
ABSTRACT
Immune checkpoint blockade (ICB) is currently evaluated in patients with glioblastoma (GBM), based on encouraging clinical data in other cancers, and results from studies with the methylcholanthrene-induced GL261 mouse glioma. In this paper, we describe a novel model faithfully recapitulating some key human GBM characteristics, including low mutational load, a factor reported as a prognostic indicator of ICB response. Consistent with this observation, SB28 is completely resistant to ICB, contrasting with treatment sensitivity of the more highly mutated GL261. Moreover, SB28 shows features of a poorly immunogenic tumor, with low MHC-I expression and modest CD8+ T-cell infiltration, suggesting that it may present similar challenges for immunotherapy as human GBM. Based on these key features for immune reactivity, SB28 may represent a treatment-resistant malignancy likely to mirror responses of many human tumors. We therefore propose that SB28 is a particularly suitable model for optimization of GBM immunotherapy.
Introduction
Glioblastoma (GBM), the most prevalent primary brain tumor, has a very poor prognosis despite standard of care treatment comprising surgical resection followed by radiochemotherapy.Citation1,Citation2 In other cancers immunotherapy is already revolutionizing therapeutic options, which has encouraged testing similar approaches in GBM.Citation3-Citation6 Nevertheless, the absence of any clinical breakthrough to date indicates that better understanding of the particularities of human GBM in response to immunotherapy is needed. Unfortunately, to achieve this, human GBM cells cannot be used for most preclinical mechanistic studies of immunotherapy because of the obligation to use a fully immunocompetent animal model. Mouse models are frequently used and two main categories can be considered: spontaneous or orthotopically implanted. Spontaneous models can be engineered to recapitulate molecular characteristics of human GBM, using known oncogenes leading to malignant transformation. These models are invaluable for many aspects of glioma biology, but variations in penetrance and kinetics do not always lend themselves to testing immunotherapy protocols.Citation7,Citation8 For orthotopically implanted models, cells from GBM cultured lines can be implanted in a syngeneic recipient mouse of a chosen age, at a chosen time, using a predetermined number of cells known to reproducibly generate tumors in vivo within a certain time.Citation9
The immunotherapies for GBM most intensively studied to date focused on T-cell mediated responses. These have been induced by therapeutic vaccines, after adoptive T-cell therapy, or indirectly through immune checkpoint blockade (ICB).Citation3,Citation6 For many years it was assumed that CD8+ T cells would be the principle effector cell, able to kill GBM cells expressing a tumor-associated peptide bound to MHC-I molecules. However, since, MHC-I can be downregulated in human GBM, this may jeopardize the efficacy of anti-tumoral immune responses unless the immunotherapeutic strategy also restores expression.Citation10,Citation11 Regarding CD4+ T cells recognizing MHC-II presented peptides, their role in GBM anti-tumor immunity is now being recognized, with IDH mutation-specific CD4+ T cells observed in patients, and the therapeutic potential of CD4+ T-cell promoting vaccines being demonstrated in brain tumor models.Citation12,Citation13
One of the most clinically impressive immunotherapy modalities reported to date is based on ICB using antibodies.Citation14,Citation15 In cancer indications for which efficacy is demonstrated in both animal models and human cancer (e.g. melanoma), the mechanisms behind ICB are becoming clearer. Specifically, the invigorated anti-tumor immunity seems to be manifested by both CD4+ and CD8+ T cells specific for neoepitopes arising from unique mutations in the tumors.Citation16 For ICB and brain tumors, there are encouraging clinical results in the case of melanoma and non-small-cell lung cancer brain metastases.Citation17,Citation18 Comparable data targeting the PD-1/PD-L1 axis in GBM has yet to be published;Citation19 moreover, there are uncertainties regarding use of PD-L1 expression as a biomarker in GBM, confounded by the lack of standardized methodology for its detection in tumor tissue.Citation20,Citation21 Clinical correlates of responsiveness to ICB include an infiltration with T cells prior to treatment, and the mutational load of the tumor.Citation22 Regarding T-cell infiltration, this is highly variable, but not considered to be abundant.Citation20,Citation23 Human GBM is mainly considered not highly mutated, even if cases of hypermutated GBM are described, as with POLE deficiency, biallelic mismatch repair deficiency, or even within areas of the same tumor.Citation24-Citation27 Based on this current knowledge of human GBM, some mouse models are starting to be analyzed for the same critical features.
Two of the most used orthotopically implanted models in GBM immunotherapy are the methylcholanthrene-induced GL261 model and the SMA-560 model, which is of spontaneous origin. The mutational landscapes of these models have been characterized in vitro, with a surprisingly high number of mutations and predicted neoepitopes in both models.Citation28 More recently, we reported the SB28 GBM model that was genetically engineered to target p53, RAS and PDGF in vivo, but which can be used as a cell line with predictable tumor formation and kinetics after orthotopic grafting in syngeneic mice.Citation29 However, to date, the mutational landscape of SB28 has not been described, nor has its suitability been assessed for testing clinically translatable ICB.
In this study, we assessed in vitro expression of key molecules involved in immune interactions, and report that SB28 may be less visible than GL261 to T-cell immunosurveillance due to absence of constitutively expressed MHC-I and MHC-II. However, both cell lines may ultimately impede immune attack due to IFNγ inducible expression of PD-L1. Whole exome sequencing and RNA sequencing of in vitro cultured, low passage SB28 cells revealed a very low mutational load for SB28 and consequently few predicted neoepitopes. Resequencing SB28 after in vivo passage revealed acquisition of further mutations, but mutational load remained low and similar to human GBM. Immunohistological analysis of SB28 showed the tumor invading normal brain parenchyma, with a sparse T-cell infiltration. An immunotherapy protocol based on anti-PD-1 and anti-CTLA-4 double ICB was curative in over 50% of GL261 bearing mice, but totally ineffective in SB28. These results suggest that SB28 will be a highly stringent model for optimizing immunotherapy that may reflect treatment resistance of certain human GBM.
Materials and methods
Cell lines
Murine SB28 and GL261 were cultured in DMEM containing 4.5 g/l glucose and 10% FCS. For cell culture, cells were detached from plastic with accutase (Sigma-Aldrich). The cell lines were tested mycoplasma-negative.
Immunophenotyping and antibodies
Where indicated, we used IFNγ (Immunotools) at 100U/ml for 48 h prior to staining. For cell surface staining, we used the following antibodies: from BD Pharmingen: anti-MHC-I (H-2Db) biotin (KH95), anti-FAS (CD95) PE-Cy7 (Jo2), anti-PD-L1 (CD274) BV421 (MIH5), anti-CD40 Biotin (3/23), anti-ICAM-1 (CD54) PE (3E2), anti-MHC-II (I-A/I-E) Biotin (2G9), Streptavidin APC. From Biolegend: anti-CD80 APC (16-10A1), anti-PD-1 (CD279) PE-Cy7 (29F.1A12), anti-CD86 AF647 (GL-1), Streptavidin PE-Cy7. And from Molecular probes: Streptavidin PE. Data were collected on a Gallios flow cytometer (Beckman Coulter) using Kaluza for Gallios software (Beckman Coulter, version1.0) and processed using Kaluza analysis software (Beckman Coulter, version 1.5)
Mice, implantation procedures, and treating antibodies
Female, 6–8 weeks C57BL/6J mice (Charles River Laboratories) were implanted intracranially with glioma cells using a stereotaxic apparatus (Stoelting). Mice were anesthetized with a mixture of Ketamine 80 mg/kg (Warner-Lambert) and Rompun 10 mg/kg (Bayer). The indicated number of GBM cells was injected in 2 μl HBSS in the pallidum (2.6 mm lateral to the bregma and 3.5 mm below the skull). Mice were monitored daily and sacrificed at a veterinary authority-stipulated endpoint (15% weight loss and/or other clinical signs and symptoms). Antibodies for in vivo experiments were: anti-PD-1 (RMP1-14), anti-CTLA-4 (9D9), rat IgG2a isotype control (2A3), and mouse IgG2b isotype control (MPC-11) all from BioXcell. All animal experimental studies were reviewed and approved by institutional and cantonal veterinary authorities in accordance with Swiss Federal law.
Genomic analysis
DNA and RNA were extracted from dry pellets with AllPrep DNA/RNA Mini Kit (Qiagen). Whole exome sequencing (WES) was performed using an Agilent protocol with 100 pair-end reads. MuTect and Haplotype Caller were used for variant calling, and SNPeff for variant annotation. Frameshift and missense mutations are noted for each cell line. To predict peptide binding we used to following algorithms: NetMHCPan 4.0/IEDBv2.18 MHCI for MHC-I, and NetMHCIIpan 3.2/IEDBv2.18 MHCII for MHC-II. Gene ontology analysis was done using PANTHER. Displayed number of mutations is always the median of all replicates (2 replicates for GL261 in vitro, 3 replicates for SB28 in vitro, 3 replicates for SB28 ex vivo). RNA-seq was performed using the TruSeq stranded RNA protocol on Illumina. The reads (50 bp) were mapped with the TopHat v2.0.13 (default parameters) software to the reference genome on new junctions and known annotations. Biological quality control and summarization were done with the PicardTools v1.80. Counts were obtained by HTSeq and Rsubread featureCounts. The normalization and differential expression analysis was performed with the R/Bioconductor package edgeR v.3.4.2, for the genes annotated in the reference genome.
Immunofluorescence
Brains were collected after trans-cardiac perfusion with Ringer’s solution, to avoid intravascular cell contamination, then immerged in 30% sucrose overnight and embedded in Tissue-Tek® OCT (Sakura) before freezing in liquid nitrogen. Samples were stored at −80°C, sectioned on a Cryostat (Leica), fixed with PFA 4% then blocked using 5% BSA and 2.5% normal goat serum (Sigma-Aldrich). Primary Ab used were: rabbit anti-GFP (Proteintech), hamster anti-CD3 (Biolegend, 145-2C11), rat anti-CD31 (Biolegend, MEC13.3), rat anti-CD8 (Thermofischer, 4SM15). Secondary Ab used were: goat anti-Rabbit AF488 (Abcam), donkey anti-Rabbit AF647 (Abcam), goat anti-hamster DyLight™ 488 (Biolegend), mouse anti-rat eFluor 660 (eBioscience, r2a-21B2). Hematoxylin and eosin staining was performed with standard protocols. Images were acquired with a Zeiss Axio Imager Z1, Axio Imager.Z2 Basis LSM 800, or a Zeiss Axioscan.Z1 microscope with either x20 or x40 objectives. Images were then processed using Zeiss Zen pro software.
Ex vivo cell sorting
Mice were sacrificed at d21 when tumor burden was certain. We isolated tumor cells (Adult Brain Dissociation kit, Miltenyi Biotec) and stained them with anti-CD45 PE (30-F11) and anti-CD11b AF700 (M1/70), both from Biolegend. 300,000 CD45−/CD11b−/GFP+ cells were sorted (FACSAria II, Becton Dickinson). DNA and RNA were purified (as described in Genomic analysis), and 3 samples were sequenced. Results shown for ex vivo samples are the median of 3 independent replicates.
Results
Flow cytometric phenotyping of SB28 and GL261 GBM cells as an indicator of immune visibility
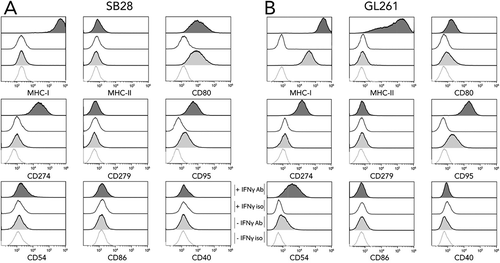
Interactions between tumor and immune cells in vivo rely upon cell-cell contact via specific molecular interactions. We used flow cytometry to examine expression of a panel of key molecules on SB28 and GL261 cells in vitro. Moreover, we also immunophenotyped cells after IFNγ stimulation to mimic a situation where IFNγ-secreting immune cells infiltrate the tumor. MHC-I and MHC-II molecules are essential for presenting peptide antigens to CD8+ and CD4+ T cells, respectively, and their expression was strikingly different between SB28 and GL261: GL261 constitutively expressed MHC-I, whereas SB28 was constitutively MHC-I negative (). After IFNγ stimulation, MHC-I expression was increased on GL261, and it was induced on SB28. Concerning MHC-II expression, this was IFNγ inducible for GL261, but negative for SB28. The immune regulatory molecule CD274 (PD-L1), was not constitutively expressed in either cell line, but was IFNγ inducible in both. CD80 and CD86 are ligands of the stimulatory CD28 receptor, and the inhibitory CTLA-4 receptorCitation30: CD80 was expressed constitutively by both lines, but more highly by SB28; CD86 was not detected. Expression of CD40 was tested because of its importance in immune interactions and its debated role in GBM,Citation31 but it was not expressed at the surface of either GBM line. An IFNγ-induced expression of the adhesion molecule CD54 (ICAM-I) was present in both models. The death receptor Fas ligand (CD95) was constitutively expressed in both lines and upregulated by IFNγ.
Figure 1. In vitro expression of key molecules for immune interaction by SB28 and GL261 glioma lines. Flow cytometry was used to assess surface expression of the indicated markers by SB28 (A) and GL261 (B). Filled curves: specific antibody staining (Ab); open curves: isotype control antibody (iso). Lower light-gray histograms show constitutive expression (-IFNγ); upper dark-gray histograms show staining of cells stimulated for 48 hours with 100 U/ml IFNγ (+ IFNγ). Representative staining profiles of 3 independent experiments.
SB28 has a lower mutational load than GL261
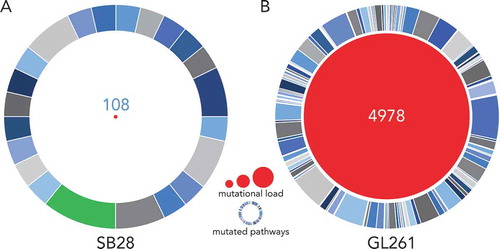
We analyzed the mutational landscape in SB28 and GL261 by whole exome sequencing (WES) of the respective cell lines. To our knowledge, such in-depth characterization of tumorigenic cell lines derived from an engineered GBM model such as SB28 has not previously been reported. We performed WES of in vitro GL261 and SB28 cells, compared with spleen cells from syngeneic mice to determine the number of tumor-specific missense and frameshift mutations. After application of all filters to the WES data (see methods), we identified 67 frameshift and 41 missense mutations for SB28, and 212 frameshift and 4766 missense mutations for GL261 (Supplementary Figure S1). The identity of all mutations identified for SB28 is available in Supplementary Table S1 (details were previously published for GL261Citation28). The difference in magnitude of the mutational load between the two models is striking, with a total of 108 somatic mutations for SB28, compared to almost 50-fold more mutations, 4978 in total, for GL261 (). Gene ontology analysis of the mutated genes from SB28 showed that most mutations were similarly distributed across multiple pathways. One exception was the PDGF signaling pathway with almost 10% of mutations involved (); this is consistent with the genetic strategy used to create the model, and reflects similar alterations commonly found in the proneural subtype of human GBMCitation32. The extremely large number of mutations detected in GL261 were predictably (in view of the carcinogen used for induction) diversely distributed, with few predominant pathways implicated (, Supplementary Table S1).Citation28
Figure 2. SB28 has a far lower mutational load than GL261 glioma. Whole exome sequencing (WES) was performed for in vitro SB28 (A) and GL261 (B), red circle size represents total number of non-synonymous somatic mutations (missense and frameshift). Outer circles represent pathways targeted by mutations, the most targeted for SB28 is highlighted in green, and corresponds to the PDGF signaling pathway. (Details of pathways targeted in Supplementary Table S1.).
SB28 is highly tumorigenic but modestly infiltrated by immune cells
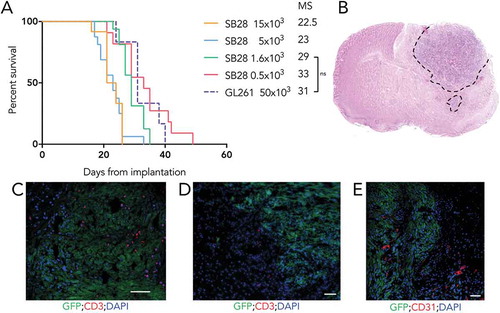
To study growth in vivo of our cell lines and assess their tumorigenicity, we implanted SB28 and GL261 cells orthotopically in syngeneic mice and determined symptom free survival (). For the SB28 model, a titration assay demonstrated an inverse correlation between the number of cells injected and the median survival. Importantly for a model assessing therapies, tumors developed in 100% of mice at all cell doses tested. Injecting less than 1,600 SB28 cells prolonged survival but gave a shallower survival curve. The group implanted with 1,600 cells had a steep survival curve, with a median survival (MS) of 29 days, offering a useable window for experimental procedures, and a predictable period of sacrifice. We also show survival after injection of mice with 50,000 GL261 cells (, dashed line); the MS was similar to that obtained with 1,600 SB28. This similar tumor growth using these cell numbers facilitates comparison of both models after therapy. To further characterize SB28 in vivo, we performed histological analysis of brains from untreated mice with advanced tumors. Hematoxylin and eosin staining showed high cellularity of the tumor area, with clear invasion of the normal parenchyma in some areas (). Immunofluorescence showed highly heterogeneous, but never abundant, T-cell infiltration (, D), comprised of both CD8+ (Supplementary Figure S2.) and CD4+ cells (not shown). Areas of hypervascularization, based on staining for CD31 were identified at the tumor margin (). Flow cytometry analysis of the infiltrating immune cells revealed a higher proportion of CD4+ T cells compared to CD8+ T cells, some of which were Foxp3+ Tregs. Around 50% of CD8 T cells expressed PD-1 and almost 60% of macrophages were PD-L1 positive (Supplementary figure S3). A modest T cell infiltrate was also detected in small tumors at day 13 after implantation (data not shown).
Figure 3. High tumorigenicity of SB28 and GL261 gliomas after orthotopic implantation. (A) Mice injected intracranially with the indicated numbers of SB28 glioma cells, or with 50,000 GL261 glioma cells; symptom-free survival is plotted, with corresponding median survival (MS) shown. (B) Hematoxylin and eosin staining (coronal section) from an SB28 implanted mouse at appearance of terminal symptoms, dashed line outlines tumor based on hypercellularity. (C, D, E) Representative photomicrographs showing tumor cells (GFP+), CD3+ T cells (C, D), and CD31+ vessels (E). Scale bar: 50 µm.
Mutations and predicted neoepitopes of SB28 after in vivo passage
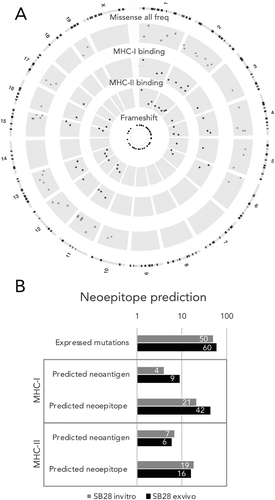
Mutations in cancer cells can give rise to neoepitopes that can render the tumors visible to adaptive anti-tumor immune responses present in an immunocompetent host. We therefore extended analyses to report not only on the SB28 cell line in vitro, but also after 21 days in vivo in syngeneic mice. We assessed whether these putative neoepitopes could be expressed, using RNA-seq. We then ran algorithms to predict peptide binding to MHC-I or MHC-II to estimate potential neoepitope formation (). In vitro, a median of 50 mutated genes were expressed; 4 neoantigens were predicted to generate neoepitopes for MHC-I and 7 for MHC-II (). To then determine whether the expressed mutanome had been sculpted after 21 days of orthotopic growth, we sorted GFP+ SB28 tumor cells. Whole exome sequencing and RNA-seq revealed a modest overall increase of mutations from a median of 108 to 132 mutations after in vivo growth (Supplementary Figure S4). Correspondingly, this increase in mutation number resulted in an increased number of neoantigens for MHC-I, with 9 neoantigens predicted, but more stable for MHC-II, with 6 predicted neoantigens ().
Figure 4. For SB28, number of predicted MHC-I neoepitopes increases after in vivo growth. (A) Circos plot showing genomic distribution of mutations and predicted neoepitopes, representative example of in vitro SB28. From outer to inner, representation of mouse karyotype, point mutation (based on allele frequency (missense all freq), inner is 0 outer is 0.5), MHC-I predicted peptides (plotted according to affinity inner 13.4 nM, outer 33880 nM), MHC-II predicted peptides (plotted according to affinity inner 213.9 nM, outer 27834.1 nM), frameshift mutations. (B, C) numbers of expressed mutated genes and predicted neoepitopes for MHC-I and MHC-II binding.
Differential sensitivity of SB28 and GL261 to anti-PD-1 and anti-CTLA-4 blockade
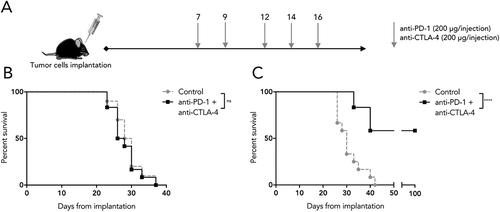
We next determined whether the highly divergent molecular profiles of SB28 and GL261 would correlate with different responses to immunotherapy. We treated mice bearing intracranial SB28 or GL261 tumors with a combination of anti-PD-1 and anti-CTLA-4 blocking antibodies (), a combination that has shown efficacy for several cancer indications in patients.Citation33,Citation34 SB28 implanted mice were totally resistant to this ICB protocol, with no cured mice, nor any prolonged survival (). In contrast, the GL261 implanted mice were highly sensitive to this treatment, with over 50% long term survivors (), consistent with previous reports for this model.Citation35,Citation36
Figure 5. Differential sensitivity of SB28 and GL261 to double ICB. (A) Mice implanted intracranially with tumor were randomized into groups to receive intraperitoneal treatment with anti-PD-1 and anti-CTLA-4 antibodies, or isotype control, at the indicated doses and time-points. (B) Symptom-free survival of mice implanted with 1,600 SB28 glioma cells. (C) Symptom-free survival of mice implanted with 50,000 GL261 glioma cells. Survival curves represent accumulated data from two independent experiments (12 mice/group).
Discussion
Major advances in understanding biological processes in neuroncology have benefitted enormously from judicious use of animal models, but for GBM, these conceptual advances have not yet led to development of new treatments that dramatically change clinical outcome for patients. The recent progress in immunotherapy for cancer, particularly ICB, is reshaping the field of oncology by transforming the way we treat patients with previously incurable malignancies.Citation37 Following promising clinical results, a large number of clinical trials are currently exploring the potential of ICB for multiple cancer indications, but some trials failed to prove any benefit, and others have yet to report their findings, as is the case for most GBM trials. In view of the human and financial investment in ICB clinical trials, together with the ethical considerations in proposing an experimental therapy with known toxicities, improving our understanding of GBM-immune interactions in relevant animal models is necessary and important.Citation38 Unfortunately, the requirements for modelling GBM-immune interactions are rather different to those for understanding other aspects of tumor biology, most notably because of the obligation to use immunocompetent animals. Nevertheless, as recent advances have shown, promising genetically engineered GBM models in immunocompetent mice have been and are still being developed, but to our knowledge have not focused on recapitulating low mutational load.Citation39 To complement current clinical research, we need to assess those features of a GBM model that matter to anti-tumor immunity. This would appear to be a good starting point to interpret data from experimental immunotherapies. Clearly, if other aspects of GBM biology are also reproduced, this would render the model even more robust, but if immune visibility is very different to human GBM, direct extrapolations will be unreliable. In awaiting the “perfect” GBM model, one pragmatic approach is to analyze currently used models for their likely interactions with the immune system.
For an anti-tumor immune response to take place, some key requirements are needed. First of all, the tumor cells should be antigenic, this feature is determined by peptides presented on MHC molecules which are recognized by T cells. These peptides are generated from antigens that can be of two origins, either tumor-associated antigens (TAA) or tumor-specific antigens (TSA). TAA are typically molecules overexpressed or aberrantly expressed by tumor cells, compared to healthy cells. They have the advantage of being expressed by many patients with a specific tumor, but their lack of specificity leading to potential off-target effects, and potential immunological tolerance to these self-antigens can sometimes limit their application for immunotherapy. In this study we focused on TSA and specifically on neoantigens derived from mutations. These mutations, if expressed, can lead to generation of neoepitopes that can potentially be presented on MHC molecules and elicit T-cell mediated immune responses. But counteracting the natural process of peptide presentation, tumor cells can exhibit a phenotype that facilitates immune escape, such as low or absent expression of MHC molecules and/or limited generation or expression of immunogenic peptides. Some of these features are described for human GBM, with low expression of MHC-ICitation10,Citation11 and a low mutational loadCitation40 leading to generation of few neoepitopes.Citation41 Regarding MHC-I, the SB28 model, with constitutive absence of expression, accurately recapitulates the characteristics of certain human GBM exhibiting MHC loss.Citation42 We also demonstrate that the estimated number of expressed neoepitopes is very low, corresponding to only 21 peptides predicted to bind to H-2Kb or H-2Db. This is clearly much lower than in the GL261 and SMA-560 GBM models that are commonly used in immunotherapy studies, for which there were a predicted 264 and 161 MHC-I binding peptides, respectively.Citation28
Expression of antigenic epitopes by a tumor is necessary, but not sufficient for inducing an anti-tumor immune response. Immunogenicity depends upon additional immune cells and mechanisms. For example, efficient priming of naïve tumor-specific T cells generally occurs most efficiently in draining lymph nodes subsequent to migration of tumor-antigen loaded professional antigen presenting cells (APCCitation43). Alternatively, naïve T cells could in some cases enter the CNS and be directly primed at the tumor site. Intriguingly, SB28 constitutively expresses CD80 which could in theory offer co-stimulation of T cells via its ligand CD28. However, we did not detect significant numbers of naïve T cells infiltrating SB28 (data not shown). Moreover, before SB28 could present antigen, local production of IFNγ would be necessary to induce MHC-I expression; the most likely source being already activated T cells or NK cells. Therefore, it is more likely that T-cell activation occurs through cross-presentation by local APC, as reported in other brain tumor models.Citation43,Citation44
Further evidence for low immunogenicity could be reflected by low infiltration by immune cells, as we show for SB28 in this study. In comparison, T-cell infiltration in human GBM is highly variable and reported to be undetectable by immunohistochemistry in around 20% of patients.Citation20 However, the overall importance of immune infiltration will likely depend upon localization and subset representation, for example, low CD8+/high CD4+ T-cell infiltration was associated with a worse prognosis.Citation45 Moreover, the presence of immunosuppressive cells and molecules will undoubtedly play a role. Indeed, analysis of SB28 tumor ex vivo by RNA-sequencing revealed expression of multiple potentially immunosuppressive molecules (PDL1, CD80, B7H3, GAL9, CD155 and TGFβ), and flow cytometry analysis of infiltrating immune cells showed presence of Tregs (Supplementary Figure S3 and data not shown). The in vivo microenvironment did influence the expressed mutanome of SB28, since ex vivo profiling indicated a different number of mutations and neoepitopes from the line analyzed in vitro and that analyzed ex vivo (108 vs. 132 expressed mutations), with a common core of mutations found from the two analyses (data not shown). To fully understand whether this in vivo reshaping of the mutanome is due to immune pressure or other mechanisms would require further study.
The CD80 that we show to be expressed by SB28 GBM cells (and by GL261 cells after IFNγ stimulation), has the potential to act as a co-inhibitory ligand for activated T cells that express CTLA-4, and to promote function of Tregs that are constitutively positive for CTLA-4.Citation46 Moreover, in the presence of IFNγ, PD-L1 expression is induced on both SB28 and GL261, which could engage and inhibit PD-1 expressing T cells; RNA-sequencing data further supported the expression of PD-L1 on SB28 ex vivo, along with several other IFNγ induced genes (data not shown). Moreover, flow cytometry analysis showed high expression of PD-1 on T cells and PD-L1 on macrophages (Supplementary figure S3). Altogether, these features support the rational to target both pathways by blocking CTLA-4 and PD-1, a strategy also tested in certain clinical trials.Citation15 We demonstrated a complete absence of response to this clinically relevant double ICB treatment in SB28, in contrast to the positive therapeutic effect in GL261, as previously reported.Citation35,Citation36 These profiles of response correlate with the difference in mutational load between the models, a factor linked to clinical responses to ICB in patients with malignancies other than GBM.Citation16,Citation47,Citation48 Regarding GBM and mutational load, previous studies have situated this malignancy in the lower half of human cancers when they are ranked according to the number of somatic mutations.Citation41 If we consider SB28 mutational load on the same scale, the figures would be close to the median value reported for GBM. This magnitude of mutational load has been estimated to “occasionally” form neoantigens.Citation41 In contrast, if we visualize GL261 mutational load, it is even more highly mutated than the median value for melanoma, but below values reported for hypermutated GBM resulting from biallelic mismatch repair deficiency.Citation49 Further mutanome analyses for GBM are now being carried out and differences can be observed between untreated and recurrent tumors, with cases of hypermutated GBM in recurrent tumors.Citation26 But even within the same untreated tumor, areas of hypermutation can be identified, as described very recently.Citation27 The different GBM models now available to us can be valuable tools for studying corresponding human GBMs with comparable mutational burdens and their response to therapy with ICB or in combination with other treatment modalities such as chemo-radiotherapy.Citation24-Citation26,Citation50
The results of our study highlight the value of different GBM models for recapitulating different types of immune interactions likely in different human gliomas. Immune visibility through neoepitope expression is only one of many aspects to be taken into account for optimal therapy, with future challenges that must include targeting other aspects of the tumor microenvironment to promote infiltration of functional immune cells with anti-tumor activity. We anticipate that the stringent SB28 model that we describe here, refractive to anti-PD-1 and anti-CTLA-4 ICB, will offer many opportunities to develop and test approaches to improve anti-glioma immunity that will be of value to GBM patients who show similar unresponsiveness to therapy.
List of abbreviations
| APC: | = | Antigen presenting cell |
| CTLA-4: | = | Cytotoxic T-lymphocyte-associated protein 4 |
| DNA: | = | Deoxyribonucleic acid |
| GBM: | = | Glioblastoma |
| GFP: | = | Green fluorescent protein |
| ICAM-1: | = | Intercellular Adhesion Molecule 1 |
| ICB: | = | Immune checkpoint blockade |
| IDH: | = | Isocitrate dehydrogenase |
| IFNγ: | = | Interferon-gamma |
| MHC-I: | = | Major histocompatibility complex-I |
| MHC-II: | = | Major histocompatibility complex-II |
| MS: | = | Median survival |
| NK cell: | = | Natural killer cell |
| PD-1: | = | Programmed death-1 |
| PD-L1: | = | Programmed death-ligand 1 |
| PDGF: | = | Platelet-derived growth factor |
| RNA-seq: | = | Ribonucleic acid sequencing |
| RNA: | = | Ribonucleic acid |
| TAA: | = | Tumor associated antigen |
| Tregs: | = | Regulatory T cell |
| TSA: | = | Tumor specific antigen |
| WES: | = | Whole exome sequencing |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Author contributions
Conceived and designed the experiments: PRW HO VG
Performed the experiments: VG EM JCC VB SIN
Analyzed the data: VG EM SIN JCC VB PRW
Wrote the paper: VG EM SIN JCC VB PYD HO PRW
Supplemental Material
Download Zip (2.8 MB)Acknowledgments
We thank Oliver Hartley and Martin Loewer for their insight, Viviane Bes and Celine Maroun Yacoub for their technical help, together with advice and assistance from the core facilities of the University of Geneva for genomics (iGE3 platform support from Mylène Docquier, Didier Chollet and Natacha Civic), flow cytometry, bioimaging, and animal housing. We thank the members of the Walker and Dietrich groups for helpful comments and discussions.
Supplemental data
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. doi:10.1056/NEJMoa043330.
- Ostrom QT, Gittleman H, Fulop J, Liu M, Blanda R, Kromer C, Wolinsky Y, Kruchko C, Barnholtz-Sloan JS. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2008-2012. Neuro Oncol. 2015;17(Suppl 4):iv1–iv62. doi:10.1093/neuonc/nov189.
- Sampson JH, Maus MV, June CH. Immunotherapy for brain tumors. J Clin Oncol. 2017;35:2450–2456. doi:10.1200/JCO.2017.72.8089.
- Finocchiaro G, Pellegatta S. Novel mechanisms and approaches in immunotherapy for brain tumors. Discov Med. 2015;20:7–15.
- Hodges TR, Ferguson SD, Heimberger AB. Immunotherapy in glioblastoma: emerging options in precision medicine. CNS Oncol. 2016;5:175–186. doi:10.2217/cns-2016-0009.
- Dutoit V, Migliorini D, Dietrich PY, Walker PR. Immunotherapy of malignant tumors in the brain: how different from other sites? Front Oncol. 2016;6:256. doi:10.3389/fonc.2016.00256.
- Lenting K, Verhaak R, Ter Laan M, Wesseling P, Leenders W. Glioma: experimental models and reality. Acta Neuropathol. 2017;133:263–282. doi:10.1007/s00401-017-1671-4.
- Huse JT, Holland EC. Genetically engineered mouse models of brain cancer and the promise of preclinical testing. Brain Pathol. 2009;19:132–143. doi:10.1111/j.1750-3639.2008.00234.x.
- Oh T, Fakurnejad S, Sayegh ET, Clark AJ, Ivan ME, Sun MZ, Safaee M, Bloch O, James CD, Parsa AT. Immunocompetent murine models for the study of glioblastoma immunotherapy. J Transl Med. 2014;12:107. doi:10.1186/1479-5876-12-107.
- Wu A, Wiesner S, Xiao J, Ericson K, Chen W, Hall WA, Low WC, Ohlfest JR. Expression of MHC I and NK ligands on human CD133+ glioma cells: possible targets of immunotherapy. J Neurooncol. 2007;83:121–131. doi:10.1007/s11060-006-9265-3.
- Di Tomaso T, Mazzoleni S, Wang E, Sovena G, Clavenna D, Franzin A, Mortini P, Ferrone S, Doglioni C, Marincola FM, et al. Immunobiological characterization of cancer stem cells isolated from glioblastoma patients. Clin Cancer Res. 2010;16:800–813. doi:10.1158/1078-0432.CCR-09-2730.
- Schumacher T, Bunse L, Pusch S, Sahm F, Wiestler B, Quandt J, Menn O, Osswald M, Oezen I, Ott M, et al. A vaccine targeting mutant IDH1 induces antitumour immunity. Nature. 2014;512:324–327. doi:10.1038/nature13387.
- Derouazi M, Di Berardino-Besson W, Belnoue E, Hoepner S, Walther R, Benkhoucha M, Teta P, Dufour Y, Maroun CY, Salazar AM, et al. Novel cell-penetrating peptide-based vaccine induces robust CD4+ and CD8+ T cell-mediated antitumor immunity. Cancer Res. 2015;75:3020–3031. doi:10.1158/0008-5472.CAN-14-3017.
- Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. 2015;27:450–461. doi:10.1016/j.ccell.2015.03.001.
- Vanpouille-Box C, Lhuillier C, Bezu L, Aranda F, Yamazaki T, Kepp O, Fucikova J, Spisek R, Demaria S, Formenti SC, et al. Trial watch: immune checkpoint blockers for cancer therapy. Oncoimmunology. 2017;6:e1373237. doi:10.1080/2162402X.2017.1373237.
- Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, Walsh LA, Postow MA, Wong P, Ho TS, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014;371:2189–2199. doi:10.1056/NEJMoa1406498.
- Goldberg SB, Gettinger SN, Mahajan A, Chiang AC, Herbst RS, Sznol M, Tsiouris AJ, Cohen J, Vortmeyer A, Jilaveanu L, et al. Pembrolizumab for patients with melanoma or non-small-cell lung cancer and untreated brain metastases: early analysis of a non-randomised, open-label, phase 2 trial. Lancet Oncol. 2016;17:976–983. doi:10.1016/S1470-2045(16)30053-5.
- Di Giacomo AM, Margolin K. Immune checkpoint blockade in patients with melanoma metastatic to the brain. Semin Oncol. 2015;42:459–465. doi:10.1053/j.seminoncol.2015.02.006.
- Chamberlain MC, Kim BT. Nivolumab for patients with recurrent glioblastoma progressing on bevacizumab: a retrospective case series. J Neurooncol. 2017;133:561–569. doi:10.1007/s11060-017-2466-0.
- Berghoff AS, Kiesel B, Widhalm G, Rajky O, Ricken G, Wohrer A, Dieckmann K, Filipits M, Brandstetter A, Weller M, et al. Programmed death ligand 1 expression and tumor-infiltrating lymphocytes in glioblastoma. Neuro Oncol. 2015;17:1064–1075.
- Wintterle S, Schreiner B, Mitsdoerffer M, Schneider D, Chen L, Meyermann R, Weller M, Wiendl H. Expression of the B7-related molecule B7-H1 by glioma cells: a potential mechanism of immune paralysis. Cancer Res. 2003;63:7462–7467.
- Fridman WH, Zitvogel L, Sautes-Fridman C, Kroemer G. The immune contexture in cancer prognosis and treatment. Nat Rev Clin Oncol. 2017;14:717–734. doi:10.1038/nrclinonc.2017.101.
- Rutledge WC, Kong J, Gao J, Gutman DA, Cooper LA, Appin C, Park Y, Scarpace L, Mikkelsen T, Cohen ML, et al. Tumor-infiltrating lymphocytes in glioblastoma are associated with specific genomic alterations and related to transcriptional class. Clin Cancer Res. 2013;19:4951–4960. doi:10.1158/1078-0432.CCR-13-0551.
- Colli LM, Machiela MJ, Myers TA, Jessop L, Yu K, Chanock SJ. Burden of nonsynonymous mutations among TCGA cancers and candidate immune checkpoint inhibitor responses. Cancer Res. 2016;76:3767–3772. doi:10.1158/0008-5472.CAN-16-0170.
- Hodges TR, Ott M, Xiu J, Gatalica Z, Swensen J, Zhou S, Huse JT, de Groot J, Li S, Overwijk WW, et al. Mutational burden, immune checkpoint expression, and mismatch repair in glioma: implications for immune checkpoint immunotherapy. Neuro Oncol. 2017;19:1047–1057. doi:10.1093/neuonc/nox026.
- Wang J, Cazzato E, Ladewig E, Frattini V, Rosenbloom DI, Zairis S, Abate F, Liu Z, Elliott O, Shin Y-J, et al. Clonal evolution of glioblastoma under therapy. Nat Genet. 2016;48:768–776. doi:10.1038/ng.3590.
- Mahlokozera T, Vellimana AK, Li T, Mao DD, Zohny ZS, Kim DH, Tran DD, Marcus DS, Fouke SJ, Campian JL, et al. Biological and therapeutic implications of multisector sequencing in newly diagnosed glioblastomas. Neuro Oncol. 2018; 20:472–483. doi:10.1093/neuonc/nox232.
- Johanns TM, Ward JP, Miller CA, Wilson C, Kobayashi DK, Bender D, Fu Y, Alexandrov A, Mardis ER, Artyomov MN, et al. Endogenous neoantigen-specific CD8 T cells identified in two glioblastoma models using a cancer immunogenomics approach. Cancer Immunol Res. 2016;4:1007–1015. doi:10.1158/2326-6066.CIR-16-0156.
- Kosaka A, Ohkuri T, Okada H. Combination of an agonistic anti-CD40 monoclonal antibody and the COX-2 inhibitor celecoxib induces anti-glioma effects by promotion of type-1 immunity in myeloid cells and T-cells. Cancer Immunol Immunother. 2014;63:847–857. doi:10.1007/s00262-014-1561-8.
- Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271:1734–1736. doi:10.1126/science.271.5256.1734.
- Walker PR, Migliorini D. The CD40/CD40L axis in glioma progression and therapy. Neuro Oncol. 2015;17:1428–1430. doi:10.1093/neuonc/nov138.
- Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi:10.1016/j.ccr.2009.12.020.
- Postow MA, Chesney J, Pavlick AC, Robert C, Grossmann K, McDermott D, Linette GP, Meyer N, Giguere JK, Agarwala SS, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med. 2015;372:2006–2017. doi:10.1056/NEJMoa1414428.
- Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, Schadendorf D, Dummer R, Smylie M, Rutkowski P, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373:23–34. doi:10.1056/NEJMoa1504030.
- Wainwright DA, Chang AL, Dey M, Balyasnikova IV, Kim CK, Tobias A, Cheng Y, Kim JW, Qiao J, Zhang L, et al. Durable therapeutic efficacy utilizing combinatorial blockade against IDO, CTLA-4, and PD-L1 in mice with brain tumors. Clin Cancer Res. 2014;20:5290–5301. doi:10.1158/1078-0432.CCR-14-0514.
- Reardon DA, Gokhale PC, Klein SR, Ligon KL, Rodig SJ, Ramkissoon SH, Jones KL, Conway AS, Liao X, Zhou J, et al. Glioblastoma eradication following immune checkpoint blockade in an orthotopic, immunocompetent model. Cancer Immunol Res. 2016;4:124–135. doi:10.1158/2326-6066.CIR-15-0151.
- Whiteside TL, Demaria S, Rodriguez-Ruiz ME, Zarour HM, Melero I. Emerging opportunities and challenges in cancer immunotherapy. Clin Cancer Res. 2016;22:1845–1855. doi:10.1158/1078-0432.CCR-16-0049.
- Garg AD, Vandenberk L, Van Woensel M, Belmans J, Schaaf M, Boon L, De Vleeschouwer S, Agostinis P. Preclinical efficacy of immune-checkpoint monotherapy does not recapitulate corresponding biomarkers-based clinical predictions in glioblastoma. Oncoimmunology. 2017;6:e1295903. doi:10.1080/2162402X.2017.1295903.
- Yeo AT, Charest A. Immune checkpoint blockade biology in mouse models of glioblastoma. J Cell Biochem. 2017;118:2516–2527. doi:10.1002/jcb.25948.
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale A-L, Rüegg C, Dietrich PY, Walker PR. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi:10.1038/nature12477.
- Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348:69–74. doi:10.1126/science.aaa4971.
- Facoetti A, Nano R, Zelini P, Morbini P, Benericetti E, Ceroni M, Campoli M, Ferrone S. Human leukocyte antigen and antigen processing machinery component defects in astrocytic tumors. Clin Cancer Res. 2005;11:8304–8311. doi:10.1158/1078-0432.CCR-04-2588.
- Calzascia T, Masson F, Di Berardino-Besson W, Contassot E, Wilmotte R, Aurrand-Lions M, et al. Homing phenotypes of tumor-specific CD8 T cells are predetermined at the tumor site by crosspresenting APCs. Immunity. 2005;22:175–184. doi:10.1016/j.immuni.2004.12.008.
- Calzascia T, Di Berardino-Besson W, Wilmotte R, Masson F, de Tribolet N, Dietrich PY, Walker PR. Cutting edge: cross-presentation as a mechanism for efficient recruitment of tumor-specific CTL to the brain. J Immunol. 2003;171:2187–2191. doi:10.4049//jimmunol.171.5.2187.
- Han S, Zhang C, Li Q, Dong J, Liu Y, Huang Y, Jiang T, Wu A. Tumour-infiltrating CD4(+) and CD8(+) lymphocytes as predictors of clinical outcome in glioma. Br J Cancer. 2014;110:2560–2568. doi:10.1038/bjc.2014.162.
- Peggs KS, Quezada SA, Korman AJ, Allison JP. Principles and use of anti-CTLA4 antibody in human cancer immunotherapy. Curr Opin Immunol. 2006;18:206–213. doi:10.1016/j.coi.2006.01.011.
- Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–128. doi:10.1126/science.aaa1348.
- Segal NH, Parsons DW, Peggs KS, Velculescu V, Kinzler KW, Vogelstein B, Allison JP. Epitope landscape in breast and colorectal cancer. Cancer Res. 2008;68:889–892. doi:10.1158/0008-5472.CAN-07-3095.
- Bouffet E, Larouche V, Campbell BB, Merico D, de Borja R, Aronson M, Durno C, Krueger J, Cabric V, Ramaswamy V, et al. Immune checkpoint inhibition for hypermutant glioblastoma multiforme resulting from germline biallelic mismatch repair deficiency. J Clin Oncol. 2016;34:2206–2211. doi:10.1200/JCO.2016.66.6552.
- Lim M, Xia Y, Bettegowda C, Weller M. Current state of immunotherapy for glioblastoma. Nat Rev Clin Oncol. 2018;15:422–442. doi:10.1038/s41571-018-0003-5.