
ABSTRACT
Cervical cancer develops as a result of infection with high-risk human papillomavirus (HPV) through persistent expression of early proteins E6 and E7. Our group pioneered a recombinant viral vector system based on Semliki Forest virus (SFV) for vaccination against cervical cancer. The most striking benefit of this alphavirus vector-based vaccine platform is its high potency. DNA vaccines on the other hand, have a major advantage with respect to ease of production. In this study, the benefits associated with both SFV-based vaccines and DNA vaccines were combined with the development of a DNA-launched RNA replicon (DREP) vaccine targeting cervical cancer. Using intradermal delivery followed by electroporation, we demonstrated that DREP encoding for E6,7 (DREP-E6,7) induced effective, therapeutic antitumor immunity. While immunizations with a conventional DNA vaccine did not prevent tumor outgrowth, immunization with a 200-fold lower equimolar dose of DREP (0.05 µg of DREP) resulted in approximately 85% of tumor-free mice. To overcome the safety concern of potential malignant transformation at the vaccination site, we evaluated the anti-tumor effect of a DREP vaccine encoding a shuffled version of E7 (DREP-E7sh). DREP-E7sh delayed tumor growth yet not to the same extent as DREP-E6,7. In addition, inclusion of a helper cassette and an ER targeting signal (sigHelp) did not significantly further enhance the suppression of tumor outgrowth in the long term, albeit exhibiting better tumor control early after immunization. Collectively, this study points towards the clinical evaluation of DREP encoding HPV antigens as a potent immunotherapy for patients with HPV16 (pre)-malignancies.
Introduction
Cervical cancer is one of the most prominent causes of deaths in women worldwide.Citation1,Citation2 The disease develops as a result of persistent infection with high-risk human papillomavirus (HPV) in (pre)malignant cervical intraepithelial lesions.Citation3 HPV16, one of the most common high-risk subtypes, is responsible for half of the global cervical cancer cases with expression of early proteins E6 and E7 essential for cellular transformation.Citation4,Citation5 Due to the persistent expression of these oncoproteins in the tumor epithelia, they are ideal targets for the various types of immunotherapeutic strategies developed for HPV-induced malignancies.Citation2
We previously explored the HPV-specific immunogenicity of a Semliki Forest virus-based cancer vaccine vector (VREP) encoding a fusion protein of HPV16 E6 and E7.Citation6 This platform resulted in 100% anti-tumor efficacy in pre-clinical studies providing a promising immunotherapeutic strategy in the clinic.Citation7 The potency of this SFV-based replicon particle strategy stems from the intrinsic adjuvant property of the encoded replicase. The replicase is responsible for the self-replication of the introduced recombinant alphaviral RNA in the infected cell. The replicase activity and the amplification of RNA results in intermediates that are recognized by pattern recognition receptors (PRR). Stimulation of PRR leads to a high production of type I interferon.Citation8,Citation9 Likely the antigenicity of the replicase is also partly due to the expression of strong helper epitopes. Subsequent to the antigen production, apoptosis of the infected cells is induced leading to activation of dendritic cells resulting in CD8 + T cell responses.Citation10,Citation11 As the production of these VREP particles for clinical use is laborious, we aimed to combine its inherent potency with the advantages associated with DNA vaccines.Citation12
Conventional DNA vectors have been poorly translated to humans with most clinical data demonstrating only weak to modest responses. Hence, several efforts have been made to improve the potency of DNA vectors. These efforts include increasing the transfection frequency in vivoCitation13,Citation14 most notably using DNA electroporation (EP).Citation15 The EP procedure itself induces inflammation while being safe and tolerable. EP further improves the potency of DNA vectors with administration in the skin. The skin is populated with antigen-presenting cells such as Langerhans cells and dermal dendritic cells for efficient priming of naïve CD8+ T cells. We have demonstrated higher intrinsic immune efficacy of our VREP vaccine via delivery in the skin as compared to the intramuscular (i.m.) route.Citation16,Citation17
Another way to enhance the potency of DNA vaccination is in the vector itself. For instance, we and others have constructed DNA vectors based on the replicase of alphaviruses such as Semliki Forest virus (SFV), Sindbis virus and Venezuelan equine encephalitis virus (VEE). These DNA replicon (DREP) vectors have been shown to elicit T cell responses superior to those of conventional plasmid DNA in preclinical studies against infectious agents such as human immunodeficiency virus or chikungunya virus.Citation18,Citation19 This has also been demonstrated within the context of cancer vaccination.Citation20,Citation21 Nevertheless, to date, no SFV-based DNA replicon vaccine has been explored as an alternative for VREP for eliciting effective anti-tumor immunity.
In this study, we developed an SFV-based DREP vaccine against HPV-induced cancers. We compared the in vivo immunogenicity of our DREP vaccine with that of conventional DNA in naïve and tumor-bearing mice using intradermal (i.d.) EP as a delivery method. This promising next-generation vaccine exhibited superior dose-sparing effects with up to a 1000-fold lower dose (0.05 µg versus 10–50 µg) required to elicit potent tumor immunity compared to the standard dose currently used for conventional DNA vaccines in most studies.
Results
HPV DNA vaccines
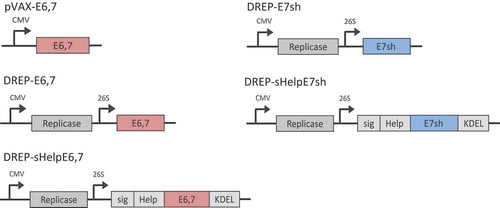
Two types of plasmids encoding HPV proteins were used in this study (). The first, pVAX1, is a conventional plasmid used in the development of DNA vaccines where the expression cassette is under the control of a cytomegalovirus (CMV) promoter. The second type is an SFV-based DNA-launched RNA replicon (DREP) vaccine encoding the SFV replicase, where the transcription of the viral replicon is under the control of a CMV promoter and where the mRNA encoding for the foreign antigens is expressed from an internal viral promoter on the replicon RNA. These vectors were constructed to express an E6,7 fusion protein. Since chromosomal integration could be a safety concern in connection with DNA vaccination, we additionally constructed a DREP vector expressing a shuffled E7 (E7sh) protein. The shuffled version of E7 loses its transforming potential but retains relevant T cell epitopes.Citation22 To potentially further enhance the immunogenicity of DREP, we included in the construct a universal “helper-cassette” with a series of T-helper epitopes as previously described (Help).Citation23 A human growth hormone signal peptide and a KDEL sequence was also added to achieve ER localization and retention (sig).Citation23 The KDEL sequences prevents the antigen from being secreted from the ER.Citation23 In previous work, within the context of HPV viral vector and DNA vaccination, we have demonstrated increased anti-HPV immunity by inclusion of sigHelp.Citation24
Figure 1. DNA vectors. Schematic representation of conventional (pVAX1) and replicon DNA (DREP) vectors and corresponding antigens (E6,7 or E7sh). CMV, cytomegalovirus promoter; 26S, subgenomic 26S promoter.
Potent immunotherapeutic effect of DREP over conventional DNA
First, we evaluated the intrinsic adjuvant property of the SFV replicase in our DNA vaccine and compared it to conventional plasmid DNA lacking the replicase. The replicase drives the amplification of replicon RNA that includes the antigen of interest. As a result, a large amount of antigen is produced. To demonstrate the enhanced expression of the transgene, E6,7, we transfected BHK-21 with either pVAX-E6,7 or DREP-E6,7 (Supplementary ). Expression of E6,7 was analyzed using flow cytometry 24 hours after transfection using an antibody targeting E7. pVAX-Luc and DREP-Luc were used as negative controls. The percentage of cells transfected with pVAX-eE6,7 and DREP-E6,7 were 8.1% and 21.1%, respectively. On a per cell basis, the expression of E6,7 was 2.5-fold higher in cells transfected with DREP-E6,7. As DREP is approximately 3x larger than pVAX, transfection with DREP results in a 7.5-fold higher expression level on a per mole basis. Hence, we demonstrate that DREP strongly enhances the expression of our antigen of interest.
Next, we determined whether the immunostimulatory properties of DREP enhances HPV-specific immunity of DREP-E6,7 above that of pVAX-eE6,7. A preferred route of immunization for DNA vaccines is the intradermal (i.d.) route aided by in vivo EP. For DREP, we have previously shown superior responses against viral targets using EP compared to intradermal administration alone.Citation18 We therefore utilized in vivo EP in this study. We immunized mice twice with an equimolar amount of DREP and pVAX (10 and 3.2 μg, respectively) with a 20-day interval. E7-specific responses were evaluated in splenocytes 7 days after the last immunization. At 10 ug, DREP induces a significantly higher percentage of E7-specific CD8+ T cells and higher number of IFN-γ-producing cells compared to pVAX immunization ().
Figure 2. HPV-specific immunity and therapeutic effect by DREP-E6,7 over conventional DNA. C57BL/6 mice (n = 3–4 per group) were immunized i.d. followed by EP at a 20-day interval with DREP-E6,7 at 10 µg or an equimolar dose of pVAX-E6,7 (3.2 µg). Phosphate buffered saline (PBS) was injected as a negative control. Seven days after the boost mice were sacrificed and spleens were isolated for assessment of percentages of E7-specific CD8+ T cells and the number of IFN-γ-producing cells with dextramer staining (A) and ELISpot analysis (B), respectively. For assessment of therapeutic effect, C57BL/6 mice were inoculated s.c. with 2 × 104 TC-1 and mice were immunized i.d. followed by EP on days 7, 14 and 21. Doses include 10, 0.2 or 0.05 µg for DREP and 3.2 µg for pVAX. Tumors were palped twice a week for 62 days. The individual growth curves for each corresponding group is depicted in (C) and the average tumor sizes and standard error of the mean per group until day 28 is shown in (D). The group size and number of mice that are still alive at day 62 are indicated in the numbers in each panel. The survival percentages are depicted in (E). The results (C-E) are pooled from two separate experiments of 7 mice/group resulting in a total of 14 mice. Blood was drawn from 3–4 mice for assessment of E7-specific CD8 + T cells with dextramer staining in mice immunized with DREP one week after the last immunization. As a cut-off for positivity for E7-specific T cells, we collected blood from mice that were still alive in the PBS control (N = 1) and pVAX group (N = 2) at that time point (F). *P < 0.05; **P < 0.01; ***P < 0.001.
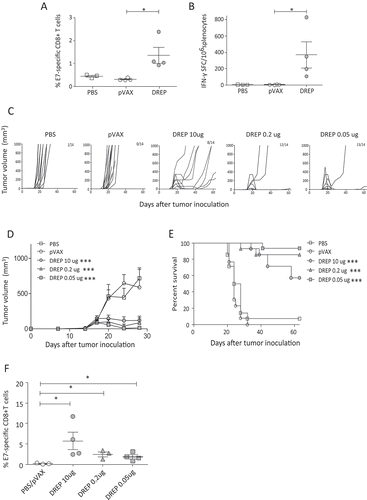
Given the enhanced E7-specific immunogenicity of DREP, we subsequently determined the therapeutic effect of DREP-E6,7 using an HPV tumor model (TC-1) (). Mice were immunized with DREP or pVAX 7, 14 and 21 days after TC-1 inoculation. In this model, palpable tumors develop approximately 14 days post inoculation. In non-vaccinated mice, tumors grow exponentially from that point onward to approximately 1000 mm3 3–4 weeks after inoculation. Due to the exponential growth of these tumors immunizations need to be scheduled weekly. This may not be the preferred schedule for replicon vaccines. We previously demonstrated that weekly injection of replicon vaccines (before contraction of the CD8 + T cell response), results in lower memory responses.Citation18,Citation25 Hence we could speculate that the frequent injections as well as the high dose could synergistically weaken the immune response. Immunization with 3.2 µg of pVAX resulted in the same tumor growth pattern as for non-vaccinated mice. An equimolar dose of DREP (10 µg) induced a significant delay in tumor growth, with approximately 43% of mice remaining tumor-free by day 60. We additionally included a group receiving a dose of 20 µg of pVAXeE6,7 as Oosterhuis et al. had applied, yet this also resulted in tumor outgrowth as seen in control mice (data not shown).Citation23 Hence, the enhanced immunogenicity with DREP also translated to effective anti-tumor immunity in this fast growing HPV tumor model.
Previous studies using DREP have also observed potent immunogenicity with doses lower than 10 µg. Reducing the dose 125 times (from 10 μg to 0.08 μg) demonstrated no significant loss of T cell responses.Citation18 Moreover, dosages higher than 10 μg results in lower antigen-specific responses.Citation18,Citation25 For this reason, we also evaluated the anti-tumor effect of DREP at doses lower than 10 μg. Using the same schedule after tumor inoculation, we immunized mice with 0.2 and 0.05 μg of DREP. With all doses, there was a significant delay in tumor growth compared to pVAX or PBS control starting at 20 days post tumor inoculation (). Immunization with either dose resulted in a similar survival rate (). One week after the last immunization, the percentage of E7-specific T cells was determined in blood, and although there was an observable, yet insignificant increase in E7-specific T cells with a high dose (10 ug), this increase was reversibly correlated with anti-tumor control ().
The inclusion of sigHelp-KDEL enhances the frequency of HPV-specific T cells but doesn’t further enhance in vivo anti-tumor effects
We next assessed whether inclusion of sigHelp-KDEL, a series of helper T cell epitopes and an ER targeting signal, enhances the immunogenicity of DREP. Mice were immunized twice with 10 µg of DREP. E7-specific responses were evaluated in splenocytes 9 days after the last immunization. At 10 µg, DREP-sHelpE6,7 induces a significantly higher percentages of E7-specific CD8+ T cells and a higher number of IFN-γ-producing cells compared to DREP-E6,7 ().
Figure 3. Inclusion of sigHelp on HPV-specific immunity and therapeutic effect by DREP-E6,7. C57BL/6 mice (n = 3 per group) were immunized i.d. followed by EP at a 20-day interval with 10 µg of DREP-eE6,7 or DREP-sHelpE6,7. Phosphate buffered saline (PBS) was injected as a negative control. Seven days after the boost mice were sacrificed and spleens were isolated for assessment of percentages of E7-specific CD8+ T cells and number of IFN-γ-producing cells with dextramer staining (A) and ELISpot analysis (B), respectively. For assessment of therapeutic effect, C57BL/6 mice (n = 7 per group) were inoculated s.c. with 2 × 104 TC-1 and mice were immunized i.d. followed by EP on days 7, 14 and 21. DREP was administered as a dose of 0.2 µg. Tumors were palped twice a week for 108 days. The individual growth curves for each corresponding group is depicted in (C) and the average tumor sizes and standard error of the mean per group is shown in (D) until day 28. The group size and number of mice that are still alive at day 108 are indicated in the numbers in each panel. The survival percentages are depicted in (E). *P < 0.05; **P < 0.01; ***P < 0.001.
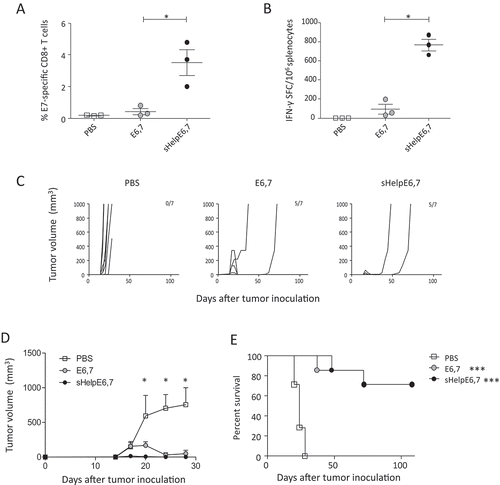
We next determined whether the inclusion of sigHelp enhancing the anti-tumor effect DREP to achieve possibly 100% of tumor-free mice (). We immunized mice with 0.2 µg of DREP with the same TC-1 model. Significant tumor control above that of the buffer control was achieved at a dosage of 0.2 µg for both DREP constructs. Inclusion of sHelp-KDEL did not significantly delay tumor growth in DREP-E6,7 resulting in similar survival rates (). This was also the case for a dosage of 0.05 µg (Supplementary Figure 2). However, albeit not significant, there is an observable decrease in the initial outgrowth of tumors with inclusion of sHelp-KDEL demonstrating early control by induced HPV immunity.
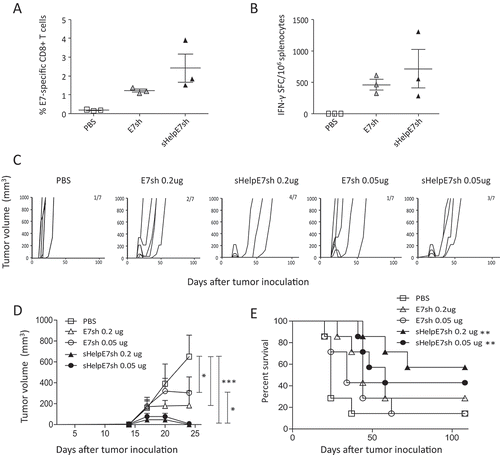
Previous studies have demonstrated prolonged survival with inclusion of sHelp-KDEL for conventional DNA vaccination targeting another HPV antigen.Citation23 This antigen, developed by Ohlschlager et al., is a shuffled version of E7 (E7sh) to avoid the risk of cellular transformation at the vaccination site.Citation22 We wanted to assess whether inclusion of sHelp-KDEL could also enhance immunity compared to DREP-E7sh. Naïve mice were immunized with 10 μg of either DREP-sHelpE7sh or DREP-E7sh (). The frequency of E7-specific CD8+ T cells in spleen was assessed at day 9 after the last immunization. At this point a trend toward an increased frequency of E7-specific T cells and number of IFN-γ-producing cells was observed for DREP-sHelpE7sh. The anti-tumor effect upon inclusion of sHelp-KDEL in DREP-E7sh was further assessed. Mice were immunized with 0.2 and 0.05 µg of DREP encoding E7sh with the same schedule as above using the TC-1 model (). DREP-sHelpE7sh resulted in tumors that regressed around day 20 with 57% and 43% of the mice being tumor-free with doses of 0.2 and 0.05 μg, respectively. This is in contrast to immunization with DREP-E7sh with suppression of tumor growth only achieved in 29% and 14% of mice immunized with doses of 0.2 and 0.05 μg, respectively (). At 28 days post tumor inoculation, there is a significant control of tumor outgrowth by all DREP constructs compared to PBS control. This was also the case for DREP-sHelpE7sh compared to DREP-E7sh at a dose of 0.05 ug (). Despite this observation, the delay in survival was insignificant between the two matched dose groups ().
Figure 4. Inclusion of sigHelp on HPV-specific immunity and therapeutic effect by DREP-E7sh. C57BL/6 mice (n = 3 per group) were immunized i.d. followed by EP at a 20-day interval with 10 µg of DREP-E7sh or DREP-sHelpE7sh. The negative control from was also used within this experiment. Seven days after the boost mice were sacrificed and spleens were isolated for assessment of percentages of E7-specific CD8+ T cells and number of IFN-γ-producing cells with dextramer staining (A) and ELISpot analysis (B). For assessment of therapeutic effect, C57BL/6 mice (n = 7 per group) were inoculated s.c. with 2 × 104 TC-1 and mice were immunized i.d. followed by EP on days 7, 14 and 21. DREP was administered at a dose of 0.2 or 0.05 ug. Tumors were palped twice a week for 108 days. The individual growth curves for each corresponding group is depicted in (C) and the average tumor sizes and standard error of the mean per group is shown in (D) until day 28. The group size and number of mice that are still alive at day 108 are indicated in the numbers in each panel. The survival percentages are depicted in (E). *P < 0.05; **P < 0.01; ***P < 0.001.
Discussion
In this study, we demonstrated potent therapeutic anti-tumor efficacy provided by a DNA replicon vector with doses as low as 0.05 µg. This is a 1000-fold lower dose as compared to most studies using conventional pDNA as application in cancer immunotherapy. Additionally, in this work, we are the first to develop a replicon DNA vaccine candidate based on Semliki Forest virus targeting HPV. Our SFV DNA-launched RNA replicon, encoding a stable fusion protein of HPV oncogenes E6 and E7 delivered by in vivo electroporation resulted in approximately 85% of mice remaining tumor-free by day 108.
The high immunogenicity of DREP over that of conventional pDNA (pVAX) is most likely due to the self-amplifying nature of the SFV replicase, resulting in high copies of viral RNA transcripts for stimulation of innate immunity.Citation9,Citation26,Citation27 The replicase also produces 5ʹ-triphosphate dsRNA of non-viral host cell RNA templates.Citation28 The RNA transcripts signal innate immunity through RNA sensing by Toll-like receptor 3 (TLR3), TLR7, TLR8, MDA-5, RIG-I and protein kinase R for a type I IFN response.Citation28 The type I IFN response induces T cell memory responses with the alphaviral replicase inducing apoptosis in targeted cells which in turn is taken up by dendritic cells, favoring antigen spreading and stimulating cross-presentation for adaptive immunity.Citation29,Citation30 The potent anti-tumor effect of DREP is likely also attributable to enhanced in vivo delivery using EP previously shown to be superior above others in the case of HPV vaccination.Citation31,Citation32 Intradermal administration of DREP-E6,7 with in vivo EP significantly increased E7-specific T cell frequencies above that of pVAX. The immunogenicity of the two DNA vectors was compared with equivalent molar doses administered. However, as seen by others pVAX was also not immunogenic at a dose of 10 µg or less, regardless of the administration method. Citation23,Citation33 In those studies there were also no detectable responses observed with repeated administrations of 20 µg of pVAX, with anti-tumor control only demonstrated within the context of an optimized DNA vector.Citation23
The tumor growth was significantly delayed when immunizing with DREP. Interestingly, lower doses of DREP induced even stronger anti-tumor efficacy with as low as 0.05 µg with 85% of mice remaining tumor-free by the last palpation. This observed inverse correlation between dose and immunogenicity of alphaviral DNA replicons is not unprecedented, as it has been previously observed in other studies with type I IFNs halting the production and amplification of the replicon due to their effect on cells undergoing an antiviral state.Citation8,Citation18 This decreased immunogenicity was observed for both humoral and cellular immune responses.Citation18,Citation34 Interestingly, upon assessment for frequencies of E7-specific T cells in blood, we hadn’t observed a significant difference between the different doses used. It is possible that the magnitude is not indicative of better tumor control. This conclusion was also made in one recent study using the TC-1 tumor model and vaccination with conventional DNA encoding E7sh.Citation35 The authors achieved 100% survival by combining vaccination with CD27 agonism and programmed death receptor 1 (PD1) blockade.Citation35 At day 15 after tumor inoculation, the combined treatments resulted in approximately 20% of E7-specific T cells compared to 60% with replacing PD1 blockade with cytotoxic T-lymphocyte-associated protein 4 (CTLA-4). However, CTLA-4 blockade with CD27 agonism and vaccination resulted in half of mice surviving by day 100. It would be of interest to determine a better correlate of therapeutic efficacy. In addition, the immunization schedule in the TC-1 tumor model includes three injections at a one-week interval as dictated by the fast growth of the tumor. In a previous study, we have demonstrated that boosting at a short interval of 1 week with replicon SFV significantly decreased cellular immunity.Citation25 It may be that injection of a short interval further decreases the strength of the immune response, yet this affects memory rather than acute responses. This decrease is not likely a result of vector-specific immune responses as we previous demonstrated in a study using an homologous prime-boost immunization with rSFV.Citation36 Other studies have also demonstrated that multiple booster immunizations do not result in weaker immune responses and no vector-specific cellular responses were observed, despite the same vector backbone used.Citation37,Citation38 Rather than the magnitude, the quality of the CD8 + T cell response may correlate with effective tumor control.Citation39 It was observed that increasing the dose of replicon particles favored T effector memory (Tem) development and inhibited T central memory (Tcm) formation.Citation25 This may have implications within the context of tumor immunity as Tcm cells possess recall proliferation capacity that are required for effective anti-tumor control.Citation40,Citation41 It has also been previously observed that homologous SFV replicon immunization results in better tumor control over homologous adenovirus immunization or heterologous immunization due to a favored central memory T cell response.Citation39,Citation42 It would also be of value to determine whether this decrease is due to T cell exhaustion and hence, assessment of exhaustion markers, such as programmed death protein 1 (PD-1) is warranted.
Removal of the transforming potential of oncogenes, such as of E6 and E7, is a prerequisite before applying DNA vaccines in particular in the clinic. For this purpose, a “shuffled” version of E7 was incorporated into DREP and compared with that of DREP-E6,7 in terms of immunogenicity. Again, despite the lower E7-specific CD8 + T cells observed in spleen with E6,7 compared to E7sh, the former presented with better tumor control. The greater anti-tumor control by DREP-E6,7 is likely due to both E6 and E7 responses for optimal anti-tumor effect as is the case for SFV VREP encoding E6 and E7. In a preclinical study with SFV replicon particles we also demonstrated higher anti-tumor immunity with the inclusion of both E6 and E7 compared to either oncogene alone.Citation43 Furthermore, in patients with HPV neoplasia both E6 and E7-specific CD8 + T cell responses have been correlated with better prognostic value.Citation44 Despite the lower immunogenicity observed with the variant of E7, administration of either a 0.2 or 0.05 ug dose of E7sh resulted in a significant delay in tumor growth compared to the control group. This is in contrast to a previous study on pVAX encoding E7sh, with no responses detected with as high dosages as 20 ug.Citation23 The low immunogenicity of pVAX-E7SH is rescued in these studies with the addition of helper epitopes and a retention signal with insertion of SigHelp-KDEL.Citation33 In a previous study using VREP particles, the insertion of SigHelp-KDEL resulted in a higher percentage of E7-specific effector T cells at both early and late time points after immunization.Citation24 Significant differences in the effector memory T cell phenotype was only significantly higher early after immunization.Citation24 In this study, we observed an increase in anti-tumor effect with inclusion DREP-sHelpE7SH, which was not the case for DREP-sHelpE6,7. This may be due to the high threshold of response set by both E6 and E7. Although it is difficult to compare this finding with the study of Oosterhuis et al.Citation23 given the different delivery methods, we nevertheless demonstrate significant anti-tumor efficacy at a 100x-lower dose of DNA upon inclusion of the SFV replicase.
In conclusion, we generated DNA-based SFV replicon vaccines which are able to elicit potent HPV-specific anti-tumor responses with unprecedented low dosages of DNA. This DNA replicon strategy could pave the way for clinical translation of vaccines applicable to a wide range of target antigens.
Materials and methods
DNA vectors
The plasmids that were used in this study are either that of conventional DNA (pVAX 1) or a DNA-launched RNA replicon (DREP) encoding HPV oncogenes E6 and/or E7. Encoded in the plasmids is foot-and-mouth disease virus 2A autoprotease inserted after the translational enhancer consisting of the first 34 amino acids of the capsid (e).Citation45 For the generation of pVAX-E6,7, the sigE7KDEL fragment was removed from pVAX1sigE7KDEL (kindly provided by Prof. T Schumacher (The Netherlands Cancer Institute The Netherlands)) by digestion with PmeI (producing PmeI-pVAX1-PmeI). pUC19e-E6,7 served as a template, digested with SmaI and HincII producing SmaI-eE6,7-HincII and treated with FastAp Alkaline Phosphatase to prevent self-ligation. SmaI-eE6,7-HincII was ligated into pVAX1 to produce pVAX-E6,7. For construction of pVAXeLuc, pVAXeE6,7 and pSFV3eLuc plasmids were used. These constructs were digested with ApaI and XbaI. ApaI-eLuc-XbaI from pSFV3 was cloned into XbaI-pVAX-ApaI to produce pVAXeLuc. DREP-OVA served as a template to produce DREP-Luc and DREP-E6,7. DREP-Luc was constructed by cloning Luc from pSFV3eLuc via digestion of both plasmids with ApaI and XmaI. DREP-E6,7 was constructed by cloning eE6,7 from pSFV20eE6,7 digestion of both plasmids with HindIII and NotI. DREP-E6,7 was used for the construction of all remaining DREP plasmids. DREP-E7sh was constructed by cloning the NotI-E7SH-XmaI fragment from the construction of pSFVeE7SH, as previously described.Citation24 The E7SH fragment was inserted into DREP upon removal of E6,7 by SmaI and PspOMI digestion. The DREP backbone was additionally ligated with sigHELP-E6,7-KDEL or sigHELP-E6,7-KDEL to create DREP-sHelpE6,7 or DREP-sHelpE7sh, respectively. The ligated inserts were both obtained from pVAX plasmids encoding either of the two antigensCitation23 by NotI and SmaI digestion upon introduction of these restriction sites with PCR. All restriction enzymes were purchased from Thermo Scientific. All plasmids were expressed and amplified in E.coli NEB10b 10, purified by an endotoxin-free DNA purification kit (Qiagen) and dissolved in phosphate-buffered saline (PBS) (Gibco, Invitrogen) and digested by enzyme restriction to confirm the correct size of the antigen. The constructs were further confirmed by sequencing.
Cell lines
Baby hamster kidney cells (BHK-21) were obtained from the American Type Culture Collection (No. CCL-10). TC-1 tumor cells, transformed with a retroviral vector expressing HPV16 E6E7, and C3, expressing the complete HPV16 genome, were received as a kind gift from Prof. C. Melief (Leiden University Medical Center, The Netherlands). These cells were cultured as previously described.Citation24
Quantification of E7-expressing cells
BHK-21 cells were transfected with 10 ug of DNA at a ratio of 1:4 with lipofectamine2000 according to the manufacturer’s instructions. Cells were harvested 24 hours later and stained with LIVE/DEAD fixable violet dead cell stain kit (Life technologies) according the manufacturer’s manuals. This was followed with an intracellular staining using a primary antibody targeting HPV16-E7 (clone 8C9) and subsequently secondary goat anti-mouse (H + L) AlexaFluor647 (Life Technologies). FACS analysis was performed with LSR-II flow cytometer (BD Bioscience) and data analyzed using FlowJo analysis software (Tree Star).
Mice and immunizations
Female inbred C57BL/6J (H-2b) female mice were bred at the Department of Microbiology, Tumor and Cell Biology, at the Karolinska Institutet or purchased from Scanbur Research. All mice were 8 to 10 weeks of age at the start of all experiments and immunized i.d. with in vivo EP as previous described.Citation18 For assessment of immunogenicity, mice were primed and boosted with 10 ug of DREP or 3.2 ug of pVAX (equimolar amounts). For assessment of anti-tumor efficacy mice were immunized three times at one week intervals at various indicated doses.
Mouse splenocyte isolation
This procedure was performed as previously described.Citation25
Blood lymphocyte isolation
Mice were bled from the tail vein and blood was collected in tubes containing heparin. Samples were lysed with red blood cell lysis buffer for 5 min after which 10 mL addition of complete RPMI medium (RPMI 1640 supplemented with 5% fetal calf serum, 2 mM L-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin (all from Gibco or Invitrogen). Then samples were centrifuged at 400 X g for 5 min, and each cell pellet was resuspended in 200 uL of complete RPMI medium.
Quantification of E7-specific cells
Splenocytes or peripheral blood collected from the tail vein were subjected to erythrocyte lysis and stained with PE-RAHYNIVTF-dextramers (Immudex) and PE-Cy7-anti-CD8a Ab (clone: 53–6.7) according to manufacturer’s protocol. FACS analysis was performed with LSR-II flow cytometer (BD Bioscience) and data analyzed using FlowJo analysis software (Tree Star, Ashland, OR).
IFN-γ elispot assay
This assay was performed as previously described.Citation18 In brief, MultiScreen-IP plates (Millipore) were activated with 70% ethanol and coated overnight with anti-IFN-gamma antibodies (AN18; Mabtech) 4°C. After washing with PBS, plates were blocked with complete RPMI medium for approx. 2 h at 37°C. Freshly isolated spleen cells then replaced the blocking medium at 0.2 million cells per well and were stimulated in triplicate with either 2 ug/ml of the E7-derived peptide (RAHYNIVTF, H-2Db) (Proimmune) or medium alone. Concanavalin A (Sigma-Aldrich) at 2 ug/ml in duplicates was used as a positive control. Plates were incubated at 37°C with 5% CO2 for 20 ± 2 h and developed as recommended by the manufacturer using biotinylated anti-IFN-gamma detector antibody (R4-6A2), streptavidin-alkaline phosphatase, and BCIP-NBT Plus substrate (Mabtech). Spots were counted using an ImmunoSpot analyzer and ImmunoSpot software (Cellular Technology) and multiplied by 5 to represent the number of spots/1 million cells.
Tumor inoculation
Mice were inoculated subcutaneously (s.c.) with 2 × 104 TC-1 cells expressed both HPV16 E6 and E7 suspended in 0.2 ml PBS. Tumor volume was determined using a caliper performed by the same skilled technician twice per week starting at day 7. Mice were sacrificed once the tumor volume exceeded 1000 mm3.
Statistical analysis
Analysis was performed using GraphPad Prism software. For comparison between two groups, the Mann-Whitney U test was used. For analysis of differences between survival curves, log-rank (Mantel-Cox) test was used. Two-way ANOVA with Bonferroni post-test was used for comparison of tumor growth curves at different time points. A P < 0.05 is considered statistically significant.
Declaration of interest
Toos Daemen and Hans Nijman are stock holders/founders of ViciniVax, a spin-off company of the UMCG, developing therapeutic cancer vaccines.
Supplemental Material
Download Zip (107.4 KB)Acknowledgments
We would like to thank Kenth Andersson for the technical assistance of the animal facility at the Department of Microbiology, Tumor and Cell Biology, Karolinska Institutet, Sweden. Additionally, we would like to thank Ton N. Schumacher, John B. Haanen and Koen Oosterhuis for providing us the plasmid DNA sigHelp-E7SH-KDEL construct. This work was supported by the Dutch Cancer Society under Grant RUG-2008-4066 and by the Swedish Research Council under Grant K2013-56X-09494-23-3.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Fitzmaurice C, Dicker D, Pain A, Hamavid H, Moradi-Lakeh M, MacIntyre MF, Allen C, Hansen G, Woodbrook R, Wolfe C, et al. The global burden of cancer 2013. JAMA Oncol [ Internet] 2015;1:505–527. [Accessed 2015 Jul 29]. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4500822&tool=pmcentrez&rendertype=abstract.
- Zur Hausen H. Papillomaviruses and cancer: from basic studies to clinical application. Nat Rev Cancer [ Internet] 2002;2:342–350. [Accessed 2014 Jul 17]. http://www.ncbi.nlm.nih.gov/pubmed/12044010.
- Bosch FX, Lorincz A, Muñoz N, Meijer CJLM, Shah KV. The causal relation between human papillomavirus and cervical cancer. J Clin Pathol [ Internet] 2002;55:244–265. [Accessed 2015 Feb 25]. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1769629&tool=pmcentrez&rendertype=abstract.
- Scheurer ME, Tortolero-Luna G, Adler-Storthz K. Human papillomavirus infection: biology, epidemiology, and prevention. Int J Gynecol Cancer [ Internet] 2005;15:727–746. [Accessed 2015 Oct 4]. http://www.ncbi.nlm.nih.gov/pubmed/16174218.
- Brinkman JA, Hughes SH, Stone P, Caffrey AS, Muderspach LI, Roman LD, Weber JS, Kast WM. Therapeutic vaccination for HPV induced cervical cancers. Dis Markers [ Internet] 2007;23:337–352. [Accessed 2015 Oct 4]. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3851105&tool=pmcentrez&rendertype=abstract.
- Daemen T, Regts J, Holtrop M, Wilschut J. Immunization strategy against cervical cancer involving an alphavirus vector expressing high levels of a stable fusion protein of human papillomavirus 16 E6 and E7. Gene Ther [ Internet] 2002;9:85–94. [Accessed 2015 Aug 17]. http://www.ncbi.nlm.nih.gov/pubmed/11857066.
- Daemen T, Riezebos-Brilman A, Regts J, Dontje B, Van Der Zee A, Wilschut J. Superior therapeutic efficacy of alphavirus-mediated immunization against human papilloma virus type 16 antigens in a murine tumour model: effects of the route of immunization. Antivir Ther. 2004;9:733–742. https://www.ncbi.nlm.nih.gov/pubmed/15535411
- Näslund TI, Kostic L, Nordström EKL, Chen M, Liljeström P. Role of innate signalling pathways in the immunogenicity of alphaviral replicon-based vaccines. Virol J. 2011;8:36. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3038947/
- Ljungberg K, Liljeström P. Self-replicating alphavirus RNA vaccines. Expert Rev Vaccines [ Internet] 2015;14:177–194. [Accessed 2015 Sep 27]. http://www.ncbi.nlm.nih.gov/pubmed/25269775.
- Chen M, Barnfield C, Näslund TI, Fleeton MN, Liljeström P. MyD88 expression is required for efficient cross-presentation of viral antigens from infected cells. J Virol [ Internet] 2005;79:2964–2972. [Accessed 2015 Dec 6]. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=548467&tool=pmcentrez&rendertype=abstract.
- Schulz O, Diebold SS, Chen M, Näslund TI, Nolte MA, Alexopoulou L, Azuma Y-T, Flavell RA, Liljeström P, Reis E Sousa C. Toll-like receptor 3 promotes cross-priming to virus-infected cells. Nature [ Internet] 2005;433:887–892. [Accessed 2017 Apr 11]. http://www.nature.com/doifinder/10.1038/nature03326.
- Kutzler MA, Weiner DB. DNA vaccines: ready for prime time? Nat Rev Genet [ Internet] 2008;9:776–788. [Accessed 2015 Oct 27]. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4317294&tool=pmcentrez&rendertype=abstract.
- van de Wall S, Nijman HW, Daemen T. HPV-specific immunotherapy: key role for immunomodulators. Anticancer Agents Med Chem [ Internet] 2014;14:265–279. http://www.ncbi.nlm.nih.gov/pubmed/24237218.
- Lee S-H, Danishmalik SN, Sin J-I. DNA Vaccines, electroporation and their applications in cancer treatment. Hum Vaccin Immunother [ Internet] 2015. http://www.tandfonline.com/doi/full/10.1080/21645515.2015.1035502.
- Abdulhaqq SA, Weiner DB. DNA vaccines: developing new strategies to enhance immune responses. Immunol Res [ Internet] 2008;42:219–232. [Accessed 2015 Dec 6]. http://www.ncbi.nlm.nih.gov/pubmed/19066740.
- van de Wall S, Walczak M, Van Rooij N, Hoogeboom B-N, Meijerhof T, Nijman H, Daemen T. Tattoo Delivery of a Semliki Forest Virus-Based Vaccine Encoding Human Papillomavirus E6 and E7. Vaccines [ Internet] 2015;3:221–238. [Accessed 2015 Aug 7]. http://www.mdpi.com/2076-393X/3/2/221/htm.
- Kis EE, Winter G, Myschik J. Devices for intradermal vaccination. Vaccine. 2012;30:523–538. doi:10.1016/j.vaccine.2011.11.020.
- Knudsen ML, Mbewe-Mvula A, Rosario M, Johansson DX, Kakoulidou M, Bridgeman A, Reyes-Sandoval A, Nicosia A, Ljungberg K, Hanke T, et al. Superior induction of T cell responses to conserved HIV-1 regions by electroporated alphavirus replicon DNA compared to that with conventional plasmid DNA vaccine. J Virol [ Internet] 2012;86:4082–4090. [Accessed 2015 Jul 13]. http://jvi.asm.org/content/86/8/4082.long.
- Knudsen ML, Ljungberg K, Tatoud R, Weber J, Esteban M, Liljeström P. Alphavirus replicon DNA expressing HIV antigens is an excellent prime for boosting with recombinant modified vaccinia Ankara (MVA) or with HIV gp140 protein antigen. PLoS One [ Internet] 2015;10:e0117042. [Accessed 2016 Feb 7]. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4314072&tool=pmcentrez&rendertype=abstract.
- Yamanaka R, Xanthopoulos KG. Induction of antigen-specific immune responses against malignant brain tumors by intramuscular injection of sindbis DNA encoding gp100 and IL-18. DNA Cell Biol [ Internet] 2005;24:317–324. [Accessed 2015 Dec 6]. http://www.ncbi.nlm.nih.gov/pubmed/15869409.
- Leslie MC, Zhao Y-J, Lachman LB, Hwu P, Wu G-J, Bar-Eli M. Immunization against MUC18/MCAM, a novel antigen that drives melanoma invasion and metastasis. Gene Ther [ Internet] 2007;14:316–323. [Accessed 2016 Feb 7]. http://www.ncbi.nlm.nih.gov/pubmed/17024104.
- Ohlschläger P, Pes M, Osen W, Dürst M, Schneider A, Gissmann L, Kaufmann AM. An improved rearranged Human Papillomavirus Type 16 E7 DNA vaccine candidate (HPV-16 E7SH) induces an E7 wildtype-specific T cell response. Vaccine [ Internet] 2006;24:2880–2893. [Accessed 2015 Aug 17]. http://www.ncbi.nlm.nih.gov/pubmed/16472545.
- Oosterhuis K, Aleyd E, Vrijland K, Schumacher TN, Haanen JB. Rational design of DNA vaccines for the induction of human papillomavirus type 16 E6- and E7-specific cytotoxic T-cell responses. Hum Gene Ther. 2012;1312:121031083217002.
- Peng IP, Boerma A, Walczak M, Oosterhuis K, Haanen JB, Schumacher TN, Nijman HW, Daemen T. Antigen design enhances the immunogenicity of Semliki Forest virus-based therapeutic human papillomavirus vaccines. Gene Ther [ Internet] 2015;560–567. http://www.nature.com/doifinder/10.1038/gt.2015.24.
- Knudsen ML, Ljungberg K, Kakoulidou M, Kostic L, Hallengärd D, García-Arriaza J, Merits A, Esteban M, Liljeström P. Kinetic and phenotypic analysis of CD8+ T cell responses after priming with alphavirus replicons and homologous or heterologous booster immunizations. J Virol [ Internet] 2014;88:12438–12451. [Accessed 2015 Jan 14]. http://www.ncbi.nlm.nih.gov/pubmed/25122792.
- Nordström EKL, Forsell MNE, Barnfield C, Bonin E, Hanke T, Sundström M, Karlsson GB, Liljeström P. Enhanced immunogenicity using an alphavirus replicon DNA vaccine against human immunodeficiency virus type 1. J Gen Virol [ Internet] 2005;86:349–354. [Accessed 2015 Jul 14]. http://www.ncbi.nlm.nih.gov/pubmed/15659754.
- Leitner WW, Ying H, Restifo NP. DNA and RNA-based vaccines: principles, progress and prospects. Vaccine [ Internet] 1999;18:765–777. [Accessed 2015 Sep 27]. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1986720&tool=pmcentrez&rendertype=abstract.
- Nikonov A, Mölder T, Sikut R, Kiiver K, Männik A, Toots U, Lulla A, Lulla V, Utt A, Merits A, et al. RIG-I and MDA-5 detection of viral RNA-dependent RNA polymerase activity restricts positive-strand RNA virus replication. PLoS Pathog [ Internet] 2013;9:e1003610. [Accessed 2016 Jun 21]. http://www.ncbi.nlm.nih.gov/pubmed/24039580.
- Diebold SS, Schulz O, Alexopoulou L, Leitner WW, Flavell RA, Reis E Sousa C. Role of TLR3 in the immunogenicity of replicon plasmid-based vaccines. Gene Ther [ Internet] 2009;16:359–366. [Accessed 2015 Jan 1]. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2655288&tool=pmcentrez&rendertype=abstract.
- Glasgow GM, McGee MM, Sheahan BJ, Atkins GJ. Death mechanisms in cultured cells infected by Semliki Forest virus. J Gen Virol [ Internet] 1997;78(Pt 7):1559–1563.[Accessed 2016 Feb 7]. http://www.ncbi.nlm.nih.gov/pubmed/9225029.
- Best SR, Peng S, Juang C-M, Hung C-F, Hannaman D, Saunders JR, Wu T-C, Pai SI. Administration of HPV DNA vaccine via electroporation elicits the strongest CD8+ T cell immune responses compared to intramuscular injection and intradermal gene gun delivery. Vaccine [ Internet] 2009;27:5450–5459. [Accessed 2015 Aug 7]. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2745985&tool=pmcentrez&rendertype=abstract.
- Trimble CL, Morrow MP, Kraynyak KA, Shen X, Dallas M, Yan J, Edwards L, Parker RL, Denny L, Giffear M, et al. Safety, efficacy, and immunogenicity of VGX-3100, a therapeutic synthetic DNA vaccine targeting human papillomavirus 16 and 18 E6 and E7 proteins for cervical intraepithelial neoplasia 2/3: a randomised, double-blind, placebo-controlled phase 2b trial. Lancet (London, England) [ Internet] 2015. [Accessed 2015 Sep 22]. http://www.ncbi.nlm.nih.gov/pubmed/26386540.
- Oosterhuis K, Ohlschläger P, Van Den Berg JH, Toebes M, Gomez R, Schumacher TN, Haanen JB. Preclinical development of highly effective and safe DNA vaccines directed against HPV 16 E6 and E7. Int J Cancer. 2011;129;397–406. doi:10.1002/ijc.25894.
- Leitner WW, Ying H, Driver DA, Dubensky TW, Restifo NP. Enhancement of tumor-specific immune response with plasmid DNA replicon vectors. Cancer Res. 2000;60:51–55.
- Ahrends T, Baba AN, Xiao Y, Yagita H, Van Eenennaam H, Borst J. CD27 agonism plus PD-1 blockade recapitulates CD4+ T cell help in therapeutic anti-cancer vaccination. Cancer Res [ Internet] 2016;1–12. http://cancerres.aacrjournals.org/cgi/doi/10.1158/0008-5472.CAN-15-3130.
- Lambeck AJA, Nijman HW, Hoogeboom BN, Regts J, De Mare A, Wilschut J, Daemen T. Role of T cell competition in the induction of cytotoxic T lymphocyte activity during viral vector-based immunization regimens. Vaccine. 2010;28;4275–4282. doi:10.1016/j.vaccine.2010.04.033.
- Berglund P, Fleeton MN, Smerdou C, Liljeström P. Immunization with recombinant Semliki Forest virus induces protection against influenza challenge in mice. Vaccine [ Internet] 1999;17:497–507. [Accessed 2016 Nov 15]. http://www.ncbi.nlm.nih.gov/pubmed/10073729.
- Uematsu Y, Vajdy M, Lian Y, Perri S, Greer CE, Legg HS, Galli G, Saletti G, Otten GR, Rappuoli R, et al. Lack of interference with immunogenicity of a chimeric alphavirus replicon particle-based influenza vaccine by preexisting antivector immunity. Clin Vaccine Immunol [ Internet] 2012;19:991–998. [Accessed 2018 May 12]. http://cvi.asm.org/lookup/doi/10.1128/CVI.00031-12.
- Näslund TI, Uyttenhove C, Nordström EKL, Colau D, Warnier G, Jondal M, Van Den Eynde BJ, Liljeström P. Comparative prime-boost vaccinations using Semliki Forest virus, adenovirus, and ALVAC vectors demonstrate differences in the generation of a protective central memory CTL response against the P815 tumor. J Immunol [ Internet] 2007;178:6761–6769. [Accessed 2017 Apr 18]. http://www.ncbi.nlm.nih.gov/pubmed/17513723.
- Klebanoff CA, Gattinoni L, Restifo NP. CD8+ T-cell memory in tumor immunology and immunotherapy. Immunol Rev [ Internet] 2006;211:214–224. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1501075&tool=pmcentrez&rendertype=abstract.
- Klebanoff CA, Gattinoni L, Torabi-Parizi P, Kerstann K, Cardones AR, Finkelstein SE, Palmer DC, Antony PA, Hwang ST, Rosenberg SA, et al. Central memory self͞tumor-reactive CD8 T cells confer superior antitumor immunity compared with effector memory T cells. 2005 [Accessed 2017 Apr 17]; http://www.pnas.org/content/102/27/9571.full.pdf
- Walczak M, De Mare A, Riezebos-Brilman A, Regts J, Hoogeboom B-N, Visser JT, Fiedler M, Jansen-Dürr P, Van Der Zee AGJ, Nijman HW, et al. Heterologous prime-boost immunizations with a virosomal and an alphavirus replicon vaccine. Mol Pharm [ Internet] 2011;8:65–77. [Accessed 2017 Apr 11]. http://www.ncbi.nlm.nih.gov/pubmed/20825215.
- Peng S, Tomson TT, Trimble C, He L, Hung C-F, Wu T-C. A combination of DNA vaccines targeting human papillomavirus type 16 E6 and E7 generates potent antitumor effects. Gene Ther [ Internet] 2006;13:257–265. [Accessed 2015 Aug 17]. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2174916&tool=pmcentrez&rendertype=abstract.
- Piersma SJ, Jordanova ES, Van Poelgeest MIE, Kwappenberg KMC, Van Der Hulst JM, Drijfhout JW, Melief CJM, Kenter GG, Fleuren GJ, Offringa R, et al. High number of intraepithelial CD8+ tumor-infiltrating lymphocytes is associated with the absence of lymph node metastases in patients with large early-stage cervical cancer. Cancer Res [ Internet] 2007;67:354–361. [Accessed 2015 Sep 30]. http://www.ncbi.nlm.nih.gov/pubmed/17210718.
- Smerdou C, Liljeström P. Two-helper RNA system for production of recombinant Semliki forest virus particles. J Virol [ Internet] 1999;73:1092–1098. [Accessed 2016 Nov 15]. http://www.ncbi.nlm.nih.gov/pubmed/9882310.