
ABSTRACT
Treatment for acute myeloid leukemia (AML) remains suboptimal and many patients remain refractory or relapse upon standard chemotherapy based on nucleoside analogs plus anthracyclines. The crosstalk between AML cells and the BM stroma is a major mechanism underlying therapy resistance in AML. Lenalidomide and pomalidomide, a new generation immunomodulatory drugs (IMiDs), possess pleiotropic anti-leukemic properties including potent immune-modulating effects and are commonly used in hematological malignances associated with intrinsic dysfunctional BM such as myelodysplastic syndromes and multiple myeloma. Whether IMiDs may improve the efficacy of current standard treatment in AML remains understudied. Here, we have exploited in vitro and in vivo preclinical AML models to analyze whether IMiDs potentiate the efficacy of AraC/Idarubicin-based standard AML chemotherapy by interfering with the BM stroma-mediated chemoresistance. We report that IMiDs do not exert cytotoxic effects on either non-del5q/5q- AML cells nor BM-MSCs, but they enhance the immunomodulatory properties of BM-MSCs. When combined with AraC/Idarubicin, IMiDs fail to circumvent BM stroma-mediated resistance of non-del5q/5q- AML cells in vitro and in vivo but induce robust extramedullary mobilization of AML cells. When administered as a single agent, lenalidomide specifically mobilizes non-del5q/5q- AML cells, but not healthy CD34+ cells, to peripheral blood (PB) through specific downregulation of CXCR4 in AML blasts. Global gene expression profiling supports a migratory/mobilization gene signature in lenalidomide-treated non-del5q/5q- AML blasts but not in CD34+ cells. Collectively, IMiDs mobilize non-del5q/5q- AML blasts to PB through CXCR4 downregulation, but fail to potentiate AraC/Idarubicin activity in preclinical models of non-del5q/5q- AML.
Introduction
Acute myeloid leukemia (AML) comprises a biologically and genetically heterogeneous group of disorders characterized by the rapid expansion of immature myeloid blasts in bone marrow (BM).Citation1,Citation2 In contrast to acute lymphoblastic leukemia (ALL), treatment of AML has not improved substantially over the last two decades and many patients still fail to respond to standard intensive chemotherapy based on nucleoside analogs such as cytarabine (AraC) or fludarabine and anthracyclines such as idarubicin and daunorubicin.Citation3,Citation4 Failure of current therapies to eradicate leukemia-initiating/propagating cells (LICs) and chemotherapy refractoriness are the major mechanisms underlying AML progression/relapse. In addition, the BM stroma has been involved in the pathogenesis of a variety of hematologic malignances including AML.Citation5 The interaction of AML cells with the BM microenvironment in functional niches is a major mechanism underlying leukemia maintenance and therapy resistance.Citation5,Citation6 Thus, the high rate of mortality/morbidity and chemo-resistance in AML guides the search for new compounds with higher efficiency and lower toxicity.
New generation of immunomodulatory drugs (IMiDs) such as lenalidomide and pomalidomide are thalidomide analogs, which possess pleiotropic anti-leukemic properties including anti-proliferative, anti-angiogenic but also immune-modulating effects.Citation7-Citation9 They are approved by the Food and Drug Administration (FDA) and the European Medicines Agency (EMA). Lenalidomide is approved for both 5q- myelodysplastic syndrome (MDS) and multiple myeloma (MM) whereas pomalidomide is approved for MM, which are hematological malignances associated with intrinsic dysfunctional BM.Citation10,Citation11 Some studies reported that lenalidomide has modest (pre)-clinical activity in lymphoma and AML with manageable toxicity.Citation9,Citation12,Citation13 Ongoing Phase I clinical trials assaying low- and high-dose lenalidomide for both newly diagnosed and relapsed/refractory AML also support that lenalidomide displays modest activity when given either alone or combined with cytarabine and anthracyclines.Citation14
Whether and how IMiDs may benefit current standard treatment in AML needs further investigation. To bring IMiDs to the forefront of AML therapeutic arsenal, a better understanding of its mechanism of action in AML is required. In fact, it has recently been shown that BM mesenchymal stromal cells (BM-MSCs) derived from diagnostic AML patients are potent immunomodulators and provide similar chemoprotection of AML cells to cytarabine/idarubicin than normal BM-MSCs.Citation5 Although IMiDs may have an anti-proliferative effect on leukemic cells, they may likely exert pleiotropic and immunomodulatory effects at the stroma-leukemic cell crosstalk, thus improving standard AML intensive chemotherapy by either potentiating AraC/anthracycline efficacy or reducing BM-MSC-derived chemoresistance.
Here, we have exploited in vitro and in vivo AML preclinical models to analyze whether IMiDs potentiate the efficacy of AraC/Idarubicin-based standard chemotherapy by lowering the BM stroma-mediated chemoresistance. Lenalidomide and pomalidomide are not cytotoxic for either non-del5q/5q- AML cells or BM-MSCs, but they enhance the immuno-modulatory capacity of BM-MSCs. When administered alone or in combination with AraC and Idarubicin, IMiDs fail to circumvent BM stroma-mediated resistance of non-del5q/5q- AML cells in vitro and in vivo but induce robust mobilization of non-del5q/5q- AML cells, but not healthy CD34+ cells, to peripheral blood (PB) and spleen likely through specific downregulation of CXCR4 in AML blasts. Global gene expression profiling supports a migratory/mobilization gene signature in lenalidomide-treated AML primary blasts. We conclude that IMiDs mobilize non-del5q/5q- AML blasts to PB through downregulation of CXCR4 but do not improve AraC/Idarubicin activity in a preclinical model of AML.
Material and methods
AML cell lines and primary leukemic and healthy cells
HL60 and MOLM-13 cells (kindly provided by Prof. Luciano Di Croce, CRG, Barcelona and Prof Michael Andreeff, MD Anderson, Houston, TX, respectively) were cultured as in RPMI medium supplemented with 10% fetal calf serum (FCS) and antibiotics (Gibco). Patient AML samples (>90% of blasts) were obtained from fresh BM from the Hospital Clínico San Carlos (Madrid, Spain) and Hospital Virgen del Rocío (Seville, Spain). AML diagnosis was based on French-American-British (FAB)Citation15 and World Health Organization (WHO) classifications,Citation16,Citation17 , summarizes patient’s main clinical/cytogenetic/molecular characteristics. Fresh peripheral blood (PB) and cord blood (CB) units were obtained from healthy donors from the Catalonia Blood Tissue Bank following the institutional guidelines approved by our local Institutional Review Board. G-SCF-mobilized PB and BM were obtained from clinical leftovers from the Hospital Clínic of Barcelona (HCB). PBMCs were isolated using Ficoll-Paque Plus (GE Healthcare) density gradient centrifugation, and CD34+ cells were MACS-purified using the human CD34 MicroBead kit and the AutoMACS device (Miltenyi Biotec, Madrid, Spain) as reported.Citation18,Citation19 The purity of the CD34+ fraction was assessed and was consistently >95%. Primary AML and CD34+ cells were cultured in Stemspam medium (Stem Cell Technologies, Vancouver, Canada) supplemented with the hematopoietic cytokines Stem Cell Factor (100 ng/mL), FLT3 ligand (100 ng/mL), IL3 (10 ng/mL, all from PeproTech) and antibiotics (Gibco). BM-MSCs were obtained, grown and characterized as extensively described by our group.Citation5,Citation20 Cultures were maintained in a humidified atmosphere with 5% CO2 at 37⁰C. The study was IRB-approved by the HCB (HCB/2014/0687) and samples received upon signed informed consent.
Table 1. Biological and cytogenetic-molecular characteristics of blasts from diagnostic AML patients.
Drugs
AraC (Cytarabine®, Pfizer), idarubicin (Zavedos®, Pfizer) and the CXCR4 antagonist AMD3100 (Sigma) were reconstituted with PBS. Lenalidomide and pomalidomide were kindly provided by Celgene Corp (San Diego, CA), reconstituted in DMSO as per supplier´s guidelines and stored in aliquots at −20⁰C.
Cytotoxicity and apoptosis assays
The cytotoxic effect of the different drugs on HL-60 and MOLM-13 cell was assessed using Cell Counting Kit-8 based on monosodium salt WST-8 according to the manufacturer’s protocol (Sigma-Aldrich). Briefly, 5.000 cells were plated in 96-well plate and incubated with increasing concentrations of the corresponding drug for 48h. After incubation time, absorbance was measured at 450 nm using a microplate reader. The cytotoxic effect of IMiDs on BM-MSCs was assessed using MTT assays as previously described.Citation21,Citation22 Briefly, 2 × 104 cells were cultured with increasing drug concentrations in 96-well plates for 48 h. To determine the chemoprotective effect of BM-MSCs a total of 2 × 104 BM-MSCs were plated in 96-well plates 18 h before addition of AML cells (2 × 104 for cell lines and 2 × 105 for primary cells). BM-MSC:AML co-cultures were treated with IC25 concentrations of the AraC (77nM) and Idarubicin (7nM) and 10 µM of lenalidomide/pomalidomide for 48–72 h. Apoptosis of CD33+ AML cells was measured using the annexin-V/7-AAD apoptosis detection kit (BD Biosciences) on a FACSCanto-II cytometer using FACSDiva software (BD Biosciences) as previously described.Citation23
Immunosuppressive assays and cytokine detection
The effect of IMiDs on the BM-MSC-mediated immunosuppressive properties and cytokine release (TNFα, IL1β, IL10, IL6 and SDF1) was determined by CSFE and Luminex Multiplex assays as previously described.Citation5,Citation24,Citation25
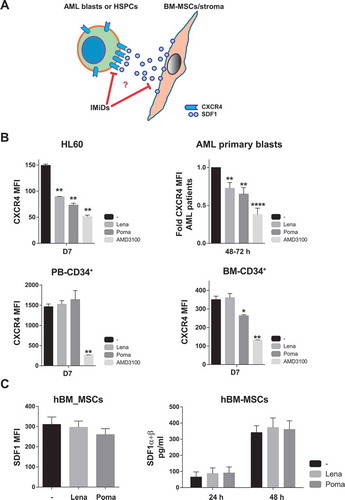
FACS analysis of CXCR4 and SDF1
CXCR4 expression was determined in AML and CD34+ cells treated with IMiDs. A total of 0.2 × 106 (for cell lines) or 1 × 106 (for primary cells) cells were incubated with 10 µM of IMiDs (the CXCR4 antagonist AMD3100 was used as a positive control). Seven days later (48 h–72 h for primary AML cells) treated cells were stained with anti-human CD45-APC.Cy7, CD33-APC, CD34-PECy7 and CD184-BV421 (BD Biosciences) and the mean fluorescence intensity (MFI) of CD184 within the blast population (CD45+CD33+) was FACS-quantified using a FACS Canto II cytometer. The expression of the CXCR4 ligand SDF1 was analyzed in BM-MSC (0.5 x 106) 48 h after treatment with 10 µM IMiDs. BM-MSCs were trypsinized and stained with anti-human CXCL12/SDF-1 (R&D Systems) using the Cell Permeabilization Kit (FIX&PERM®).Citation26
NSG mice xenotransplantation and analysis of engraftment
All experimental procedures were approved by the Animal Care Committee of The Barcelona Biomedical Research Park (HRH-16–0037). Eight-to-14-week-old NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG) mice housed under pathogen free conditions were used. In order to recapitulate a BM stroma milieu, 1 × 105 HL60 (n = 31 mice) or MOLM-13 (n = 33 mice) cells were intra-BM transplanted into non-irradiated NSG mice together with 3 × 105 irradiated BM-MSCs.Citation25,Citation27 HL60 AML grafts were monitored by BM aspiration and mice were randomized into treatment groups when graft was >1% in the contralateral BM (~2–3 weeks post-transplant). For MOLM-13 AML grafts, mice were randomized for treatment 3 days after intra-BM transplant due to its aggressiveness. At the end of the treatment period, mice were sacrificed and cells from the BM, spleen and PB were stained with anti-human HLA-ABC-FITC, CD33-PE and CD45-APC (BD Biosciences) to analyze human leukemic engraftment by flow cytometry. Similarly, healthy CD34+ HSPCs (0.1 × 106 cells) were intra-BM-transplanted into sublethally irradiated (2.25 Gy) NSG mice (n = 10) together with 3 × 105 irradiated BM-MSCs. Six weeks later, when human healthy chimerism was evident, mice were treated with lenalidomide. At the end of lenalidomide treatment, mice were sacrificed and cells from the BM, spleen and PB were stained as above to analyze human normal chimerism by flow cytometry. Multilineage engraftment was assessed using CD33-APC, CD19-PE and CD34-PE.Cy7 (BD Biosciences). Spleen infiltration of AML cells was analyzed by immunohistochemistry using an anti-human-CD45.
In vivo chemotherapy and imids treatment
Ara-C and idarubicin were administrated following a well-established 5 + 3 treatment schedule, consisting on intravenous (i.v.) co-delivery of 30 mg/Kg of Ara-C and 0.1 mg/Kg of idarubicin for 3 days followed by intraperitoneal injection (i.p.) of Ara-C for two further days (A + I treatment).Citation28 Lenalidomide (25 mg/kg) given as single agent was administrated i.p. for 10 days. When combined with A + I treatment, lenalidomide (25 mg/kg) was administrated alone during days 1–3 and 9–10 and with A + I on days 4–8. 1% DMSO/PBS solution was always used as vehicle treatment. Mice were weighted throughout treatment and drug doses were recalculated as necessary.
Microarray gene expression profiling (GEP)
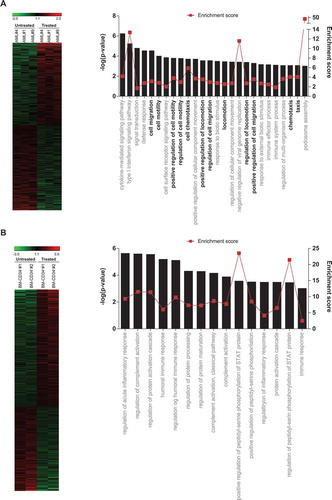
Non-del5q/5q- AML primary samples (n = 3) and BM-CD34+ HSPCs (n = 2) were FACS purified (CD45+CD33+ purity >98%) and cultured with 10 µM lenalidomide for 48h before global gene expression profiling (GEP) as described.Citation29 Total RNA was then extracted using a Maxwell® RSC simplyRNA Cells Kit in a Maxwell® RSC Instrument (Promega) and its quality checked in the Agilent 2100 Bioanalyzer. Total RNA samples were labeled with Cy3 using the Quick-Amp Labeling Kit and hybridized with the Gene Expression Hybridization Kit to the GeneChip Human Gene 2.0 ST (Affymetrix) following Manufacturer’s instructions. Hierarchical clustering of genes was performed with the one minus correlation metric and the unweighted average distance. Only genes showing >1.5-fold change expression and p-value<0.05 were considered differentially expressed and were subjected to gene ontology (GO) term analysis using GorillaCitation30,Citation31 publicly available at http://cbl-gorilla.cs.technion.ac.il. Microarray data were deposited in the public Gene Expression Omnibus database, accession number GSE106748.
Results
BM stroma/milieu confers resistance of AML cells to AraC/idarubicin
Chemotherapy based on the combination of Ara-C with anthracyclines, either idarubicin or daunorubicin, is the first-line induction treatment in diagnostic AML.Citation3,Citation4 Here, viability in vitro assays revealed that both cytotoxic drugs, Ara-C and idarubicin, are equally toxic in different AML cell lines with an IC50 of ~10−7M (Fig 1S). Exposure to Ara-C plus idarubicin (Ara-C/Ida) resulted in 80%–95% cell death of AML cell lines, measured by Annexin V staining (). However, when co-cultured with BM-MSCs, AML cell lines were chemo-resistant with only 40–50% cell death (). To further test whether the BM stroma/milieu protects AML cells from Ara-C/Ida treatment, AML cell lines were co-transplanted via intra BM transplant (IBMT) into NSG mice with human BM-MSCs which largely increase AML engraftment (Fig 2S), as reported in several hematologic malignances.Citation32-Citation35 When engraftment was >1% in non-injected tibia, mice were treated with Ara-C/Ida administrated following the well-established 5 + 3 treatment schedule.Citation28 Similar to the in vitro results, Ara-C/Ida significantly (2-3-fold) reduced AML engraftment/survival in extramedular BM hematopoietic tissues and in contralateral BM (CL) whereas the co-transplanted human BM-MSCs conferred chemoresistant to AML cells in injected tibia (IT) ().
Figure 1. BM microenvironment protects HL60 and MOLM-13 cells from AraC+Idarubicin-based chemotherapy in vitro (A) and in vivo (B). (n = 3). *p < 0.05. **p < 0.01.
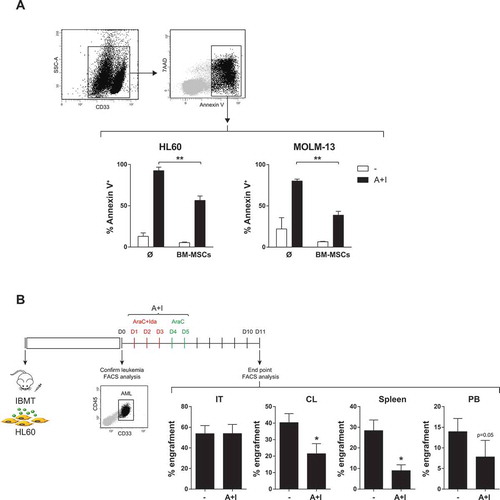
Imids exert no toxicity on non-del5q/5q- AML cells but increase immunosuppressive properties of BM-MSCs
The immune-modulating properties of BM stroma/BM-MSCs are a major mechanism underlying BM stroma-mediated chemoresistance in hematological malignances.Citation5 Lenalidomide and pomalidomide possess pleiotropic anti-tumor properties including also tumor-controlling immune-modulating effects in other hematological BM malignances such as MDS and MM.Citation9-Citation11 We therefore set out to assess the anti-leukemia effects of lenalidomide and pomalidomide in AML. Strikingly, as single agents, lenalidomide and pomalidomide display no cytotoxic effects (assayed at 48h and 72h after drug exposure) on neither AML cell lines () nor diagnostic primary AMLs (n = 9) covering different cytogenetic/molecular AML subtypes other than del5q/5q- AMLs (, ). Importantly, despite lenalidomide/pomalidomide had neither cytotoxic effects on BM-MSCs () they did enhance the immunosuppressive capacity of BM-MSCs (). In fact, IMiDs boosted the BM-MSC-mediated inhibition of activated T cells () through blocking T-cell release of pro-inflammatory cytokines such as TNFα and IL1β while enhancing the production of the master anti-inflammatory cytokine IL10 (). Lenalidomide/pomalidomide also inhibited the release of the pro-inflammatory cytokine IL6 by BM-MSC (). The potent immune-modulatory properties of IMiDs on BM-MSCsCitation24 led us to combine IMiDs with Ara-C/Ida-based standard AML treatment as a potential therapeutic approach to bypass the BM-MSC-mediated chemoresistance to Ara-C/Ida.
Figure 2. Effects of IMiDs on primary AML blasts and BM-MSC. (A) Lenalidomide and pomalidomide do not have a cytotoxic effect on either AML cell lines (n = 3) (top panel), primary AML blasts (n = 9, bottom left panels) or BM-MSC (n = 3) (right bottom panels). (B) Left panel: IMiDs potentiate the immunosuppressive effect of BM-MSCs, measured as percentage of CFSE+ non-proliferating cells. CSFE-labeled PBMCs were stimulated with PHA and then co-cultured with BM-MSCs in the presence of lenalidomide or pomalidomide for 5 days (n = 3). Middle panels: representative flow-cytometry histograms of cycling (CSFElow) PBMCs. Right panels: Concentration of the indicated cytokines in cell-culture supernatants determined by Luminex Multiplex assays. PBMCs were co-cultured with BM-MSCs in the presence/absence of IMiDs. Error bars indicate the SEM values of the n = 3 biological replicates. (C) IMiDs diminish IL6 production by BM-MSC (n = 3). *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
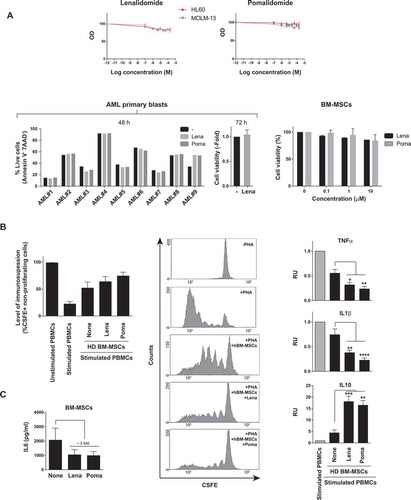
Imids fail to overcome BM-MSC/stroma-mediated chemoresistance of non-del5q/5q- AML cells but induce specific mobilization of AML cells to PB
Neither lenalidomide nor pomalidomide were unable to overcome the BM-MSC-mediated resistance of non-del5q/5q- AML cells to Ara-C/Ida chemotherapy either in vitro () or in vivo (). Surprisingly, when lenalidomide was combined with chemotherapy we observed a robust (3–4-fold) mobilization of non-del5q/5q- AML cells into extramedular BM hematopoietic sites including PB and spleen (). Next, we performed similar in vivo experiments by treating AML-engrafted NSG mice with lenalidomide as single agent and confirmed that it induces a robust mobilization of non-del5q/5q- AML cells into PB () and spleen ().
Figure 3. IMiDs combined with AraC-Idarubicin do not circumvent BM- microenvironment-mediated resistance of AML cells but induce robust extramedullary mobilization of AML cells. (A) Lenalidomide and pomalidomide do not overcome BM-MSC-mediated resistance of AML cell lines to intensive chemotherapy (n = 3). (B) IMiDs combined with AraC+Idarubicin highly mobilize AML blasts to PB and spleen in AML xenograft models (n = 3). *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
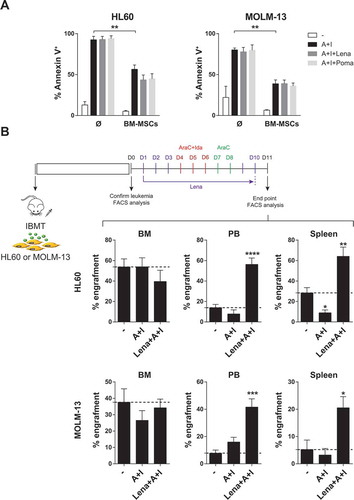
Figure 4. When administered as a single agent, lenalidomide highly mobilizes AML cells, but not healthy CD34+ cells, to PB. (A,B) Lenalidomide administered alone mobilizes AML cells to PB (A) and spleen (B). AML infiltration in PB and spleen was detected by FACS and immunohistochemistry, respectively. (C) Lenalidomide fails to mobilize healthy CD34+ cells (n = 3). *p < 0.05. **p < 0.01.
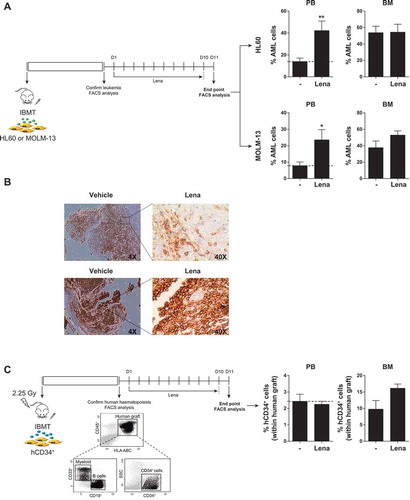
Specific mobilization of leukemic cells from BM to PB is commonly desirable in the clinical setting because circulating blasts are more sensitive to treatment in part due to their detachment from the chemoprotective BM niche.Citation4,Citation36,Citation37 However, mobilization of normal hematopoietic stem/progenitor cells (HSPCs) to PB is not desired because it would accelerate BM aplasia and subsequent hematopoietic defects. Thus, we next analyzed whether lenalidomide also mobilizes normal HSPCs to PB. CB CD34+ cells were IBM-transplanted in NSG mice and total (CD45+HLA-ABC+) and immature (CD45+HLA-ABC+CD34+) engraftment was analyzed 6–7 weeks later. Interestingly, lenalidomide failed to mobilize normal HSPC indicating that lenalidomide-driven mobilization into PB is specific for AML cells ().
Lenalidomide-mediated mobilization of AML cells to PB is associated to downregulation of CXCR4 and a migratory transcriptomic signature in AML cells
We next sought to gain insights into the mechanisms underlying lenalidomide-mediated mobilization of AML cells. Clearly, the CXCR4/SDF1 axis plays a major role in the interaction of HSPCs and leukemic cells within BM niches. CXCR4 is highly expressed in HSPCs/leukemic cells whereas SDF1 is a cytokine commonly released by BM-MSCs,Citation37,Citation38 (). Consequently, we first treated both AML cell lines and primary non-del5q/5q- AML blasts with lenalidomide and pomalidomide and found a significant (25%–50%) reduction in the levels of CXCR4 in AML cells similar to treatment with the specific CXCR4 inhibitor AMD3100 (). In line with the IMiDs-mediated specific mobilization of AML blasts (), we confirmed that lenalidomide/pomalidomide do not down-regulate CXCR4 expression in healthy PB- or BM-derived HSPCs (). In contrast, lenalidomide and pomalidomide treatment did not impact the either expression levels or cytokine release of SDF1 by human BM-MSCs (). Together, these results suggest that IMiDs specifically mediate the mobilization of AML cells to PB through downregulation of CXCR4.
Figure 5. IMiDs mobilizes AML cells to PB through CXCR4 downregulation. (A) Cartoon of the CXCR4-SDF1 crosstalk between leukemic blasts and BM stromal cells. (B) Effect of IMiDs on the expression levels of CXCR4 in AML cell lines, primary AML cells (top panels), PB-derived CD34+ cells and BM-derived CD34+ cells (bottom panels). (C) Effect of IMiDs on the expression (Left panel) and production levels (right panel) of SDF1 by BM-MSCs. The specific CXCR4 inhibitor AMD3100 was used as positive control.
To identify patterns of gene expression that might provide a molecular explanation for the IMiDs-induced mobilization of AML cells, we performed whole-genome GEP in lenalidomide-treated versus DMSO-treated AML blasts (blast purity>95%, n = 3 de novo non-del5q/5q- AMLs). A heatmap representation of hierarchical clustering of genes differentially expressed (1.5-fold regulation, p-value<0.05) between lenalidomide- versus DMSO-treated AML cells is represented in . A total of 323 genes were differentially expressed between lenalidomide- and DMSO-treated AML cells. Of these, 196 (61%) were upregulated and 127 (39%) downregulated in lenalidomide-treated AML cells, establishing a lenaledomide-specific transcriptomic signature. To get insights into the biological functions affected by differentially expressed genes, we used the Gorilla software,Citation30,Citation31 and found that many significant biological processes predicted to be activated in the lenalidomide-treated AML cells were associated with “migration/motility/mobilization/chemotaxis” (). In contrast, exposure of CB-CD34+ HSPCs to lenalidomide did not affect such biological functions (). This lenalidomide-induced transcriptomic signature supports a CXCR4-mediated mechanism underlying the mobilization of non-del5q/5q- AML cells to PB in response to IMiDs.
Figure 6. Global GEP reveals a migratory signature in lenalidomide-treated AML blasts. (A, B) Left panels: Heatmap representation of hierarchical clustering of genes differentially expressed between lenalidomide-treated and untreated AML blasts (n = 3 leukemias) (top panel) and BM-CD34+ cells (n = 2 healthy donors) (bottom panel). Right panel: Statistically significant biological functions identified using IPA on genes differentially expressed in lenalidomide-treated versus untreated AML blasts (top panel) and CD34+ cells (bottom panel). They are ranked by z-score. A z-score >2 indicates a predicted activation of that biological function. Biological functions associated with ‘cell migration/movement/motility’ are shown in black.
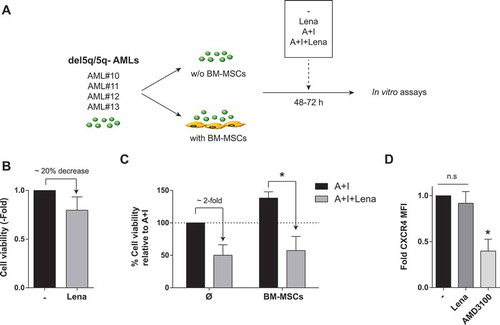
Lenalidomide overcomes BM-MSC/stroma-mediated chemoresistance of AML cells from del5q/5q- patients
A phase II clinical trial has recently reported promising results for lenalidomide combined with AraC/daunorubicine in both MDS and AML patients with 5q alterations (del5q/5q-).Citation39 Thus, we next focused our efforts on this cytogenetic subgroup of AML patients as potential candidate patients benefiting from the combination of IMiDs with AraC/idarubicine. Primary cells from del5q/5q- AML patients were treated with lenalidomide, AraC/Ida or their combination for 48-72h, in the presence/absence of BM-MSCs (). Opposite to non-del5q/5q- AML cells (), lenaledomide used as single agent displayed a slight, but consistent, 20% toxicity in del5q/5q- AML cells (). More importantly, lenaledomide significantly boosted the toxicity of del5q/5q- AML cells when combined with AraC/Ida, overcoming the BM-MSC/stroma-mediated chemoresistance of AML cells from del5q/5q- patients (). Interestingly, lenalidomide did not down-regulate CXCR4 expression on del5q/5q- AML cells (), indicating that lenalidomide partially overcomes BM-MSC/stroma-mediated chemoresistance of AML cells from del5q/5q- patients likely at the expense of blast mobilization.
Figure 7. Effects of Lenalidomide on primary blasts from patients with del5q/5q- AML. (A) Scheme depicting the experimental design. (B) Lenalidomide toxicity on primary del5q/5q- AML cells (n = 4). (C) When combined with standard chemotherapy lenalidomide largely overcomes BM-MSC-mediated resistance of del5q/5q- AMLs to AraC+Idarubicin (n = 4). (D) Effect of lenalidomide on the expression levels of CXCR4 in del5q/5q- AML cells (n = 4). AMD3100 was used as positive control. *p < 0.05; n.s, no statistical differences.
Discussion
AML represents a clinically and genetically very heterogeneous group of diseases characterized by an accumulation of proliferative, abnormally differentiated hematopoietic cells in the BM and other tissues, leading to interference of normal hematopoiesis and BM failure. AML is one of the most common hematopoietic malignances with ~25.000 new cases per year in United States and Europe.Citation40 Overall, AML has an unfavorable clinical outcome, largely determined by age, cytogenetic/genetic lesions as well as patient´s initial response to standard treatment.Citation2 The large genetic/cytogenetic and phenotypic heterogeneity together with a median age at diagnosis of ~65–67 years old have long represented a limitation towards the development of better treatments.Citation4 Despite many clinical trials have been conducted over the last two decades, the standard current treatment in AML remains unchanged, and relies on intensive induction chemotherapy based on nucleoside analogs such as Ara-C or fludarabine and anthracyclines such as idarubicin and daunorubicin followed by high doses Ara-C consolidation.Citation41 Although complete remission rates (CRR) of ~80% are commonly achieved with conventional chemotherapy,Citation4,Citation42 the median survival time is short and the outcome of relapsed patients is dismal.Citation43-Citation45 Novel agents or better treatment combinations/schedules with higher efficacy and less toxicity are therefore in high demand for de novo and relapsed AML.
Failure of current therapies to eradicate LICs and chemotherapy refractoriness are the major mechanisms underlying AML progression/relapse, being in part because the BM provides a protective microenvironment to support the survival of LICs/bulk leukemic cells, hence promoting drug resistance and influencing clinical outcome. In AML, the high rate of relapse may in part be a result of the inability of current treatments to effectively overcome the protective influence of the BM niche.Citation5,Citation46 In fact, it has been recently shown that BM mesenchymal stromal cells (BM-MSCs) derived from diagnostic AML patients are strong immunomodulators and provide similar or even higher chemoprotection of AML cells to cytarabine/idarubicin than normal BM-MSCs.Citation5 Therefore, new compounds or improved combinatory regimens targeting the interaction between the BM microenvironment and AML leukemic cells are very attractive candidates.
The new generation of immunomodulatory drugs (IMiDs) such as lenalidomide and pomalidomide are thalidomide analogs, which possess pleiotropic anti-leukemic properties including anti-proliferative, anti-angiogenic but also immune-modulating effects.Citation7-Citation9 They are approved by the FDA and EMA for 5q- MDS and/or MM which are hematological malignances associated with intrinsic dysfunctional BM with remarkable clinical responses.Citation10,Citation11 However, whether IMiDs may potentiate current standard treatment in AML remains an issue of debate. To bring IMiDs to the forefront of AML therapeutic arsenal, a better understanding of its mechanism of action in AML cells and in the interphase tumor-stroma is required. In this study, we hypothesized that the testing of IMiDs may be facilitated by in vitro and in vivo assays that model the tumor-BM stroma interactions.Citation47-Citation49 Hence, we used human primary BM-MSCs and intra BM co-transplantation with AML cells into NSG mice to mimic the interaction of AML cells with the BM stroma as a platform to assess whether IMiDs add efficiency to current standard AML chemotherapy. We report that lenalidomide and pomalidomide, when administered as single agents, have cytotoxic effects on neither non-del5q/5q AML cells nor BM-MSCs, even at doses as high as 100 µM. This contrasts the anti-proliferative effects reported for IMiDs in other hematopoietic malignances, evidencing the immature nature of AML cells (progenitor-like cells) and the complex biology of AML.Citation9-Citation11 However, IMiDs enhanced the immunomodulatory properties of BM-MSCs as demonstrated by the suppression activated T cells proliferation, inhibition of the production of pro-inflammatory cytokines including TNFα, IL1β and IL6 and increased production of the anti-inflammatory cytokine IL10, suggesting that IMiDs may represent safe candidate drugs interfering with the protective BM stroma for combination with current AML chemotherapy.
Unfortunately, when combined with AraC and Idarubicin, both lenalidomide and pomalidomide fail to circumvent BM stroma-mediated resistance of non-del5q/5q AML cells in in vitro and in vivo AML models. The clinical efficacy of lenalidomide has been explored as either low-dose or high-dose lenalidomide as monotherapyCitation50-Citation54 and in combination with azacitidineCitation55,Citation56 for both newly diagnosed and relapsed/refractory AML. Overall, these clinical trials were somewhat disappointing with CRR as low as 11%–20% and remission duration below 5–7 months.Citation14 Our preclinical studies mechanistically support previous modest clinical activity reported for IMiDs in AML, and discourages future clinical trials using either lenalidomide or pomalidomide for non-del5q/5q AML either as monotherapy or polytherapy. Of note, a recent Phase II clinical study of lenalidomide combined with intensive chemotherapy in AML has reported that only del5q/5q- AML patients show promising CRR.Citation39 Here, our pre-clinical studies confirm that lenaledomide used as single agent displays ~20% toxicity only in del5q/5q- AML cells and it overcomes BM-MSC/stroma-mediated chemoresistance of AML cells exclusively from del5q/5q- patients. A potential mechanism of action of lenalidomide involves the degradation of casein kinase 1A1 (CK1α).Citation57 In fact, del5q MDS patients possess a heterozygous deletion CK1α.Citation58 Collectively, our data is in line with Ades and colleaguesCitation39 and suggest that the mechanisms of action of lenalidomide on AML may depend on cell intrinsic/autonomous genetic makeup, further pinpointing genetic and biological heterogeneity in AML. Despite the overall limited activity of lenalidomide during induction/consolidation therapy its efficiency during AML maintenance therapy has yet to be tested.
Intriguingly, we found that lenalidomide, administered either as monotherapy or combined with conventional chemotherapy, induces robust mobilization of non-del5q/5q AML cells to extramedullary hematopoietic tissues including PB and spleen, suggesting that IMiDs might function as plerixafor, a small molecule inhibitor of the CXCR4/SDF1 axis, extensively used for chemosensitazion of AML blasts by disrupting the interaction of leukemic blasts with the environment.Citation4,Citation36,Citation37,Citation59 In fact, lenalidomide mediated the mobilization of AML cells to PB through downregulation of CXCR4 in non-del5q/5q AML cells but does not affect either the expression or the production of its ligand SDF1 by BM-MSCs. In contrast to plerixafor, lenalidomide does not mobilize normal CD34+ HSPCs to PB. Whole-genome GEP revealed a transcriptomic migratory signature of lenalidomide-treated non-del5q/5q AML primary blasts but not lenalidomide-treated CD34+ cells, confirming that the IMiD-mediated mobilization is specific of AML blasts. We cannot rule out that other signalling pathways/mechanisms contribute to the IMiD-induced mobilization phenotype. Importantly, in contrast to plerixafor, IMiDs do not chemosensitize AML blasts to chemotherapy, further supporting that the addition of IMiDs to cytotoxic chemotherapy seems not feasible in AML.Citation36,Citation59 Lenalidomide-attenuated pro-inflammatory cytokines including TNFα, γIFN, IL1β and macrophage inhibitory factor (MIF) have been previously described to interact with master pathways/molecules such as CXCR4-SDF1 axis, collagenases, integrins and eicosanoids in controlling cell migration and mobilization.Citation60-Citation63 Thus, the immunomodulatory effects exerted by IMiDs seem to influence the BM niche in a way non-del5q/5q AML cells physically detach from BM stroma. Future in vivo imaging studies will precisely define the mechanisms underlying the lenalidomide-mediated AML chemotaxis.
Author contributions
BLM: designed and performed experiments, analyzed data and wrote the manuscript. HRH, RDG, DRM, CP, FG, PC, AI, CB: designed and performed experiments, and analyzed data. EA, JAPS, JABG, FAA, MR, PM, AUI: provided leukemic samples. PM: conceived the study, designed experiments, analyzed data and wrote the manuscript.
Supplemental Material
Download Zip (48.3 KB)Acknowledgments
This work was supported by the European Research Council (CoG-2014-646903 to P.M), the Spanish Ministry of Economy and Competitiveness (SAF-SAF2013-43065, RTC-2016-4603-1 to P.M), the Asociación Española Contra el Cáncer (AECC-CI-2015), FERO Foundation, and the ISCIII/FEDER (PI14-01191) to C.B and the ‘‘Fundación Hay Esperanza’’ to E.A. P.M also acknowledges financial support from The Obra Social La Caixa-Fundaciò Josep Carreras, The Inocente Inocente Foundation and The Generalitat de Catalunya (SGR330). P.M an investigator of the Spanish Cell Therapy cooperative network (TERCEL). We thank Celgene Corporation (San Diego, CA) for providing IMiDs. We thank Judit Sopena and Mariano Graupera (IDIBELL, Barcelona) for their technical assistance with immunohistochamical analysis
Disclosure statement
The authors have nothing to disclose.
Supplemental Material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Porwit A, Bene MC. 2015. Acute leukemias of ambiguous origin. Am J Clin Pathol 144:361–376. doi:10.1309/AJCPSTU55DRQEGTE.
- Grimwade D, Ivey A, Huntly BJ. 2016. Molecular landscape of acute myeloid leukemia in younger adults and its clinical relevance. Blood 127:29–41. doi:10.1182/blood-2015-07-604496.
- Burnett AK. 2012. New induction and postinduction strategies in acute myeloid leukemia. Curr Opin Hematol 19:76–81. doi:10.1097/MOH.0b013e3283500a92.
- Dohner H, Estey E, Grimwade D. 2017. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 129:424–447. doi:10.1182/blood-2016-08-733196.
- Diaz de la Guardia R, Lopez-Millan B, Lavoie JR. 2017. Detailed characterization of mesenchymal stem/stromal cells from a large cohort of AML patients demonstrates a definitive link to treatment outcomes. Stem Cell Rep 8:1573–1586. doi:10.1016/j.stemcr.2017.04.019.
- Korn C, Mendez-Ferrer S. 2017. Myeloid malignancies and the microenvironment. Blood 129:811–822. doi:10.1182/blood-2016-09-670224.
- Jelinek T, Kufova Z, Hajek R. 2016. Immunomodulatory drugs in AL amyloidosis. Crit Rev Oncol Hematol 99:249–260. doi:10.1016/j.critrevonc.2016.01.004.
- Maffei R, Colaci E, Fiorcari S. 2016. Lenalidomide in chronic lymphocytic leukemia: the present and future in the era of tyrosine kinase inhibitors. Crit Rev Oncol Hematol 97:291–302. doi:10.1016/j.critrevonc.2015.09.003.
- Ghosh N, Grunwald MR, Fasan O, Bhutani M. 2015. Expanding role of lenalidomide in hematologic malignancies. Cancer Manag Res 7:105–119. doi:10.2147/CMAR.
- 2006. New treatment for myelodysplastic syndrome. FDA Consum 40:4.
- Schey SA, Fields P, Bartlett JB. 2004. Phase I study of an immunomodulatory thalidomide analog, CC-4047, in relapsed or refractory multiple myeloma. J Clin Oncol 22:3269–3276. doi:10.1200/JCO.2004.10.052.
- Moros A, Bustany S, Cahu J. 2014. Antitumoral activity of lenalidomide in in vitro and in vivo models of mantle cell lymphoma involves the destabilization of cyclin D1/p27KIP1 complexes. Clin Cancer Res 20:393–403. doi:10.1158/1078-0432.CCR-13-1569.
- Moros A, Rodriguez V, Saborit-Villarroya I. 2014. Synergistic antitumor activity of lenalidomide with the BET bromodomain inhibitor CPI203 in bortezomib-resistant mantle cell lymphoma. Leukemia 28:2049–2059. doi:10.1038/leu.2014.106.
- Chen Y, Borthakur G. 2013. Lenalidomide as a novel treatment of acute myeloid leukemia. Expert Opin Investig Drugs 22:389–397. doi:10.1517/13543784.2013.758712.
- Bennett JM, Catovsky D, Daniel MT. 1985. Proposed revised criteria for the classification of acute myeloid leukemia. Report French-American-British Cooperative Group Annals Internal Medicine 103:620–625.
- Dohner H, Estey EH, Amadori S. 2010. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood 115:453–474. doi:10.1182/blood-2009-07-235358.
- Grimwade D, Lo Coco F. 2002. Acute promyelocytic leukemia: a model for the role of molecular diagnosis and residual disease monitoring in directing treatment approach in acute myeloid leukemia. Leukemia 16:1959–1973. doi:10.1038/sj.leu.2402721.
- Bueno C, Montes R, Martin L. 2008. NG2 antigen is expressed in CD34+ HPCs and plasmacytoid dendritic cell precursors: is NG2 expression in leukemia dependent on the target cell where leukemogenesis is triggered?. Leukemia 22:1475–1478. doi:10.1038/leu.2008.134.
- Bueno C, Montes R, Menendez P. 2010. The ROCK inhibitor Y-27632 negatively affects the expansion/survival of both fresh and cryopreserved cord blood-derived CD34+ hematopoietic progenitor cells: Y-27632 negatively affects the expansion/survival of CD34+HSPCs. Stem Cell Rev 6:215–223. doi:10.1007/s12015-010-9118-5.
- de la Guardia RD, Correa JG, Lopez-Millan B. 2017. Detection of inflammatory monocytes but not mesenchymal stem/stromal cells in peripheral blood of patients with myelofibrosis. Br J Haematol. 2018 Apr;181(1):133–137.
- Serrano-Heras G, Cuenca-Lopez MD, Montero JC. 2015. Phospho-kinase profile of colorectal tumors guides in the selection of multi-kinase inhibitors. Oncotarget 6:31272–31283. doi:10.18632/oncotarget.5211.
- Cuenca-Lopez MD, Serrano-Heras G, Montero JC. 2015. Antitumor activity of the novel multi-kinase inhibitor EC-70124 in triple negative breast cancer. Oncotarget 6:27923–27937. doi:10.18632/oncotarget.4736.
- Romero-Moya D, Bueno C, Montes R. 2013. Cord blood-derived CD34+ hematopoietic cells with low mitochondrial mass are enriched in hematopoietic repopulating stem cell function. Haematologica 98:1022–1029. doi:10.3324/haematol.2012.079244.
- Lopez-Millan B, Diaz De La Guardia R, Roca-Ho H. 2017. Therapeutic effect of the immunomodulatory drug lenalidomide, but not pomalidomide, in experimental models of rheumatoid arthritis and inflammatory bowel disease. Exp Mol Med 49:e290. doi:10.1038/emm.2016.143.
- Rodriguez R, Rosu-Myles M, Arauzo-Bravo M. 2014. Human bone marrow stromal cells lose immunosuppressive and anti-inflammatory properties upon oncogenic transformation. Stem Cell Rep 3:606–619. doi:10.1016/j.stemcr.2014.08.005.
- Gratama JW, Menendez P, Kraan J, Orfao A. 2000. Loss of CD34(+) hematopoietic progenitor cells due to washing can be reduced by the use of fixative-free erythrocyte lysing reagents. J Immunol Methods 239:13–23. doi:10.1016/S0022-1759(00)00154-X.
- Prieto C, Lopez-Millan B, Roca-Ho H. 2017. NG2 antigen is involved in leukemia invasiveness and central nervous system infiltration in MLL-rearranged infant B-ALL. Leukemia. 2018 Mar;32(3):633–644.
- Wunderlich M, Mizukawa B, Chou FS. 2013. AML cells are differentially sensitive to chemotherapy treatment in a human xenograft model. Blood 121:e90–7. doi:10.1182/blood-2012-10-464677.
- Prieto C, Stam RW, Agraz-Doblas A. 2016. Activated KRAS cooperates with MLL-AF4 to promote extramedullary engraftment and migration of cord blood CD34+ HSPC but is insufficient to initiate leukemia. Cancer Res 76:2478–2489. doi:10.1158/0008-5472.CAN-15-2769.
- Bueno C, Montes R, Melen GJ. 2012. A human ESC model for MLL-AF4 leukemic fusion gene reveals an impaired early hematopoietic-endothelial specification. Cell Res 22:986–1002. doi:10.1038/cr.2012.4.
- Rubio R, Gutierrez-Aranda I, Saez-Castillo AI. 2013. The differentiation stage of p53-Rb-deficient bone marrow mesenchymal stem cells imposes the phenotype of in vivo sarcoma development. Oncogene 32:4970–4980. doi:10.1038/onc.2012.507.
- Bueno C, Roldan M, Anguita E. 2014. Bone marrow mesenchymal stem cells from patients with aplastic anemia maintain functional and immune properties and do not contribute to the pathogenesis of the disease. Haematologica 99:1168–1175. doi:10.3324/haematol.2014.103580.
- Battula VL, Le PM, Sun JC. 2017. AML-induced osteogenic differentiation in mesenchymal stromal cells supports leukemia growth. JCI Insight. 2017 Jul 6;2(13):e90036.
- Konopleva MY, Jordan CT. 2011. Leukemia stem cells and microenvironment: biology and therapeutic targeting. J Clin Oncol 29:591–599. doi:10.1200/JCO.2010.31.0904.
- Medyouf H, Mossner M, Jann JC. 2014. Myelodysplastic cells in patients reprogram mesenchymal stromal cells to establish a transplantable stem cell niche disease unit. Cell Stem Cell 14:824–837. doi:10.1016/j.stem.2014.02.014.
- Nervi B, Ramirez P, Rettig MP. 2009. Chemosensitization of acute myeloid leukemia (AML) following mobilization by the CXCR4 antagonist AMD3100. Blood 113:6206–6214. doi:10.1182/blood-2008-06-162123.
- Bendall L. 2017. Extracellular molecules in hematopoietic stem cell mobilisation. Int J Hematol 105:118–128. doi:10.1007/s12185-016-2123-y.
- Aiuti A, Webb IJ, Bleul C, Springer T, Gutierrez-Ramos JC. 1997. The chemokine SDF-1 is a chemoattractant for human CD34+ hematopoietic progenitor cells and provides a new mechanism to explain the mobilization of CD34+ progenitors to peripheral blood. J Exp Med 185:111–120. doi:10.1084/jem.185.1.111.
- Ades L, Prebet T, Stamatoullas A. 2017. Lenalidomide combined with intensive chemotherapy in acute myeloid leukemia and higher-risk myelodysplastic syndrome with 5q deletion. Results of a phase II study by the Groupe Francophone Des Myelodysplasies. Haematologica 102:728–735. doi:10.3324/haematol.2016.151894.
- Jemal A, Siegel R, Xu J, Ward E. 2010. Cancer statistics, 2010. CA Cancer J Clin 60:277–300.
- Burnett A, Wetzler M, Lowenberg B. 2011. Therapeutic advances in acute myeloid leukemia. J Clin Oncol 29:487–494. doi:10.1200/JCO.2010.30.1820.
- Dombret H, Gardin C. 2016. An update of current treatments for adult acute myeloid leukemia. Blood 127:53–61. doi:10.1182/blood-2015-08-604520.
- Goldstone AH, Burnett AK, Wheatley K. 2001. Attempts to improve treatment outcomes in acute myeloid leukemia (AML) in older patients: the results of the United Kingdom Medical Research Council AML11 trial. Blood 98:1302–1311. doi:10.1182/blood.V98.5.1302.
- Bradstock KF, Matthews JP, Lowenthal RM. 2005. A randomized trial of high-versus conventional-dose cytarabine in consolidation chemotherapy for adult de novo acute myeloid leukemia in first remission after induction therapy containing high-dose cytarabine. Blood 105:481–488. doi:10.1182/blood-2004-01-0326.
- Lowenberg B, Pabst T, Vellenga E. 2011. Cytarabine dose for acute myeloid leukemia. N Engl J Med 364:1027–1036. doi:10.1056/NEJMoa1010222.
- Karjalainen R, Pemovska T, Popa M. 2017. JAK1/2 and BCL2 inhibitors synergize to counteract bone marrow stromal cell-induced protection of AML. Blood 130:789–802. doi:10.1182/blood-2016-02-699363.
- Hartwell KA, Miller PG, Mukherjee S. 2013. Niche-based screening identifies small-molecule inhibitors of leukemia stem cells. Nat Chem Biol 9:840–848. doi:10.1038/nchembio.1367.
- McMillin DW, Delmore J, Weisberg E. 2010. Tumor cell-specific bioluminescence platform to identify stroma-induced changes to anticancer drug activity. Nat Med 16:483–489. doi:10.1038/nm.2112.
- McMillin DW, Negri JM, Mitsiades CS. 2013. The role of tumour-stromal interactions in modifying drug response: challenges and opportunities. Nat Rev Drug Discovery 12:217–228. doi:10.1038/nrd3870.
- Blum W, Klisovic RB, Becker H. 2010. Dose escalation of lenalidomide in relapsed or refractory acute leukemias. J Clin Oncol 28:4919–4925. doi:10.1200/JCO.2010.30.3339.
- Chen Y, Kantarjian H, Estrov Z. 2012. A phase II study of lenalidomide alone in relapsed/refractory acute myeloid leukemia or high-risk myelodysplastic syndromes with chromosome 5 abnormalities. Clin Lymphoma Myeloma Leuk 12:341–344. doi:10.1016/j.clml.2012.04.001.
- Fehniger TA, Uy GL, Trinkaus K. 2011. A phase 2 study of high-dose lenalidomide as initial therapy for older patients with acute myeloid leukemia. Blood 117:1828–1833. doi:10.1182/blood-2010-07-297143.
- Mollgard L, Saft L, Treppendahl MB. 2011. Clinical effect of increasing doses of lenalidomide in high-risk myelodysplastic syndrome and acute myeloid leukemia with chromosome 5 abnormalities. Haematologica 96:963–971. doi:10.3324/haematol.2010.039669.
- Sekeres MA, Gundacker H, Lancet J. 2011. A phase 2 study of lenalidomide monotherapy in patients with deletion 5q acute myeloid leukemia: southwest Oncology Group Study S0605. Blood 118:523–528. doi:10.1182/blood-2011-02-337303.
- Pollyea DA, Kohrt HE, Gallegos L. 2012. Safety, efficacy and biological predictors of response to sequential azacitidine and lenalidomide for elderly patients with acute myeloid leukemia. Leukemia 26:893–901. doi:10.1038/leu.2011.294.
- Ramsingh G, Westervelt P, Cashen AF. 2013. A phase 1 study of concomitant high-dose lenalidomide and 5-azacitidine induction in the treatment of AML. Leukemia 27:725–728. doi:10.1038/leu.2012.214.
- Kronke J, Fink EC, Hollenbach PW. 2015. Lenalidomide induces ubiquitination and degradation of CK1alpha in del(5q) MDS. Nature 523:183–188. doi:10.1038/nature14610.
- Smith AE, Kulasekararaj AG, Jiang J. 2015. CSNK1A1 mutations and isolated del(5q) abnormality in myelodysplastic syndrome: a retrospective mutational analysis. Lancet Haematol 2:e212–21. doi:10.1016/S2352-3026(15)00050-2.
- Uy GL, Rettig MP, Motabi IH. 2012. A phase 1/2 study of chemosensitization with the CXCR4 antagonist plerixafor in relapsed or refractory acute myeloid leukemia. Blood 119:3917–3924. doi:10.1182/blood-2011-10-383406.
- Sanchez L, Gutierrez-Aranda I, Ligero G. 2011. Enrichment of human ESC-derived multipotent mesenchymal stem cells with immunosuppressive and anti-inflammatory properties capable to protect against experimental inflammatory bowel disease. Stem Cells 29:251–262. doi:10.1002/stem.569.
- Choudhuri A, Fast EM, Zon LI. 2017. Using zebrafish to study pathways that regulate hematopoietic stem cell self-renewal and migration. Stem Cell Rep 8:1465–1471. doi:10.1016/j.stemcr.2017.05.018.
- Rettig MP, Ansstas G, DiPersio JF. 2012. Mobilization of hematopoietic stem and progenitor cells using inhibitors of CXCR4 and VLA-4. Leukemia 26:34–53. doi:10.1038/leu.2011.197.
- Hoggatt J, Pelus LM. 2010. Eicosanoid regulation of hematopoiesis and hematopoietic stem and progenitor trafficking. Leukemia 24:1993–2002. doi:10.1038/leu.2010.216.