) disease of mixed proliferation of hematopoietic cells. Mice #44C and #114A were sacrificed due to life threatening rapid weight loss. They exhibited scattered infiltration of leukocytes in liver mostly with myeloid but occasional lymphoid infiltrates with morphology suggestive of autonomous proliferation (, ). Mice #117C and # 111A were selected in the disease cohort due to splenomegaly and hematopoietic liver infiltrates. Immune cell clusters in these mice showed perivascular location, consistent with histological changes in aged liver that is associated with carcinogenesis.Citation28
When compiling data, we found that although the frequency of deaths was almost double in p53+/-WASp− mice compared to p53+/-WASp+ mice, the distribution of various cancer types was similar between all p53+/- mice (). One out of 9 irradiated WT (p53+/+WASp+) control mice developed histiocytic sarcoma. In contrast, lymphomas only occurred in p53+/- mice (). The death rate at 12 weeks was 10.6% of p53±/-WASp± mice, 28.6% of p53±/-WASp− mice (21.4% confirmed malignant), and 0% of p53±/-WASp-XLN mice and p53±/-WASp± mice (). The death rate of confirmed malignancies (, indicated in bold font) during 24 weeks was 35.7% of p53±/-WASp− mice and 25.0% of p53±/-WASp± mice. This suggests that the onset of tumor development differed between the groups. To compare the onset of deaths in each group, we calculated the survival time and confidence intervals at 10th and 20th survival percentiles (in each cohort corresponding to 90% and 80% live mice, respectively) from . We found that p53±/-WASp− mice had earlier onset of deaths when compared to p53±/-WASp± mice ( top panel). In contrast, p53±/-WASp-XLN mice had had later onset of deaths when compared to p53±/-WASp± mice ( bottom panel) in these survival percentiles.
Figure 4. Frequency of deaths in p53+/-WASp mutant tumor model. (A) Frequency of deaths per genotype. Bold text indicates malignant cause of death. (B) Time of death after irradiation. Causes of death are color coded as on Figure 4A. (C) Survival time and 95% confidence intervals at 10th and 20th survival percentiles was calculated from the survival curve with Laplace regression. (D, E) Killing of YAC-1-luciferase tumor cells by NK cells was quantified with chemiluminescence. Mean±SD; Two-way ANOVA (D) and ANOVA with Bonferroni’s multiple comparisons test (E). **: p ≤ 0.01, ***: p ≤ 0.001, ****: p ≤ 0.0001.
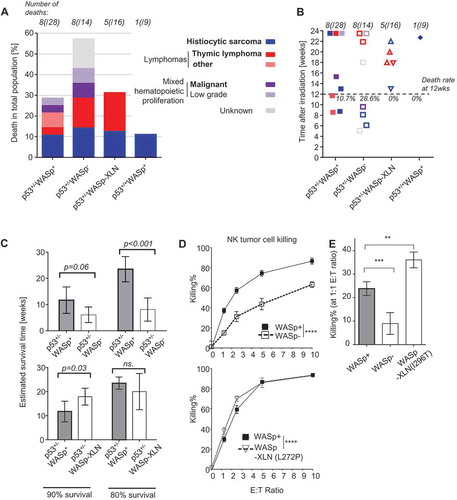
The finding that there was no dominance of a specific tumor in the different WASp genotypes, suggested that altered tumor surveillance due to WASp mutations may contribute to the tumor incidence. To test tumor cell killing, NK cells were incubated with YAC-1 lymphoma cells expressing luciferase and the loss of luciferase signal quantified. WASp deficient NK cells showed reduced killing of YAC-1 lymphoma cells when compared to wildtype NK cells (-) whereas WASp-XLN NK cells showed increased killing of YAC-1 lymphoma cells (-). Together, this data suggests that reduced tumor immune surveillance by WASp deficient NK cells contribute to poor survival of p53±/-WASp− mice and on the contrary, that overactive WASp in WASp-XLN NK cells lead to delayed tumor onset of p53±/-WASp-XLN mice.
Discussion
Together, we describe a spontaneous murine tumor model where sub-lethally irradiated p53+/-WASp mutant mice developed solid tumors, lymphoid malignancies, and other pathological conditions. Although predominant in WAS patients, no B cell lymphomas developed in the p53+/-WASp− colony. Targeted deletion of p53 gene expression results an earlier occurrence of thymic lymphomas and only a small percentage of p53-deficient mice succumb to B cell lymphomas.Citation29 If p53 is conditionally targeted in the B cell lineage, a very late onset of B cell lymphomas occurs and these lack immunoglobulin translocationsCitation29. Importantly, we show here that WASp deletion itself does not alter the T cell lymphoma propensity bias of p53 mutant mice toward B cell malignancies. This suggests that WAS patient lymphomas do not stem from intrinsic failure of WAS B cells to suppress tumor transformation but rather an external factor such as the overall tumor surveillance defect of the WASp deficient immune system. We show here that WASp deficient NK cells have reduced capacity to kill YAC-1 lymphoma cells. B cell lymphoma predominance in WAS patients is likely to originate from oncogenic transformation of EBV infection or long exposure – due to long lifespan – of human B cells to proliferation signals. Noticeably, p53+/-WASp− mice had a significantly decreased survival rate and they developed various early onset malignancies sooner than p53+/-WASp+ mice. It has previously been shown that WASp− mice bred to tumor susceptible Cdkn2a knockout background also show earlier death and poor rejection of injected B16 melanoma cells when compared to WASp+ mice.Citation30 Poor tumor survival and rejection capacity is at least partly caused by reduced tumor surveillance by WASp− NK cells and cytotoxic T cells as we and others have shown.Citation30-Citation34 It is therefore likely that poor tumor prognosis in p53+/-WASp− could be explained by aberrant tumor surveillance by WASp− cytotoxic cells.
Similarly to p53+/-WASp− mice that do not develop B cell lymphomas, p53+/-WASp-XLN mice do not develop AML. Severe congenital neutropenias (SCNs), such as XLN are considered pre-leukemic conditions due to bone marrow failure. Although the exact pathophysiology of XLN is not known yet it is likely that AML emerges in XLN patients due to chronic neutropenia and the occasional G-CSF treatment. XLN mutations have been suggested to cause chromosomal instability due to increased F-actin load in the cytoplasm and elevated cytoplasmic viscosity which mechanically disrupt proper cell division.Citation16 We found here that WASp-XLN myeloid cells proliferated extensively in vitro and that two XLN-WASp mice showed marked myeloid expansion at 2 weeks after irradiation that was quickly resolved. Despite the dysregulated myeloid proliferation, XLN-WASp had late onset tumors. This suggests the myeloid expansion do not lead to transformation in vivo using this model. Importantly, p53 only responds to particular types of genotoxic stress and our experiments suggest that chromosomal abnormalities emerging in XLN fail to synergize with p53 deletion .Citation29 In contrast to the p53+/-WASp− colony, the p53+/-WASp-XLN mice developed cancer mostly with late onset leading to delayed mortality. This implies that p53+/-WASp-XLN mice were more resistant against tumor development when compared to p53+/-WASp+ mice and that tumor immunosurveillance was not impaired in p53+/-WASp-XLN mice. In fact, WASp-XLN NK cells showed increased killing capacity of YAC-1 lymphoma cells.
WASp mutations compromise the immune system at multiple levels.Citation35 In our model, tumor development depends on the combination of genetic predisposition to DNA mutations by p53 deficiency, environmental stress by gamma radiation, and compromised immune system by WASp mutations. Our results help to understand to multifaceted nature of carcinogenic effects of WASp mutations.
Abbreviations
| SD | = | standard deviation |
| WAS | = | Wiskott-Aldrich syndrome |
| WASp | = | WAS protein |
| WT | = | wild type |
| XLN | = | X-linked neutropenia |
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was reported by the authors.
Acknowledgments
We thank Tarja Schröder and Agneta Andersson at the Karolinska Institutet core facility for Morphologic Phenotype Analysis to perform histotechnique and immunohistochemistry. We thank Andrea Discacciati at the Karolinska Institutet Biostatistics Core Facility for biostatistics consultation.
Supplemental Material
Download MS Power Point (66.4 KB)Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Mortaz E, Tabarsi P, Mansouri D, Khosravi A, Garssen J, Velayati A, Adcock IM. Cancers related to immunodeficiencies: update and perspectives. Front Immunol. 2016;7:365. doi:10.3389/fimmu.2016.00365.
- Imai K, Morio T, Zhu Y, Jin Y, Itoh S, Kajiwara M, Yata J, Mizutani S, Ochs HD, Nonoyama S. Clinical course of patients with WASP gene mutations. Blood. 2004;103:456–464. doi:10.1182/blood-2003-05-1480.
- Sullivan KE, Mullen CA, Blaese RM, Winkelstein JA. A multiinstitutional survey of the Wiskott-Aldrich syndrome. J Pediatr. 1994;125:876–885. doi:10.1016/S0022-3476(05)82002-5.
- Tran H, Nourse J, Hall S, Green M, Griffiths L, Gandhi MK. Immunodeficiency-associated lymphomas. Blood Rev. 2008;22:261–281. doi:10.1016/j.blre.2008.03.009.
- Senapati J, Devasia AJ, David S, Manipadam MT, Nair S, Jayandharan GR, George B. Diffuse large B cell lymphoma in wiskott-Aldrich syndrome: a case report and review of literature. Indian J Hematol Blood Transfus. 2014;30:309–313. doi:10.1007/s12288-014-0377-1.
- Du S, Scuderi R, Malicki DM, Willert J, Bastian J, Weidner N. Hodgkin’s and non-Hodgkin’s lymphomas occurring in two brothers with Wiskott-Aldrich syndrome and review of the literature. Pediatr Dev Pathol: Off J Soc Pediatr Pathol Paediatric Pathol Soc. 2011;14:64–70. doi:10.2350/10-01-0787-CR.1.
- Filipovich AH, Mathur A, Kamat D, Shapiro RS. Primary immunodeficiencies: genetic risk factors for lymphoma. Cancer Res. 1992;52:5465s–7s.
- Salavoura K, Kolialexi A, Tsangaris G, Mavrou A. Development of cancer in patients with primary immunodeficiencies. Anticancer Res. 2008;28:1263–1269.
- Worth AJ, Thrasher AJ. Current and emerging treatment options for Wiskott-Aldrich syndrome. Expert Rev Clin Immunol. 2015;11:1015–1032. doi:10.1586/1744666X.2015.1062366.
- Yoshimi A, Kamachi Y, Imai K, Watanabe N, Nakadate H, Kanazawa T, Ozono S, Kobayashi R, Yoshida M, Kobayashi C, et al. Wiskott-Aldrich syndrome presenting with a clinical picture mimicking juvenile myelomonocytic leukaemia. Pediatr Blood Cancer. 2013;60:836–841. doi:10.1002/pbc.v60.5.
- Beel K, Cotter MM, Blatny J, Bond J, Lucas G, Green F, Vanduppen V, Leung DW, Rooney S, Smith OP, et al. A large kindred with X-linked neutropenia with an I294T mutation of the Wiskott-Aldrich syndrome gene. Br J Haematol. 2009;144:120–126. doi:10.1111/bjh.2008.144.issue-1.
- Devriendt K, Kim AS, Mathijs G, Frints SG, Schwartz M, Van Den Oord JJ, Verhoef GEG, Boogaerts MA, Fryns J-P, You D, et al. Constitutively activating mutation in WASP causes X-linked severe congenital neutropenia. Nat Genet. 2001;27:313–317. doi:10.1038/85886.
- Ancliff PJ, Blundell MP, Cory GO, et al. Two novel activating mutations in the Wiskott-Aldrich syndrome protein result in congenital neutropenia. Blood. 2006;108:2182–2189. doi:10.1182/blood-2006-01-010249.
- Moulding DA, Blundell MP, Spiller DG, White MR, Cory GO, Calle Y, Kempski H, Sinclair J, Ancliff PJ, Kinnon C, et al. Unregulated actin polymerization by WASp causes defects of mitosis and cytokinesis in X-linked neutropenia. J Exp Med. 2007;204:2213–2224. doi:10.1084/jem.20062324.
- Westerberg LS, Meelu P, Baptista M, Eston MA, Adamovich DA, Cotta-de-Almeida V, Seed B, Rosen MK, Vandenberghe P, Thrasher AJ, et al. Activating WASP mutations associated with X-linked neutropenia result in enhanced actin polymerization, altered cytoskeletal responses, and genomic instability in lymphocytes. J Exp Med. 2010;207:1145–1152. doi:10.1084/jem.20091245.
- Moulding DA, Moeendarbary E, Valon L, Record J, Charras GT, Thrasher AJ. Excess F-actin mechanically impedes mitosis leading to cytokinesis failure in X-linked neutropenia by exceeding Aurora B kinase error correction capacity. Blood. 2012;120:3803–3811. doi:10.1182/blood-2012-03-419663.
- Coppe A, Nogara L, Pizzuto MS, Cani A, Cesaro S, Masetti R, Locatelli F, Te Kronnie G, Basso G, Bortoluzzi S, et al. Somatic mutations activating Wiskott-Aldrich syndrome protein concomitant with RAS pathway mutations in juvenile myelomonocytic leukemia patients. Hum Mutat. 2018;39:579–587. doi:10.1002/humu.23399.
- Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA Jr., Butel JS, Bradley A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215–221. doi:10.1038/356215a0.
- Kemp CJ, Wheldon T, Balmain A. p53-deficient mice are extremely susceptible to radiation-induced tumorigenesis. Nat Genet. 1994;8:66–69. doi:10.1038/ng0994-66.
- Bouffler SD, Kemp CJ, Balmain A, Cox R. Spontaneous and ionizing radiation-induced chromosomal abnormalities in p53-deficient mice. Cancer Res. 1995;55:3883–3889.
- Lee JM, Abrahamson JL, Kandel R, Donehower LA, Bernstein A. Susceptibility to radiation-carcinogenesis and accumulation of chromosomal breakage in p53 deficient mice. Oncogene. 1994;9:3731–3736.
- Zhao Z, Zuber J, Diaz-Flores E, Lintault L, Kogan SC, Shannon K, Lowe SW. p53 loss promotes acute myeloid leukemia by enabling aberrant self-renewal. Genes Dev. 2010;24:1389–1402. doi:10.1101/gad.1940710.
- Adams CM, Eischen CM. Inactivation of p53 is insufficient to allow B cells and B-cell lymphomas to survive without Dicer. Cancer Res. 2014;74:3923–3934. doi:10.1158/0008-5472.CAN-13-1866.
- Safran M, Kim WY, O’Connell F, Flippin L, Gunzler V, Horner JW, DePinho RA, Kaelin WG. Mouse model for noninvasive imaging of HIF prolyl hydroxylase activity: assessment of an oral agent that stimulates erythropoietin production. Proc Natl Acad Sci USA. 2006;103:105–110. doi:10.1073/pnas.0509459103.
- Gupta D, Shah HP, Malu K, Berliner N, Gaines P. Differentiation and characterization of myeloid cells. Curr Protoc Immunol. 2014;104:Unit 22F 5.
- Bottai M, Zhang J. Laplace regression with censored data. Biom J Biometrische Zeitschrift. 2010;52:487–503. doi:10.1002/bimj.200900310.
- Lacroix-Triki M, Lacoste-Collin L, Jozan S, Charlet JP, Caratero C, Courtade M. Histiocytic sarcoma in C57BL/6J female mice is associated with liver hematopoiesis: review of 41 cases. Toxicol Pathol. 2003;31:304–309. doi:10.1080/01926230390204342.
- Singh P, Coskun ZZ, Goode C, Dean A, Thompson-Snipes L, Darlington G. Lymphoid neogenesis and immune infiltration in aged liver. Hepatology (Baltimore, Md). 2008;47:1680–1690. doi:10.1002/hep.22224.
- Gostissa M, Bianco JM, Malkin DJ, Kutok JL, Rodig SJ, Morse HC 3rd, Bassing CH, Alt FW. Conditional inactivation of p53 in mature B cells promotes generation of nongerminal center-derived B-cell lymphomas. Proc Natl Acad Sci USA. 2013;110:2934–2939. doi:10.1073/pnas.1222570110.
- Catucci M, Zanoni I, Draghici E, Bosticardo M, Castiello MC, Venturini M, Cesana D, Montini E, Ponzoni M, Granucci F, et al. Wiskott-Aldrich syndrome protein deficiency in natural killer and dendritic cells affects antitumor immunity. Eur J Immunol. 2014;44:1039–1045. doi:10.1002/eji.201343935.
- Orange JS, Roy-Ghanta S, Mace EM, Maru S, Rak GD, Sanborn KB, Fasth A, Saltzman R, Paisley A, Monaco-Shawver L, et al. IL-2 induces a WAVE2-dependent pathway for actin reorganization that enables WASp-independent human NK cell function. J Clin Invest. 2011;121:1535–1548. doi:10.1172/JCI44862.
- Kritikou JS, Dahlberg CI, Baptista MA, Wagner AK, Banerjee PP, Gwalani LA, Poli C, Panda SK, Kärre K, Kaech SM, et al. IL-2 in the tumor microenvironment is necessary for Wiskott-Aldrich syndrome protein deficient NK cells to respond to tumors in vivo. Sci Rep. 2016;6:30636. doi:10.1038/srep30636.
- De Meester J, Calvez R, Valitutti S, Dupre L. The Wiskott-Aldrich syndrome protein regulates CTL cytotoxicity and is required for efficient killing of B cell lymphoma targets. J Leukoc Biol. 2010;88:1031–1040. doi:10.1189/jlb.0410197.
- Ishihara D, Dovas A, Hernandez L, Pozzuto M, Wyckoff J, Segall JE, Condeelis JS, Bresnick AR, Cox D. Wiskott-Aldrich syndrome protein regulates leukocyte-dependent breast cancer metastasis. Cell Rep. 2013;4:429–436. doi:10.1016/j.celrep.2013.07.007.
- Thrasher AJ, Burns SO. WASP: a key immunological multitasker. Nat Reviews Immunol. 2010;10:182–192. doi:10.1038/nri2724.