
ABSTRACT
Rationale: Regulatory T cells (Treg) play a pivotal role in the immunosuppressive tumor micro-environment in cancer, including mesothelioma. Recently, the combination of autologous tumor lysate-pulsed dendritic cells (DC) and metronomic cyclophosphamide (mCTX) was reported as a feasible and well-tolerated treatment in malignant pleural mesothelioma patients and further as a method to reduce circulating Tregs.
Objectives: The aim of this study was to establish the immunological effects of mCTX alone and in combination with DC-based immunotherapy on circulating Treg and other T cell subsets in mesothelioma patients.
Methods: Ten patients received mCTX and DC-based immunotherapy after chemotherapy (n = 5) or chemotherapy and debulking surgery (n = 5). Peripheral blood mononuclear cells before, during and after treatment were analyzed for various Treg and other lymphocyte subsets by flow cytometry.
Results: After one week treatment with mCTX, both activated FoxP3hi and naïve CD45RA+ Tregs were effectively decreased in all patients. In addition, a shift from naïve and central memory towards effector memory and effector T cells was observed. Survival analysis showed that overall Treg levels before treatment were not correlated with survival, however, nTreg levels before treatment were positively correlated with survival. After completion of mCTX and DC-based immunotherapy treatment, all cell subsets returned to baseline levels, except for the proportions of proliferating EM CD8 T cells, which increased.
Conclusions: mCTX treatment effectively reduced the proportions of circulating Tregs, both aTregs and nTregs, thereby favoring EM T cell subsets in mesothelioma patients. Interestingly, baseline levels of nTregs were positively correlated to overall survival upon complete treatment.
Introduction
Malignant pleural mesothelioma (MPM) is a rare but aggressive form of cancer most often caused by asbestos fiber inhalation. The incidence of MPM is rising,Citation1,Citation2 and the prognosis is infaust; with the best standard of care, antifolate and platinum combination chemotherapy, overall survival is 13.3 months.Citation3 In the last ten years no major breakthroughs have been reported and consequently, this systemic therapy has remained unchanged.Citation3 Recently, addition of bevacizumab to the chemotherapy-backbone showed a positive effect on survival.Citation4 Extensive investigation of the effects of implementation of radiotherapy and/or debulking surgery in standard treatment revealed variable success, but only when applied to select patient subgroups.Citation1,Citation5–Citation7
Complementary to the current standard anti-cancer treatment options, immunotherapy is gaining momentum.Citation8–Citation10 The potential of cancer immunotherapy lies in the ability of the immune system to recognize tumor cells, without harming healthy tissue. There are various methods to either induce or enhance an anti-tumor immune response, including adoptive transfer of immune cells, peptide or tumor cell vaccines and immune checkpoint blockade.Citation10 Recently, there have been major leaps in development of blocking the immunosuppressive tumor microenvironment (TME) by checkpoint inhibitors.Citation11–Citation15 Importantly, for immunotherapy, including checkpoint inhibition, to become successful, tumor recognition by the immune system is necessary.Citation16 For an effective anti-tumor immune response, both a CD4 and CD8 T cell response is required, which can be enhanced by immune checkpoint blockade.Citation17 By loading them ex vivo with tumor antigens, they can be used as cellular immune therapy. DC-based immunotherapy is, in contrast to other immunotherapies including adoptive T cell transfer and peptide-based vaccines, not human lymphocyte antigen (HLA)-restricted and can induce an immune response to a wide array of antigens. In a recent meta-analysis, it was shown that cellular immunotherapy seems to be more effective than tumor vaccines in non-small cell lung carcinoma (NSCLC).Citation18 Furthermore, in an earlier phase I clinical trial with MPM patients DC-based immunotherapy, in which DCs were loaded with autologous tumor lysate, has been proven safe, feasible and capable of inducing an anti-tumor response, which was detectable in peripheral blood of patients.Citation19
Aside from inhibitory receptor expression, efficacy of immunotherapy can also be hampered by the immunosuppressive TME induced by the tumor.Citation20 In particular, the tumor affects regulatory T cell (Treg) function, quenches pro-inflammatory signals and inhibits antigen presentation,Citation21,Citation22 all of which ultimately prevent successful execution of antitumor immune responses. As illustrated by the study of Bjoern et al.,Citation23 melanoma patients treated with DC vaccination and low-dose interleukin 2 (IL-2) that progressed under this therapy had significantly higher levels of CD25high CD4 T cells than patients with stable disease. Miyara et al.Citation24 have shown that the classically defined Treg population of FoxP3+ CD4 T cells, comprise three functionally different subpopulations: suppressive naïve Tregs (nTreg; CD45RA+FoxP3med), activated Tregs (aTreg; CD45RA−FoxP3hi) and the cytokine-secreting activated T cells (aTcell; CD45RA−FoxP3med). Santegoets et al.Citation25 showed that the frequency of aTregs and proliferating Ki67+ FoxP3+CD25+CD127low Tregs prior to treatment were associated with worse survival in recurrent ovarium carcinoma patients undergoing chemo-immunotherapeutic treatment, whilst frequencies of classically defined Tregs prior to treatment were not associated with survival. In mesothelioma, Tregs contribute to an impaired T cell functionCitation26,Citation27 and are associated with tumor progression and poor prognosis.Citation28 Low-dose (metronomic) cyclophosphamide (mCTX) regimens have beneficial immunomodulatory effects by inducing Treg apoptosis or by reducing their functionality.Citation29–Citation32 In mice we have previously shown that mCTX induced beneficial immunomodulatory effects, by decreasing the Tregs numbers and thereby improving CD8 T cell function.Citation33 It is unknown what the effects of debulking surgery and mCTX are on the different subpopulations of Tregs.
To improve DC-based immunotherapy, the immunosuppressive TME, specifically Tregs, was targeted by mCTX to the treatment in a phase I/II clinical trial.Citation34 This therapy has also been proven safe, feasible and moreover, effective in depleting Tregs. Radiographic disease control was obtained in 8 out of 10 patients and the median overall survival was 26 months.Citation34
The aim of this study was to determine whether mCTX treatment, has beneficial effects on subpopulations of circulating Tregs or other peripheral blood mononuclear cell subsets that could explain the enhanced survival observed in MPM patients treated with DC/mCTX-based immunotherapy. To this end, an in depth immunological analysis was performed on peripheral blood of patients included in a phase I/II clinical trial.Citation34
Results
Patient characteristics and toxicity
Ten patients with MPM suitable for extended pleurectomy/decortication (P/D) and a stable disease or response after an antifolate-based regimen of chemotherapy were enrolled in this study between August 2009 and October 2011. The DC/mCTX treatment was preceded by P/D in five of the ten patients (); all patients completed the full treatment schedule and were available for immunological analysis. Patient characteristics, safety and toxicity data, as well as clinical response were previously reported.Citation34 There was no significant difference in survival between patients that did or did not undergo the P/D (data not shown). To establish the effect of P/D on T cells, peripheral blood mononuclear cells (PBMC) obtained at t = 0 were compared between the P/D group and the no P/D group by flow cytometry. The gating strategy for characterizing nTregs and aTregs using CD45RA and FoxP3, as well as the differentiation status for CD4 and CD8 T cell subpopulations, using CD45RA and CCR7 to distinguish between naïve (CD45RA+CCR7+), central memory (CM; CD45RA−CCR7+), effector memory (EM; CD45RA−CCR7−) and effector (EMRA; CD45RA+CCR7−) was performed according Supplementary Figure S1.Citation24
Figure 1. Schematic overview of the clinical trial. Scheme of clinical trial. Patients were included if they had partial response or stable disease after pemetrexed-based chemotherapy. Five patients underwent additional P/D 7–15 weeks after chemotherapy. DC/mCTX therapy started 10–17 weeks after either the last chemotherapeutic treatment or P/D. Blood samples were obtained at t = 0 (baseline); t = 2 (mCTX); t = 4; t = 6; t = 8 (2 wk after DC/mCTX therapy); t = 18.
Within the circulating T cell compartment, there is a trend to an increase in T cells and a decrease in monocytes in P/D patients, however these changes were not significant, and neither were changes in other T cell subsets, including Tregs (Supplementary Figure S2A and S2B). In addition, no significant differences were found in the proportions of total CD4 and CD8 T cells, B cells, natural killer (NK) cells, NK T cells, γδ T cells and monocytes and IFNγ-producing or Granzyme B (GrB) containing T cells (Supplementary Figure S2C).
Thus, in peripheral blood of the P/D group all measured circulating immune subsets were comparable to mesothelioma patients without debulking surgery and for further analyses data from P/D and no P/D patients were pooled.
mCTX treatment affected both aTregs and nTregs, while increasing effector memory populations
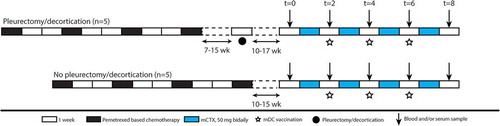
To determine the effect of mCTX on circulating Tregs, and other T cells subsets, PBMCs obtained at t = 0 and t = 2 weeks were analyzed by flow cytometry. Compared with nTregs, the aTregs showed higher expression of CCR4, CTLA-4 and Ki67, confirming their active and immunosuppressive state (Supplementary Figure S3A).
After two weeks (with one week of mCTX treatment; ), total T cells and CD4 T cells decreased upon mCTX treatment, whereas CD8 T cells increased and both the proportions of nTregs and aTregs (as percentage of total CD4 T cells) were significantly decreased (-). Within the FoxP3− CD4 T cells and CD8 T cells the naïve and central memory subsets decreased, while the effector subsets increased (-). The percentages of proliferating FoxP3−CD4 T cells significantly increased in all subsets, except for the TEMRA subset (). In the circulating CD8 T cells an increase in proliferation was observed in all subsets, except for the CM subset (). In addition, even though the percentage of both Treg populations decreased, the percentage of proliferating nTregs increased upon treatment with mCTX, while the percentage of proliferating aTregs did not change (). Also the CTLA4 expression increased in the nTregs, however, in the aTregs the expression of CTLA4 decreased (). The proportions of IFNγ-producing and GrB-containing CD4 and CD8 T cells seemed to increase, although nog significantly (). Correlation analysis indicated that the change IFNγ-producing and GrB-containing CD4 T cells and the GrB-containing CD8 T cells induced by mCTX might inversely correlate with the change in Tregs (Supplementary Figures S4), however, this correlation was not significant. The proportions of B cells, NK cells, NKT cells and monocytes (as percentage of total PBMCs), did not change upon mCTX treatment, the proportions of γδ T cells slightly increased (Supplementary Figure S5).
Figure 2. Both aTregs and nTregs, and other naïve cell subsets decreased upon mCTX administration, meanwhile the percentages of proliferating T cells increased. To determine the effect of mCTX administration on activated and naïve Tregs, and other T cell populations, flowcytometric analysis was performed on PBMCs obtained at t = 0 and t = 2, and thereby comparing baseline proportions with the proportions after mCTX administration. To determine IFN·-production, T cells were stimulated 4hrs with PMA/ionomycin in the presence of monensin. A. The proportion of CD3 T cells decreased (41.14%±5.80 to 27.5%±5.0) significantly upon mCTX treatment, as did CD4 T cells (40.99%±6.81 to 21.28%±4.08). The percentage of CD8 T cells increased (54.73%±6.91 to 74.88%±4.28) significantly. B. Both percentages of naïve and activated Tregs decreased significantly from 1.97%±0.40 to 0.86%±0.17, and from 3.23%±0.87 to 1.52%±0.36 respectively. C. The percentages of naïve (28.12%±6.43 to 7.39%±2.21), CM (15.27%±2.41 to 7.74%±2.35) and activated (6.60%±1.58 to 3.64%±0.73) CD4 T cells decreased significantly, while the percentages of EM (38.25%±5.40 to 56.39%±4.85) and TEMRA (6.38%±0.85 to 21.89%±3.91) CD4 T cells increased significantly upon mCTX treatment. D. The percentages of naïve (12.49%±4.41 to 2.62%±1.87) and CM (1.73%±0.57 to 0.35%±0.13) CD8 T cells decreased significantly, while the percentages of TEMRA (57.6%±5.94 to 71.84%±5.67) CD8 T cells increased significantly upon mCTX treatment. The percentage of EM CD8 T cells did not change (28.19%±3.83 vs 25.19%±5.43). E. Upon treatment with mCTX the percentages of proliferating CD4 T cells increased in the naïve (1.50%±0.34 to 14.4%±4.58), CM (4.12%±0.67 to 11.76%±2.61), EM (7.21%±1.14 to 16.22%±4.63) and activated (11.12%±1.15 to 22.99%±3.17) CD4 T cells subset, but not in the TEMRA (10.23%±3.75 vs 25.22%±6.37) subset. The percentage of proliferating nTregs increased (5.89%±1.48 to 37.80%±9.27), but not of aTregs (33.74%±3.11 vs 46.3%±9.77). In CD8 T cells, the naïve (2.13%±1.15 to 10.07%±2.16), EM (5.71%±1.30 to 13.51%±2.95) and TEMRA (3.28%±0.76 to 12.82%±3.42) cells had increased proliferatioin, but not the CM (4.23%±0.88 vs 11.71%±4.11) subset. F. CTLA4 expression in nTegs and aTregs. The dashed line represents the MFI (mean fluorescence intensity) of CTLA4 in a healthy individual. The MFI of CTLA4 increased significantly in nTregs and decreased significantly in aTregs upon treatment with mCTX. G. The proportions of IFN·-producing CD4 (16.99%±3.83 vs 40.15%±7.05) and CD8 (41.17%±6.77 vs 63.57%±6.04) T cells did not change significantly, neither did the percentage of GrB+ CD4 (11.26%±3.55 vs 34.61%±8.17) and CD8 (45.33%±7.25 vs 69.24%±10.01) T cells. Results represent mean ± Standard Error of the Mean (SEM). *p < 0.05, **p > 0.01 (Wilcoxon matched-pairs signed rank test), differences were considered significant when p < 0.05.
From these findings, we conclude that mCTX effectively reduced the proportions of both nTregs and aTregs within the CD4 T cell population, with a decreased CTLA4 expression in aTregs and an increased expression in nTregs. In addition, within the total population of T cells the proportions of CD4 T cells decreased and CD8 T cells increased, a shift was observed from the naïve and CM subsets to the EM and TEMRA subsets and the majority of circulating CD4 and CD8 T cell subsets had an increased proliferation.
DC/mCTX-based immunotherapy increased proliferation of central memory CD8 T cells
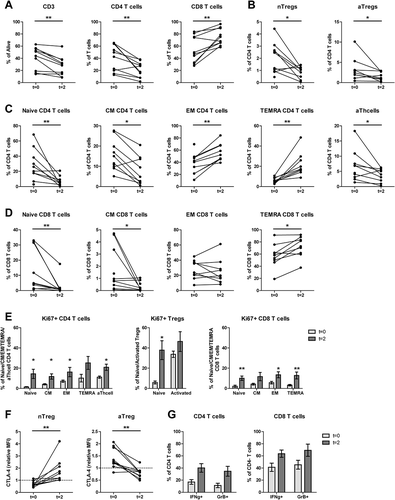
To examine the effect of combined DC/mCTX-based immunotherapy on T cells, flowcytometric analysis of PBMCs obtained at t = 0 were compared with those of t = 8, corresponding to the time point after completion of DC/mCTX-based immunotherapy ().
Whereas after one week of mCTX percentages of total T cells and CD4 T cells were decreased and CD8 T cells were increased, these percentages returned to baseline after the complete treatment (). Also, Treg levels and all differentiated T cell subsets (-) returned back to their levels before therapy. At t = 8, also the percentages of CD4 T cell subsets, and proliferating nTregs were comparable to baseline, nevertheless, the percentages of proliferating CM CD8 T cells were significantly increased and the TEMRA CD8 T cell population showed a trend towards an increase (). The CTLA4 expression in both nTregs and aTregs were comparable to baseline (), as were the percentages of IFNγ-producing and GrB-containing CD4 and CD8 T cells () and the proportions of B cell, NK cell, NKT cell, γδ T cell and monocytes (from total PBMC) (Supplementary Figure S6).
Figure 3. After completion of DC/mCTX-based immunotherapy, all Treg and other T cell populations were returned to baseline levels, the percentage of proliferating CM CD8 T was increased compared to baseline. To determine the effects of DC/mCTX-based immunotherapy, flowcytometric analysis of PBMCs obtained at t = 0 (baseline) and t = 8 (after completing DC/mCTX-based immunotherapy) were compared. To determine IFN·-production, T cells were stimulated 4hrs with PMA/ionomycin in the presence of monensin. A. The proportion of CD3 (41.14%±5.80 vs 39.24%±5.332), CD4 (40.99%±6.81 vs 34.23%±5.38) and CD8 (54.73%±6.91 vs 60.82%±5.67) T cells after DC/mCTX-based immunotherapy were comparable to baseline. B. The percentages of nTregs (1.97%±0.40 vs 1.60%±0.32) and aTregs (3.23%±0.87 vs 3.92%±0.89) did not change significantly. C. The percentages of naïve (28.12%±6.43 vs 20.24%±4.34), CM (15.27%±2.41 vs 13.97%±1.80), EM (38.25%±5.40 vs 45.68%±4010), TEMRA (6.38%±0.85 vs 8.82%±1.53) and activated (6.60%±1.58 vs 5.78%±1.21) CD4 T cells did not change significantly D. Neither did the different subsets in CD8 T cells; naïve (12.49%±4.41 vs 8.19%±3.17), CM (1.73%±0.57 vs 1.29%±0.44), EM (28.19%±3.83 vs 31.42%±4.55) and TEMRA (57.6%±5.94 to 59.12%±5.93). E. Upon treatment with DC/mCTX-based immunotherapy the percentages of proliferating CD4 T cells did not change in the different subsets; naïve (1.50%±0.34 vs 1.73%±0.48), CM (4.12%±0.67 vs 4.77%±0.58), EM (7.21%±1.14 vs 9.21%±1.39), TEMRA (10.23%±3.75 vs 9.75%±3.06) and activated (11.12%±1.15 vs 12.69%±1.30). Neither did the proportion of proliferating nTregs (5.89%±1.48 vs 7.54%±2.04) and aTregs (33.74%±3.11 vs 35.9%±2.55). In CD8 T cells the proportion of proliferating cells was higher in the CM (4.23%±0.88 vs 7.19%±1.50) subset and there was a trend towards more proliferating TEMRA (3.28%±0.76 to 4.98%±1.29). In the naïve (2.13%±1.15 to 3.07%±1.35) and EM (5.71%±1.30 to 6.93%±1.96) subset the proportion of proliferating cells was equal before and after DC/mCTX-based immunotherapy. F. The proportions of IFN·-producing CD4 (16.99%±3.83 vs 15.69%±3.17) and CD8 (41.17%±6.77 vs 34.39%±7.67) T cells did not change significantly, neither did the percentage of GrB+ CD4 (11.26%±3.55 vs 17.41%±6.37) and CD8 (45.33%±7.25 vs 49.31%±10.11) T cells. Results represent mean ± Standard Error of the Mean (SEM). *p < 0.05 (Wilcoxon matched-pairs signed rank test), differences were considered significant when p < 0.05.
In conclusion, after completed DC/mCTX-based immunotherapy at t = 8, all immune cell subsets, including Tregs, returned to baseline. Only the CM CD8 T cell subset showed an increased proportion of proliferating cells compared to baseline.
Correlation of pre-treatment proportions of Treg subsets with overall survival
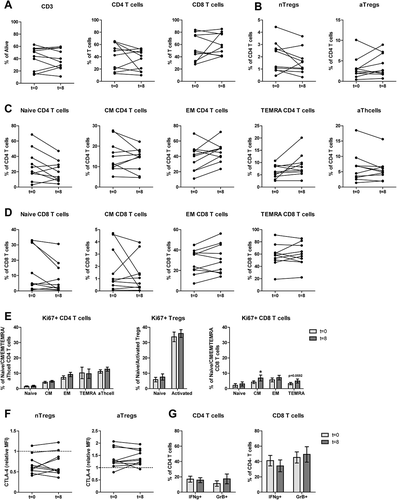
To determine whether the proportions of Tregs before treatment (at t = 0) were correlated with survival in MPM patients treated with DC/mCTX-based immunotherapy, linear regression analyses were performed. The pre-treatment percentage of total Tregs (nTregs and aTregs), did not show a correlation with survival (). Subsequently, the same analysis was performed for aTregs and nTregs separately. Interestingly, the pre-treatment percentage of nTregs correlated positively with survival, whereas the percentage of aTregs did not show any correlation (). On the basis of the positive correlation of nTregs and overall survival, two patient clusters could be distinguished: six patients with low proportions of nTregs and a low overall survival, and four patients with high pre-treatment nTreg proportions and a relatively high overall survival. For these groups – patients with a pre-treatment nTreg percentage below 2% of total CD4 T cells (n = 5; range 0.46%-1.19%) and higher than 2% of CD4 T cells (n = 5; range 2.46%-4.43%) – a survival analysis was performed. This confirmed that proportions of nTregs above 2% of total CD4 T cells were associated with a higher overall survival ().
Figure 4. Pretreatment frequencies of nTregs correlated with overall survival in mesothelioma patients treated with DC/mCTX-based immunotherapy. A. To determine whether pretreatment frequencies of total Tregs (the percentage of nTregs and aTregs of total CD4 T cells), aTregs or nTregscorrelated with survival in mesothelioma patients treated with DC/mCTX-based immunotherapy, linear regression was performed. No significant correlation was observed between total Tregs (A, left) or aTregs (A, middle) and survival. Linear regression showed a significant positive correlation between pretreatment nTreg frequencies and survival (A, left). B. To determine whether patients with lower pretreatment nTreg frequencies (below 2% of total CD4 T cells, n = 5) had a different survival from patients with higher nTreg frequencies (above 2% of total CD4 T cells, n = 5), survival analysis was performed. Patients with a nTreg percentage above 2% of CD4 T cells, had a better overall survival. Statistical analysis was performed by log-rank (Mantel-Cox) testing, and differences were considered significant when p < 0.05.
Thus, we concluded that in mesothelioma patients treated with DC/mCTX-based immunotherapy, the pretreatment percentage of circulating nTregs had a positive correlation with overall survival.
Discussion
The primary aims of this study were to determine the immunological effects of mCTX and the effects of DC/mCTX based therapy specifically on subpopulations of Tregs and other immune cell subsets in peripheral blood of mesothelioma patients. Patients with partial response (PR) or stable disease (SD) after standard chemotherapeutic treatment were included in this study. As we have previously shown,Citation34 mCTX effectively reduced the number of FoxP3+CD25+CD127low CD4 T cells. In this study, we showed that both the nTreg and aTreg subsets were reduced by mCTX treatment, and that pre-treatment proportions of nTregs positively correlated with survival. P/D had no effect on nTregs or aTregs, nor on other circulating lymphocyte and monocyte subsets. Total Tregs and aTregs were not correlated with survival. At t = 2, after mCTX treatment, FoxP3− CD4 T cells were also decreased in quantity, while CD8 T cells increased and a shift from naïve to effector T cell populations was observed. Other lymphocyte and monocyte subsets were not affected. At t = 8, after completion of DC/mCTX therapy, all examined cell subsets returned to the patients’ baseline levels before the therapy, with the exception of proliferating CM CD8 T cells. We found an increase in the proportions of these cells, which were not increased by mCTX treatment alone, indicating that these CD8 T cells started proliferating upon the DC vaccinations.
A key factor for inducing an effective immune response is an immune-stimulatory environment. In mesothelioma, Tregs are a major contributor to creating an immunosuppressive environment.Citation26,Citation27 To reduce the number of Tregs in mesothelioma patients, debulking surgery and mCTX administration were investigated. It is hypothesized that debulking surgery can reduce Tregs locally by decreasing the tumor load and mCTX systemically by directly targeting Tregs.Citation30 In NSCLC, circulating Treg levels in thoracotomy patients were reduced up until postoperative day (POD) 30 and had normalized at POD 90.Citation35 In another study in NSCLC patients, circulating Treg levels were also reduced 1–3 months after pneumectomy or lobectomy.Citation36 In contrast to these studies, in our study the Treg levels in blood were not significantly different between the P/D and no P/D groups three months after surgery. However, we analyzed only a limited number of patients, we could not study the effect of P/D on Tregs within the TME, preoperative data were not available and the P/D patients were at least three months after P/D. This could indicate that mCTX might be an effective treatment strategy to reduce Tregs in both of these groups.
Administration of mCTX transiently depleted Tregs, as has been shown before.Citation30 However, to the best of our knowledge, we are the first to show that both naïve and activated Tregs are depleted. In addition, upon mCTX the CTLA4 expression was reduced specifically on aTregs. Conflicting results have been published about the effect of mCTX on the suppressive capacity of Tregs; in metastasized breast cancer patients 50 mg cyclophosphamide daily for three months resulted in an initial Treg reduction but a preservation of their suppressive function.Citation37 Another study in end stage cancer patients treated with 50 mg cyclophosphamide twice daily, one week on and one week off for one month, also found a selective reduction of Tregs, but also a suppression of their inhibitory functions.Citation30 Since we observed a downregulation of CTLA4 in aTregs, but an upregulation in nTregs, these inconsistent results could be explained by a subset dependent effect of mCTX. Another explanation could be the dosing schedule of cyclophosphamide,Citation38 in a murine model cyclophosphamide treatments with drug free intervals of 6, 9 and 12 days were tested and only the 6 day drug free interval showed induction of tumor specific CD8 T cells. It is hypothesized that if the interval is too short, activated CD8 T cells and NK cells can also be depleted, but if the interval is too long, the cells could acquire drug resistance and the therapy would lose its effect.Citation39
Complementary to the decrease of CTLA4 in aTregs, and indicative of reduced immune suppression, we detected a shift from naïve and CM subsets towards effector memory and effector subsets. The previously mentioned clinical studies by Ghiringhelli, et al.Citation30 and Ge, at al.,Citation37 also described an increase in effector T cells upon mCTX treatment. In animal models skewing towards a Th1 profile with increased type I interferons and IL-2 upon mCTX treatment was observed,Citation40,Citation41 which could correspond to the increase in IFNγ+ CD4 and CD8 T cells observed in this study. The IL-2 secretion by Th1 cells could induce proliferative expansion of CD8 T cells,Citation41 which could have led to the increased effector T cells. In addition, these cells could be enhanced by the decrease of Treg mediated immune suppression.Citation39 And lastly, a cytokine storm and thereafter homeostatic proliferation, could be caused by the lympho-depletion due to the mCTX treatment.Citation42 This would be in concordance with the increased proportion of proliferating naïve Tregs, CD4 T cells and CD8 T cells. No change was observed in the proportions of B cells, NK cells, γδ T cells and monocytes. Comparable results have been reported in other studies.Citation37,Citation43
The analysis of Tregs showed that pretreatment circulating Treg levels did not correlate with survival when patients were treated with DC/mCTX based immunotherapy. In fact, the patient with the highest proportion of both nTregs and aTregs had a survival of more than 6 years after diagnosis and is still alive at the time of writing. Survival analysis and correlation of pretreatment percentages of the two Treg subsets with overall survival rate, showed that patients with higher percentages of nTregs had a better survival. nTregs differentiate into aTregs upon T-cell receptor stimulation by antigen recognition,Citation24,Citation44,Citation45 which could imply that patients having a relatively high percentage of peripheral nTregs have less tumor-specific Tregs. Due to their naïve phenotype, these nTregs are inefficient in infiltrating the tumor,Citation46 as is also illustrated by their low CCR4 expression, and thus these cells cannot exert immunosuppressive activity. Therefore, immunotherapy might be more effective in these patients, which is a possible explanation for this counterintuitive finding. Moreover, Treg diversity, including the pool of nTregs, is controlled by homeostasis,Citation44 thus having a higher percentage of nTregs might indicate a healthier immune system.
However, from this study alone we cannot deduce whether patients with a higher percentage of nTregs have a better survival due to the mCTX or the DC-based immunotherapy, the combination therapy or have an initial better survival. In contrast to our study, Kwa et al,Citation47 found that elevated baseline levels of nTregs were a negative predictive factor for survival in metastatic breast cancer patients treated with exemestane and mCTX. However, Kwa et al used a different definition of nTregs (CD4+CD45RO-FoxP3+Helios+) and the mCTX treatment was combined with hormone therapy instead of immunotherapy, which might have resulted in a different outcome. In addition, they did not establish an effect of mCTX alone on either memory or naïve Tregs, so it cannot be excluded that the observed effects were caused by the combination of mCTX and hormone therapy, which possibly increases Tregs and their function.Citation48
In light of the recent developments in the tumor immunology field, the approved checkpoint inhibitors, against CTLA-4 or PD-(L)1,Citation15,Citation49,Citation50 or anti-CCR4 antibodies to inhibit aTregs,Citation51,Citation52 could be interesting methods to reduce the immunosuppressive TME as a synergistic addition to DC-based immunotherapy in mesothelioma, instead of or complementary to surgery and mCTX.
Our study has several limitations. First, to make the autologous tumor lysate used to pulse the DCs with, in the non-P/D group only patients that had sufficient amounts of tumor cells in the pleural fluid were included. For the P/D group, patients had to be fit enough to be able to undergo surgery. Both of these factors might have caused a selection bias. In addition, this study was exploratory and only ten patients were enrolled in this study, which might not be enough to objectify smaller differences and establish significant results and thus larger patient groups are needed to validate findings in this study. For example, the positive correlation between higher pretreatment levels of nTregs and overall survival should be validated in a larger patient cohort.
In summary, in this small patient cohort DC/mCTX-based immunotherapy in mesothelioma patients seems to improve survival;Citation34 this therapy simultaneously countered tumor-induced immune suppression and induced a distinct adaptive immune response. Based on these results and the improved overall survival compared to DC-based immunotherapy alone,Citation19 mCTX seems to add to solely DC-based immunotherapy in mesothelioma patients with stable disease after the standard chemotherapy regimen, and seems to specifically benefit patients with a high pretreatment level of nTregs. It would be very interesting to explore synergistic therapies to reduce immunosuppression, such as checkpoint inhibitors, to complement DC/mCTX-based immunotherapy.
Materials & Methods
Study design
The institutional ethical committee of the Erasmus MC (MEC-2008–109) and the Central Committee on Research involving Human Subjects (CCMO; NL24050-000–08) as defined by the WMO (Medical Research Involving Human Subjects Act) approved the phase I study.Citation34 Procedures followed were in accordance with the ethical standards of these committees on human experimentation and with the Helsinki Declaration of 1975. The study is registered at http://www.clinicaltrials.gov with identifier NCT01241682.
Patients and treatment
An extensive description of the patient eligibility and treatment is given by Cornelissen et al.Citation34 In short, patients with mesothelioma suitable for P/D and partial response (PR) or stable disease (SD) after standard chemotherapeutic treatment were included. Before inclusion a delayed type hypersensitivity (DTH) test with tetanus toxoid as a positive control and saline as a negative was performed to confirm immunological competence. DC-based immunotherapy in combination with mCTX was planned 8 to 10 weeks after completion of chemotherapy (n = 5) or chemotherapy and P/D (n = 5). Patients received at least three vaccinations consisting of 50 × 106 mature DC (mDC) pulsed with autologous tumor lysate with a 2-week interval, every immunization one-third of the dosage was administered intradermally in the forearm and two-thirds was administered intravenously. Patients were treated with 50 mg tablet twice daily of CTX (Endoxan; Baxter B.V., Utrecht, The Netherlands) for a week, followed by a week interval in which the vaccination was administered, starting a week before the first vaccination and ending one week after the third vaccination. The treatment schedule is depicted in .
Survival data were determined on March 1st, 2018. Blood and serum samples were obtained before immunotherapy treatment initiation (t = 0), just before administration of the vaccinations (t = 2, t = 4 and t = 6), two weeks after the third vaccination (t = 8), and three months after the third vaccination (t = 18), as illustrated in . Peripheral blood mononuclear cells (PBMC) were isolated using Ficoll density gradient centrifugation, cryopreserved in 50% RPMI-1640 medium (Gibco Life Technologies), 40% Fetal Calf Serum (FCS),and 10% DMSO and stored in liquid nitrogen until use. Serum samples were stored at −80°C until use.
Immunological evaluation
Cellular immune response upon DC/mCTX therapy
Flowcytometric analysis
Flow cytometric analyses of Tregs was based on markers that differentiate between activated and naïve Tregs, as previously described by Miyara et al.Citation24 and Santegoets, et al..Citation25 Cryopreserved PBMCs were thawed and washed twice in cold PBS. Dead cells were stained using LIVE/DEAD Fixable Aqua Dead Cell Stain (Invitrogen life technologies, Cat# L34957). Antibodies used in stainings are specified in Supplementary Table S1 (“Treg” and “Immune Subsets (IS)” panel). The eBioscience FoxP3/Transcription factor staining buffer kit (eBioscience, Cat# 00–5521-00) was used for fixation and permeabilization of cells in the Treg panel for detection of FoxP3, Ki67 and CTLA-4, cells in the IS panel were not fixated and permeabilized. Cells were measured on the LSR-II flow cytometer (BD Biosciences) and analyzed using FlowJo software (version 10.1r5, FlowJo). All populations with less than 100 cells were excluded for further analysis (Ki67 and CTLA4 expression and CD4 T cell differentiation). Since experiments were performed on several days, expression of CTLA-4 in nTregs and aTregs ( and ) was normalized to the expression of CTLA-4 in those respective populations in a healthy individual that was included in every experiment, the dashed line in the figures represents the MFI of CTLA-4 in the nTreg (left panel) and aTreg (right panel) population of the healthy individual.
Effector t cell responses
The cryopreserved PBMCs obtained at t = 0 (all patients), t = 2 (4 patients; 2–3 and 9–10), t = 4 (all patients), t = 6 (9 patients; 1–7 and 9–10) and t = 8 (7 patients; 2, 4–8 and 10) were thawed and per sample 1 × 106 PBMCs were stimulated with phorbol myristate acetate (PMA; Sigma-Aldrich) and ionomycin (Sigma-Aldrich) in the presence of Golgistop (BD Biosciences) in RPMI supplemented with 10% pooled human AB serum (Human Culture Medium; HCM) at 37°C for 4 hours. Following the stimulation, cells were stained using LIVE/DEAD Fixable Aqua Dead Cell Stain and antibodies that are specified in Supplementary Table S1 (“Cytokines” panel). IFNγ and GrB were detected following fixation and permeabilization using 2% paraformaldehyde (PFA) in PBS and subsequently 0.5% saponin in PBS. Cells were measured on the LSR-II flow cytometer (BD Biosciences) and analyzed using FlowJo software (version 10.1r5, FlowJo).
Statistical analysis
Statistical calculations were performed using GraphPad Prism (version 6.0c for Mac OS X, GraphPad Software, La Jolla California USA). For unpaired samples, the Mann Whitney test was used and for paired samples the Wilcoxon matched paired test was used, as indicated in the figures. There was no correction performed for multiple testing. For survival analysis the Mantel-Cox log-rank test was used. Statistical significance was established at the p < 0.05 level, and analysis was two-sided.
Abbreviations
Activated regulatory T cell (aTreg), central memory (CM), dendritic cell (DC), delayed type hypersensitivity (DTH), effector T cell (TEMRA), effector memory (EM), Granzyme B (GrB), Human Culture Medium (HCM; RPMI supplemented with 10% pooled human AB serum), interferon gamma (IFNγ), metronomic cyclophosphamide (mCTX), mature dendritic cell (mDC), major histocompability complex (MHC), malignant pleural mesothelioma (MPM), natural killer cell (NK cell), natural killer T cell (NKT cells), non-small cell lung cancer (NSCLC), naïve regulatory T cell (nTreg), pleurectomy/decortication (P/D; debulking surgery), peripheral blood mononuclear cell (PBMC), postoperative day (POD), partial response (PR), stable disease (SD), regulatory T cell (Treg).
Author Contributions
Conception and design clinical trial: JPJJH and JGJVA. Patient selection and patient data collection: RC, LN, HCH and JGJVA. Patient treatment: RC, APWMM and JGJVA. Experimental design: LN, HV, JPJJH and MEHK-L. Acquisition and analysis of data: LN, MEHK-L, JPJJH and KB. Interpretation of data: LN, HV, RW, JPJJH and MEHK-L. Writing of the manuscript: LN and HV. Review and revision of the manuscript and final approval: LN, MEHK-L, KB, RC, APWMM, HCH, JGJVA, RWH, JPJJH and HV.
Additional information
Funding
References
- Raja S, Murthy SC, Mason DP. Malignant pleural mesothelioma. Curr Oncol Rep. 2011;13(4):259–264. 10.1007/s11912-011-0177-9
- Robinson BM. Malignant pleural mesothelioma: an epidemiological perspective. Ann Cardiothorac Surg. 2012;1(4):491–496.
- Yap TA, Aerts JG, Popat S, Fennell DA. Novel insights into mesothelioma biology and implications for therapy. Nat Rev Cancer. 2017;17(8):475–488. 10.1038/nrc.2017.42
- Zalcman G, Mazieres J, Margery J, Greillier L, Audigier-Valette C, Moro-Sibilot D, Molinier O, Corre R, Monnet I, Gounant V, et al. Bevacizumab for newly diagnosed pleural mesothelioma in the mesothelioma avastin cisplatin pemetrexed study (MAPS): a randomised, controlled, open-label, phase 3 trial.Lancet. 2016;387(10026):1405–1414. 10.1016/S0140-6736(15)01238-6
- Haas AR, Sterman DH. Malignant pleural mesothelioma: update on treatment options with a focus on novel therapies. Clin Chest Med. 2013;34(1):99–111. 10.1016/j.ccm.2012.12.005
- Vogelzang NJ, Rusthoven JJ, Symanowski J, Denham C, Kaukel E, Ruffie P, Gatzemeier U, Boyer M, Emri S, Manegold C, et al. Phase III study of pemetrexed in combination with cisplatin versus cisplatin alone in patients with malignant pleural mesothelioma.J Clinical Oncology: Official Journal Am Soc Clin Oncol. 2003;21(14):2636–2644. 10.1200/JCO.2003.11.136
- Treasure T, Lang-Lazdunski L, Waller D, Bliss JM, Tan C, Entwisle J, Snee M, O’Brien M, Thomas G, Senan S, et al. Extra-pleural pneumonectomy versus no extra-pleural pneumonectomy for patients with malignant pleural mesothelioma: clinical outcomes of the mesothelioma and radical surgery (MARS) randomised feasibility study.Lancet Oncol. 2011;12(8):763–772. 10.1016/S1470-2045(11)70149-8
- Coulie PG, van den Eynde BJ, van der Bruggen P, Boon T. Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nature Reviews Cancer. 2014;14(2):135–146. 10.1038/nrc3670
- van der Burg SH, Arens R, Ossendorp F, van Hall T, Melief CJM. Vaccines for established cancer: overcoming the challenges posed by immune evasion. Nature Reviews Cancer. 2016;16(4):219–233. 10.1038/nrc.2016.16
- Lievense LA, Sterman DH, Cornelissen R, Aerts JG. Checkpoint blockade in lung cancer and mesothelioma. Am J Respir Crit Care Med. 2017;196:274–282. 10.1164/rccm.201608-1755CI
- Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, Chow LQ, Vokes EE, Felip E, Holgado E, et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer.N Engl J Med. 2015;373(17):1627–1639. 10.1056/NEJMoa1507643
- Brahmer J, Reckamp KL, Baas P, Crino L, Eberhardt WE, Poddubskaya E, Antonia S, Pluzanski A, Vokes EE, Holgado E, et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer.N Engl J Med. 2015;373(2):123–135. 10.1056/NEJMoa1504627
- Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, Tykodi SS, Sosman JA, Procopio G, Plimack ER, et al. Nivolumab versus everolimus in advanced renal-cell carcinoma.N Engl J Med. 2015;373(19):1803–1813. 10.1056/NEJMoa1510665
- Reck M, Rodriguez-Abreu D, Robinson AG, Hui R, Csoszi T, Fulop A, Gottfried M, Peled N Tafreshi A, Cuffe S, et al. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer.N Engl J Med. 2016;375(19):1823–1833. 10.1056/NEJMoa1606774
- Maverakis E, Cornelius LA, Bowen GM, Phan T, Patel FB, Fitzmaurice S, He Y, Burrall B, Duong C, Kloxin AM, et al. Metastatic melanoma - a review of current and future treatment options.Acta Derm Venereol. 2015;95(5):516–524. 10.2340/00015555-2035
- Housseau F, Llosa NJ. Immune checkpoint blockade in microsatellite instable colorectal cancers: back to the clinic. Oncoimmunology. 2015;4(6):e1008858. 10.1080/2162402X.2015.1008858
- Dobrzanski MJ. Expanding roles for CD4 T cells and their subpopulations in tumor immunity and therapy. Front Oncol. 2013;3:63. 10.3389/fonc.2013.00063
- Dammeijer F, Lievense LA, Veerman GD, Hoogsteden HC, Hegmans JP, Arends LR, Aerts JG. Efficacy of tumor vaccines and cellular immunotherapies in non-small-cell lung cancer: a systematic review and meta-analysis.J Clin Oncol. 2016;34(26):3204–3212. 10.1200/JCO.2015.66.3955
- Hegmans JP, Veltman JD, Lambers ME, De Vries IJ, Figdor CG, Hendriks RW, et al. Consolidative dendritic cell-based immunotherapy elicits cytotoxicity against malignant mesothelioma.Am J Respir Crit Care Med. 2010;181(12):1383–1390. 10.1164/rccm.200909-1465OC
- Hegmans JP, Aerts JG. Immunomodulation in cancer. Curr Opin Pharmacol. 2014;17:17–21. 10.1016/j.coph.2014.06.007
- Drake CG, Jaffee E, Pardoll DM. Mechanisms of immune evasion by tumors. Adv Immunol. 2006;90:51–81
- Vinay DS, Ryan EP, Pawelec G, Talib WH, Stagg J, Elkord E, et al. Immune evasion in cancer: mechanistic basis and therapeutic strategies. Semin Cancer Biol. 2015;35:Suppl:S185-98. 10.1016/j.semcancer.2015.03.004
- Bjoern J, Brimnes MK, Andersen MH, Thor Straten P, Svane IM. Changes in peripheral blood level of regulatory T cells in patients with malignant melanoma during treatment with dendritic cell vaccination and low-dose IL-2. Scand J Immunol. 2011;73(3):222–233. 10.1111/j.1365-3083.2010.02494.x
- Miyara M, Yoshioka Y, Kitoh A, Shima T, Wing K, Niwa A, et al. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity. 2009;30(6):899–911. 10.1016/j.immuni.2009.03.019
- Santegoets SJ, Dijkgraaf EM, Battaglia A, Beckhove P, Britten CM, Gallimore A, Godkin A, Gouttefangeas C, de Gruijl TD, Koenen HJ, et al. Monitoring regulatory T cells in clinical samples: consensus on an essential marker set and gating strategy for regulatory T cell analysis by flow cytometry.Cancer Immunology, Immunotherapy: CII. 2015;64(10):1271–1286. 10.1007/s00262-015-1729-x
- Hegmans JP, Hemmes A, Hammad H, Boon L, Hoogsteden HC, Lambrecht BN. Mesothelioma environment comprises cytokines and T-regulatory cells that suppress immune responses. Eur Respir J. 2006;27(6):1086–1095. 10.1183/09031936.06.00135305
- Ireland DJ, Kissick HT, Beilharz MW, Ireland DJ, Kissick HT, Beilharz MW. The role of regulatory T cells in mesothelioma. Cancer Microenvironment: Official Journal of the International Cancer Microenvironment Society. 2012;5(2):165–172. 10.1007/s12307-012-0100-4
- McCoy MJ, Nowak AK, VanDer Most RG, Dick IM, Lake RA. Peripheral CD8(+) T cell proliferation is prognostic for patients with advanced thoracic malignancies. Cancer Immunology, Immunotherapy: CII. 2013;62(3):529–539. 10.1007/s00262-012-1360-z
- Motoyoshi Y, Kaminoda K, Saitoh O, Hamasaki K, Nakao K, Ishii N, Nagayama Y, Eguchi K. Different mechanisms for anti-tumor effects of low- and high-dose cyclophosphamide. Oncol Rep. 2006;16(1):141–146.
- Ghiringhelli F, Menard C, Puig PE, Ladoire S, Roux S, Martin F, Solary E, Le Cesne A, Zitvogel L, Chauffert B, et al. Metronomic cyclophosphamide regimen selectively depletes CD4+CD25+ regulatory T cells and restores T and NK effector functions in end stage cancer patients.Cancer Immunology, Immunotherapy: CII. 2007;56(5):641–648. 10.1007/s00262-006-0225-8
- Kan S, Hazama S, Maeda K, Inoue Y, Homma S, Koido S, Okamoto M, Oka Oka. Suppressive effects of cyclophosphamide and gemcitabine on regulatory T-cell induction in vitro. Anticancer Res. 2012;32(12):5363–5369.
- Heylmann D, Bauer M, Becker H, VanGool S, Bacher N, Steinbrink K, Kaina B. Human CD4+CD25+ regulatory T cells are sensitive to low dose cyclophosphamide: implications for the immune response.PloS One. 2013;8(12):e83384. 10.1371/journal.pone.0083384
- Veltman JD, Lambers ME, VanNimwegen M, De Jong S, Hendriks RW, Hoogsteden HC, Aerts JG, Hegmans JP. Low-dose cyclophosphamide synergizes with dendritic cell-based immunotherapy in antitumor activity. J Biomed Biotechnol. 2010;2010:798467. 10.1155/2010/798467
- Cornelissen R, Hegmans JP, Maat AP, Kaijen-Lambers ME, Bezemer K, Hendriks RW, Hoogsteden HC, Aerts JG. Extended tumor control after dendritic cell vaccination with low dose cyclophosphamide as adjuvant treatment in patients with malignant pleural mesothelioma. Am J Respir Crit Care Med. 2015
- Zhang S, Pan SB, Lyu QH, Wu P, Qin GM, Wang Q, He ZL, He XM, Wu M, Chen G. Postoperative regulatory T-cells and natural killer cells in stage i nonsmall cell lung cancer underwent video-assisted thoracoscopic lobectomy or thoracotomy.Chin Med J (Engl). 2015;128(11):1502–1509. 10.4103/0366-6999.157672
- Chen C, Chen D, Zhang Y, Chen Z, Zhu W, Zhang B, Wang Z, Le H. Changes of CD4+CD25+FOXP3+ and CD8+CD28- regulatory T cells in non-small cell lung cancer patients undergoing surgery.Int Immunopharmacol. 2014;18(2):255–261. 10.1016/j.intimp.2013.12.004
- Ge Y, Domschke C, Stoiber N, Schott S, Heil J, Rom J, Blumenstein M, Thum J, Sohn C, Schneeweiss A, et al. Metronomic cyclophosphamide treatment in metastasized breast cancer patients: immunological effects and clinical outcome.Cancer Immunol Immunother. 2012;61(3):353–362. 10.1007/s00262-011-1106-3
- Wu J, Waxman DJ. Metronomic cyclophosphamide schedule-dependence of innate immune cell recruitment and tumor regression in an implanted glioma model. Cancer Lett. 2014;353(2):272–280. 10.1016/j.canlet.2014.07.033
- Madondo MT, Quinn M, Plebanski M. Low dose cyclophosphamide: mechanisms of T cell modulation. Cancer Treat Rev. 2016;42:3–9. 10.1016/j.ctrv.2015.11.005
- Matar P, Rozados VR, Gervasoni SI, Scharovsky GO. Th2/Th1 switch induced by a single low dose of cyclophosphamide in a rat metastatic lymphoma model. Cancer Immunol Immunother. 2002;50(11):588–596. 10.1007/s00262-001-0237-3
- Schiavoni G, Mattei F, Di Pucchio T, Santini SM, Bracci L, Belardelli F, Proietti E. Cyclophosphamide induces type I interferon and augments the number of CD44(hi) T lymphocytes in mice: implications for strategies of chemoimmunotherapy of cancer. Blood. 2000;95(6):2024–2030.
- Bracci L, Moschella F, Sestili P, La Sorsa V, Valentini M, Canini I, Baccarini S, Maccari S, Ramoni C, Belardelli F, et al. Cyclophosphamide enhances the antitumor efficacy of adoptively transferred immune cells through the induction of cytokine expression, B-cell and T-cell homeostatic proliferation, and specific tumor infiltration.Clin Cancer Res. 2007;13(2 Pt 1):644–653. 10.1158/1078-0432.CCR-06-1209
- Podrazil M, Horvath R, Becht E, Rozkova D, Bilkova P, Sochorova K, Hromadkova H, Kayserova J, Vavrova K, Lastovicka Jet al. Phase I/II clinical trial of dendritic-cell based immunotherapy (DCVAC/PCa) combined with chemotherapy in patients with metastatic, castration-resistant prostate cancer.Oncotarget. 2015;6(20):18192–18205. 10.18632/oncotarget.4145
- Liston A, Gray DHD. Homeostatic control of regulatory T cell diversity. Nat Rev Immunol. 2014;14(3):154–165. 10.1038/nri3605
- Booth NJ, McQuaid AJ, Sobande T, Kissane S, Agius E, Jackson SE, Salmon M, Falciani F, Yong K, Rustin MH, et al. Different proliferative potential and migratory characteristics of human CD4+ regulatory T cells that express either CD45RA or CD45RO.J Immunol. 2010;184(8):4317–4326. 10.4049/jimmunol.0903781
- Wang C, Lee JH, Kim CH. Optimal population of FoxP3+ T cells in tumors requires an antigen priming-dependent trafficking receptor switch. PloS One. 2012;7(1):e30793. 10.1371/journal.pone.0030793
- Kwa M, Li X, Novik Y, Oratz R, Jhaveri K, Wu J, Gu P, Meyers M, Muggia F, Speyer J, et al. Serial immunological parameters in a phase II trial of exemestane and low-dose oral cyclophosphamide in advanced hormone receptor-positive breast cancer.Breast Cancer Res Treat. 2018;168(1):57–67. 10.1007/s10549-017-4570-4
- Prieto GA, Rosenstein Y. Oestradiol potentiates the suppressive function of human CD4 CD25 regulatory T cells by promoting their proliferation. Immunology. 2006;118(1):58–65. 10.1111/j.1365-2567.2006.02339.x
- Sundar R, Cho B-C, Brahmer JR, Soo RA. Nivolumab in NSCLC: latest evidence and clinical potential. Ther Adv Med Oncol. 2015;7(2):85–96. 10.1177/1758834014567470
- Johnson DB, Peng C, Sosman JA. Nivolumab in melanoma: latest evidence and clinical potential. Ther Adv Med Oncol. 2015;7(2):97–106. 10.1177/1758834014567469
- Sugiyama D, Nishikawa H, Maeda Y, Nishioka M, Tanemura A, Katayama I, E E, Kanakura Y, Sato E, Fukumori Yet al. Anti-CCR4 mAb selectively depletes effector-type FoxP3+CD4+ regulatory T cells, evoking antitumor immune responses in humans.Proc Natl Acad Sci U S A. 2013;110(44):17945–17950. 10.1073/pnas.1316796110
- Kurose K, Ohue Y, Sato E, Yamauchi A, Eikawa S, Isobe M, Nishio Y, Uenaka A, Oka M, Nakayama E. Increase in activated Treg in TIL in lung cancer and in vitro depletion of Treg by ADCC using an antihuman CCR4 mAb (KM2760).J Thorac Oncol. 2015;10(1):74–83. 10.1097/JTO.0000000000000364