
ABSTRACT
Adoptive cell therapy (ACT) with retargeted T cells has produced remarkable clinical responses against cancer, but also serious toxicity. Telomerase is overexpressed in most cancers, but also expressed in some normal cells, raising safety concerns. We hypothesize that ACT with T-helper cell receptors may overcome tumour tolerance, mobilize host immune cells and induce epitope spreading, with limited toxicity. From long term survivors after cancer vaccination, we have isolated telomerase-specific T cell receptors (TCRs) from T-helper cells. Herein, we report the development of transient retargeting of T cells with mRNA-based TCRs. This strategy allows for safer clinical testing and meaningful dose escalation. DP4 is the most common HLA molecule. We cloned two telomerase-specific, DP4-restricted TCRs into the mRNA expression vector pCIpA102, together with the sorter/marker/suicide gene RQR8. Donor T cells were electroporated with mRNA encoding TCR_RQR8. The results showed that both TCR_RQR8 constructs were expressed in >90% of T cells. The transfected T cells specifically recognized the relevant peptide, as well as naturally processed epitopes from a 177aa telomerase protein fragment, and remained functional for six days. A polyfunctional and Th1-like cytokine profile was observed. The TCRs were functional in both CD4+and CD8+recipient T cells, even though DP4-restricted. The findings demonstrate that the cloned TCRs confer recipient T cells with the desired telomerase-specificity and functionality. Preclinical experiments may provide limited information on the efficacy and toxicity of T-helper TCRs, as these mobilize the host immune system. We therefore intend to use the mRNA-based TCRs for a first-in-man trial.
Introduction
Adoptive cell therapy (ACT) with retargeted T cells shows robust clinical efficacy against several hematological malignancies. The most successful trials to date have employed T cells transduced with chimeric antigen receptors (CARs) against the B-cell antigen CD19.Citation1–Citation4 This has led to FDA approval in 2017 for CAR T cell therapy against leukaemia and lymphoma. In other cancer forms, there is a need to target other antigens. Further, there are concerns about how to overcome established immune evasion in solid tumours.Citation5,Citation6 We address these challenges by developing T helper cell receptors against the universal cancer antigen human telomerase reverse transcriptase (hTERT), based on T cell receptors (TCRs) from long term survivors after cancer vaccination.
Most TCRs for ACT, and most cancer vaccines, have focused on CD8+cytotoxic T cells (CTL). The importance of CD4+ T helper (Th) cells for anti-cancer responses has, however, become increasingly emphasized.Citation7–Citation14 Our proposed strategy is to transfer large numbers of hTERT-specific Th cells to the patient, in order to engraft the tumour and lymphoid organs. The Th cells may both exert direct anti-tumour killing and modulate the microenvironment in tumour, lymph nodes and bone marrow, thereby overcoming tumour-induced immune suppression. The Th cells may drive cancer into senescenceCitation15 and are capable of mobilizing both the innate and adaptive immune system. It is well documented that Th Cells are important for CTL memory responses.Citation16,Citation17 Further, Th cells may promote type 1 polarization of DCs and macrophages and counter immunosuppression from Tregs and MDSC.Citation11,Citation18,Citation19 These anti-tumour effects are partly considered to depend on a Th1-like cytokine profile of the Th cells.Citation11,Citation15,Citation18 We hypothesize that Th cells redirected against hTERT may induce epitope spreadingCitation20–Citation22 and lead to a broad and “personalized” immune response, including responses against neoantigens.Citation23,Citation24
We have previously reported the retroviral cloning and testing of two TCRs, called C13 and D71, against the hTERT-epitope GV1001.Citation25 Both C13 and D71 recognize peptides presented on DP4, which is the most prevalent HLA-molecule and expressed by >70% of Caucasians.Citation26 Their common HLA-restriction and the universal expression of hTERT suggest that these TCRs may be widely applicable in a clinical setting. TCR C13 and D71 were isolated from two long term survivors in a vaccine trial that we conducted in advanced NSCLC.Citation27,Citation28 In this trial, patients that developed an immune response against the vaccine peptide GV1001 had significantly extended survival compared with non-immune responders (median 19 months versus 3.5 months; p < 0.001).Citation28 TCR C13 and D71 were obtained from patient C and D, respectively. These patients both developed a clinical and immunological response, without signs of adverse effects. The immune responses were durable. Patient C developed complete tumor regression and was alive without evidence of relapse at the last clinical follow-up after 12 years. His GV1001-specific T cell immune response remained detectable at the last PBMC sampling after 8 years. Patient D eventually developed disease progression. She survived for 46 month.Citation28
Herein, we describe the development of mRNA-based transient retargeting of T cells with the two hTERT–specific TCRs, C13 and D71.Telomerase is expressed by most cancer form.Citation29–Citation32 Because hTERT expression is often mandatory to tumor growth, the risk of tumor escape is presumably limited. On the other hand, hTERT is expressed to some extent in normal tissues, leading to a concern for potential side effects. ACT with TCRs/CARs against several new targets has produced severe, and in some cases fatal, toxicity in first-in-man studies.Citation33–Citation36 In several cases, these adverse effects have not been predicted in preclinical models. The Th-cells are likely to be less toxic than CTLs against hTERT, as HLA class II expression is limited to certain cell types. This aspect, combined with the absence of toxicity in the patients from which TCR C13 and D71 were derived, supports the rationale for developing these TCRs for therapeutic use. However, the clinical testing of new hTERT-specific TCRs in patients still carries a considerable safety risk. The conventional viral vectors permanently integrate the TCR into the T cell genome. This may give durable anti-cancer activity, but also confer a risk for sustained and increasing toxicity, as the T cells proliferate in the patient after infusion. In the present study, we address the safety issue by developing biodegradable TCRs, based on transfecting T cells with mRNA encoding the TCR. This approach may be a feasible strategy for clinical testing of novel TCRs, as discussed below.
Results
Molecular cloning of TCR C13 and D71 with RQR8 into mRNA expression vector
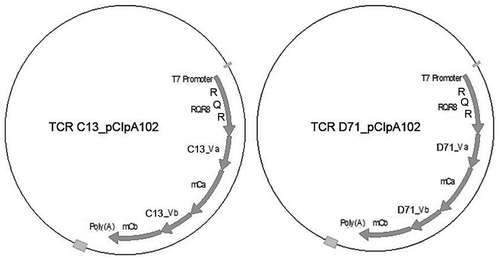
We cloned the α- and β-chains from TCR C13 and D71 into the mRNA expression vector pCIpA10Citation37 (). Both TCRs are DP4-restricted, specific for the hTERT peptide GV1001 and were isolated from long term survivors after cancer vaccination, as described above. The TCR sequences were codon optimized and modified to express mouse TCR constant regions, in order to improve surface expression and facilitate easy detection of the expressed TCR on human T cells. Both TCRs were cloned into pCIpA102 together with the suicide-marker-sorter gene RQR8. The RQR8 construct consists of a minimal binding epitope from CD34 flanked by CD20 epitopes that bind to the commonly used mAb rituximab.Citation38The RQR8 fusion protein thus allows for large scale purification of transduced T cells with the clinically approved CliniMACS CD34 system, and for depletion of transduced cells with rituximab.
Figure 1. TCR cloning.
The sequences for TCR C13 and D71 were codon optimized and modified to express mouse constant regions (mC). Both TCRs were cloned into vector pCIpA102 downstream of the suicide-marker-sorter gene RQR8. The RQR8 construct consists of a minimal binding epitope from CD34 (Q) flanked by CD20 epitopes (R) that bind rituximab.Citation38 mCb: Murine constant beta TCR region; mCa: Murine constant alpha TCR region; Va: Variable alpha TCR region; Vb: variable beta TCR region.
TCR/RQR8 expression in primary T cells after transfection
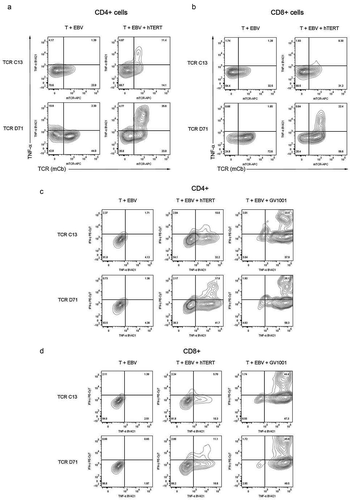
Primary T cells from peripheral blood were expanded for 10 days and transfected by electroporation with mRNA encoding TCR C13_RQR8 or D71_RQR8. The transfection efficacy was assessed using a mAb against the murine constant beta region (mCb) incorporated in the TCRs, and by a mAb against the CD34 epitope in RQR8. A representative experiment is shown in . Both C13 and D71 were expressed well by primary T cells after transfection. The TCRs were expressed in both CD4+ and CD8+ T cells. The fraction of positive cells tended to be higher for CD8+ T cells ( and ).
Figure 2. TCR expression in primary T cells.
Primary T cells from peripheral blood were expanded for 10 days and transfected by electroporation with mRNA encoding TCR C13_RQR8 or TCR D71_RQR8. The cells were cultured and stained for analysis by flow cytometry 17h after electroporation. The expression of the TCRs (C13 or D71) was measured with a mAb recognizing the murine constant beta region incorporated in the TCRs. The expression of marker/suicide gene RQR8 was measured with the mAb QBen10. The contour plots represent non-transfected (NT) T cells (left), T cells transfected with TCR C13 (middle) or TCR D71 (right). Top panels show CD4+cells, bottom panels show CD8+cells. The percentage of cells in each quartile is given. The figure shows representative data from three experiments.
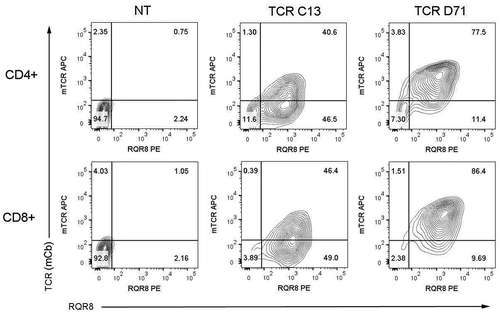
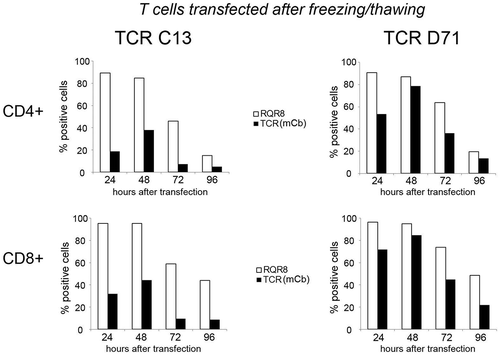
Figure 3. TCR expression over time in T cells transfected after freezing and thawing.
PBMCs were expanded for 10 days, then frozen in liquid nitrogen and thawed before electroporation with mRNA encoding TCR C13_RQR8 or TCR D71_RQR8. The figure shows representative data from three experiments. The cells were cultured after electroporation and stained for analysis by flow cytometry at four time points; 24 h, 48 h, 72 h and 96 h after electroporation. The expression of the TCRs (C13 or D71) was measured with a mAb recognizing the murine constant beta region (mCb) incorporated in the TCRs. The expression of marker/suicide gene RQR8 was measured with the mAb QBen10. The expression of C13_RQR8 (left panels) and D71_RQR8 (right panels) are shown in , separately for CD4+ and CD8+ T cells. Time after electroporation is indicated on the x-axis. The number of positive cells was calculated based quartiles in dot plots, set from non-transfected controls.
Both TCR C13 and D71 were efficiently co-expressed with the marker protein RQR8, which was expressed in 85–95% of cells. TCR D71 repeatedly showed a higher expression than TCR C13. In the experiment in , TCR D71 was detected in about 85% of recipient cells, compared to 45% for C13. The data suggested that RQR8 was equally well expressed in C13 and D71 transfected cells, i.e. that the construct/mRNA quality and the electroporation were similar. The difference in expression between C13 and D71 thus appeared to be inherent to the TCR. By contrast, the difference between CD4+ CD8+ cells was generally mirrored between RQR8 and the mCb-readout ( and ).
TCR expression in T cells transfected after freezing and thawing
In the clinical setting, it may be logistically simpler to transfect frozen/thawed T cells, rather than fresh cells. We tested the transfection efficacy in cells that were expanded for 10 days, then frozen, stored in liquid nitrogen and thawed before mRNA-electroporation. A representative experiment, from three serial tests, is shown in . The TCR expression was not substantially different than observed in other experiments with fresh cells. The RQR8 data generally indicated successful transfection in >90% of thawed T cells (). The direct measurement of the transfected TCRs by anti-mCb indicated that D71 was expressed better than C13, as in our experiments with fresh cells. TCR D71 (Cb) was detected in >80% of cells after 48h, compared to ̴40% for C13 (). As with fresh cells, we observed higher TCR expression in CD8 +than CD4+cells ().
Dynamics of TCR and RQR8 expression after mRNA transfection
The protein expression after mRNA transfection will gradually decrease as determined by the T1/2 of the mRNA and the TCR. We assessed the dynamics by measuring the expression of the TCR-mCb and RQR8 by flow cytometry at consecutive time points. The direct Cb-measurements suggested that the TCR expression increased for the first 48h and thereafter gradually decreased (). In the experiment in , TCR D71 was detected in >80% of T cells after 48h, about 40% of T cells after 72h and 18% after 96h. TCR C13 was expressed at lower levels and also decreased faster. The percentage of transfected cells based on RQR8 measurements was higher at all time points for both construct C13_RQR8 and D71_RQR8, and higher for CD8+ T cells. The RQR8-level declined at a slower rate than Cb in most experiments (̴45% of CD8+after 96h in ), suggesting that this protein may be more stable than the TCRs.
T-helper TCRs C13 and D71 are functional in both CD4+ CD8+recipient cells
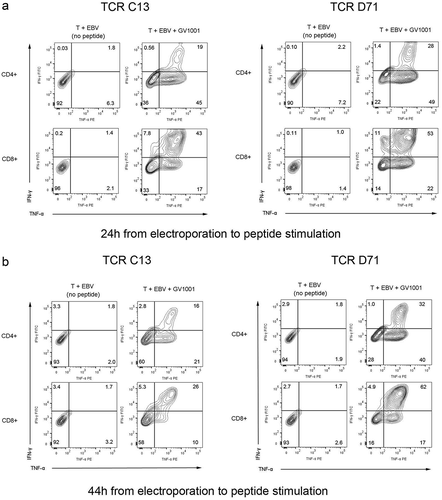
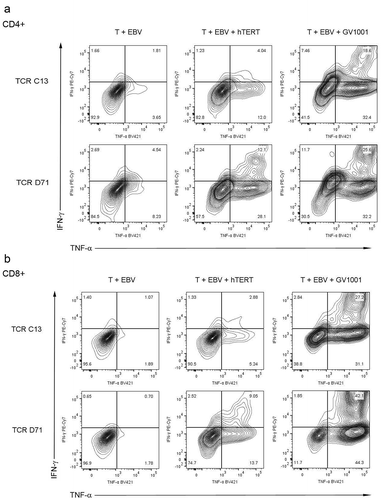
We next tested the functionality of the mRNA-transfected T cells. Primary PBMCs were expanded for 10 days and transfected with TCR C13_RQR8 or D71_RQR8. After electroporation, the T cells were rested for different time intervals, ranging from 24h-115h, before stimulation with DP4-positive target cells with/without peptide GV1001. The results showed that both C13 and D71 transfected T cells acquired GV1001-specific functionality. This applied both to CD4+and CD8+ T cells, even though TCR C13 and D712 were both HLA class II restricted and derived from CD4+ T cells. A representative experiment is shown in . Here, the T cells were rested for either 24 or 44 hours between electroporation and peptide stimulation. For the D71-transfected cells, we observed that 78% of CD4+and 86% of CD8+ T cells secreted TNFα and/or IFNγ after stimulation with GV1001 (). For C13, we detected a TNFα/IFNγ-response in 64% of CD4+and 67% of CD8+ T cells. Only low background was observed in cultures without peptide () and in non-transfected control T cells with peptide (not shown). includes all T cells, not only the RQR8+cells. The TNFα/IFNγ response rate among RQR8+ T cells was >90% for D71 and ̴80% for C13 (not shown). Nearly all responding T cells secreted TNFα, while IFNγ secretion was detected in more CD8+ than CD4+ T cells ().
Figure 4. TCR C13 and D71 confer recipient T cells with GV1001-specific functionality.
Primary T cells were transfected with mRNA encoding TCR C13_RQR8 or D71_RQR8. The TCR expression and functionality was assessed at two different time points, on T cells rested for either 24h or 44 hours between electroporation and peptide stimulation. The transfected T cells and non-transfected control T cells were stimulated with irradiated DP04+EBV-transformed cells (EBV) ± hTERT peptide GV1001. The cultures were incubated overnight and analysed by flow cytometry. Figs. A and B show INFγ/TNFα staining of cells rested for 24h or 44h, respectively. Percentage of cells in each quartile is given. The transfection efficacy was about 60% for CD4+cells and 90% for CD8+cells, as measured by RQR8 expression (not shown). (a) and (b) include all CD4+and CD8+cells, regardless of RQR8 expression. The transfected TCRs were derived from CD4+ T cells, but conferred GV1001-specific functionality to both CD4+and CD8+recipient cells. The figure shows representative data from three experiments in which there was some variation in the second time point measured.
Durability of redirected TCR functionality after mRNA-transfection
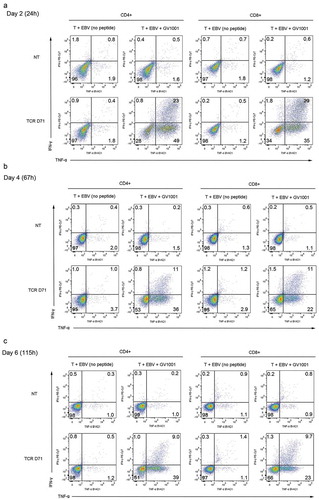
The durability of redirected TCR functionality is important for the clinical application. In the experiment shown in , the D71 T cells had retained functionality at the second test (44h), with ̴80% TNFα and/or IFNγ positive cells (). The majority of C13 T cells also retained their functionality, but the responsive fraction decreased from ̴65% to ̴40%.
shows a representative experiment more extensively testing the duration of TCR functionality. Here, the T cells were rested for 24 h, 67 h or 115h after transfection with D71, before stimulation with DP4+EBV cells ± peptide. About 70% of T cells responded after 24h, while non-transfected controls and controls without peptide were negative (). Nearly all responding cells secreted TNFα, while about half of them also stained positive for IFNγ. The responsive T cell fraction decreased moderately over time, but 49% of the CD4% T cells and 34% of CD8 retained a GV1001-response when tested on day 6 (). The proportion of responding cells was higher than the proportion where TCR expression was detected on flow cytometry. This observation suggests that T cells expressing low levels of the TCR may still be functional.
Figure 5. Durability of GV1001-specific functionality after mRNA transfection.
Primary T cells were transfected with mRNA encoding TCR D71_RQR8. The TCR expression and functionality was assessed at three different time points, on T cells rested for 24 h, 67 h or 115 h between electroporation and peptide stimulation. The transfected T cells and non-transfected control T cells were stimulated with irradiated EBV-transformed cells ± hTERT peptide GV1001. The cultures were incubated for 5 h and analysed by flow cytometry. Figs. A, B and C show INFγ/TNFα staining of cells rested for 24 h, 67 h or 115 h, respectively. Percentage of cells in each quartile is given. The transfection efficacy was about 50% for CD4+cells and 90% for CD8+cells, as measured by RQR8 expression (not shown). The figures include all CD4+and CD8+cells, regardless of RQR8 expression.
Recognition of naturally processed hTERT protein
An immune response against peptides is of sparse relevance if naturally processed epitopes are not recognize.Citation39 We evaluated if the redirected T cells responded to naturally processed epitopes by stimulating the T cells with APCs loaded with a 173aa hTERT protein fragment, that contained the GV1001 sequence. To keep the large hTERT protein fragment in solution, there is a need to include chemicals that are toxic to cells.Citation40 For this reason, we had to titrate down the concentration of the protein in the T cell assays to 2μM. The TCR-transected T cells still responded to stimulation with hTERT protein. A representative experiment is shown in . Here, a TNFα/IFNγ response was detected for 42% of CD4+ and 25% of CD8+D71 T cells (). The responsive fraction was lower for C13 cells.
Figure 6. TCR transfected T cells respond to naturally processed hTERT epitopes.
Primary T cells were expanded for 10 days and frozen at liquid nitrogen. The cells were thawed and electroporated with mRNA encoding TCR C13_RQR8 or D71_RQR8. After 24 h in culture, the cells were stimulated with irradiated EBV-transformed cells ± hTERT protein (2 μM) or peptide GV1001 (20μM). The cultures were incubated overnight and analysed by flow cytometry. (a) and (b) show INFγ/TNFα staining for CD4+and CD8+ T cells, respectively. Top panels in (a) and (b) show data for C13-transfected T cells, bottom panels show data for D71 T cells. The figures include all electroporated cells, also those where RQR8 and the TCR were not detectable.
Correlation between TCR expression and hTERT response
The individual T cells displayed varying levels of TCR expression after transfection, as assessed by anti-mCb. We repeatedly observed that there was a strong correlation between the TCR expression and the functional response. This applied both to CD4+and CD8+ T cells. A representative experiment is shown in . Here, after stimulation with hTERT protein (2μM), we detected a TNFα-response in 61% of CD4+ T cells staining D71 positive at flow cytometry, compared to 12% of those in which D71 expression was not detectable (). For CD8+ T cells, the corresponding figures were 28% versus 3.0% for D71+ and D71− cells, respectively (). Further, the few D71− cells in which a cytokine response was detected, had only low-moderate cytokine levels (). Within the D71+ cells, those with a higher expression of the TCR also tended to have the highest cytokine response (). Similar correlations between TCR expression and functional response were observed for T cells transfected with construct C13, but at lower levels (, b). Among the TCR-expressing T cells, the responsive cell proportion after GV1001-stimulation was >90% for both TCR C13 and D71 ().
Figure 7. Correlation between TCR expression and response to hTERT.
Same experiment as in . Primary T cells were transfected with C13 or D71 and stimulated with irradiated EBV-transformed cells ± hTERT protein (2 μM) or peptide GV1001 (20 μM). The cultures were incubated overnight and analysed by flow cytometry. The expression of the transfected TCR (C13 or D71) was measured with a mAb recognizing the murine constant beta region (mCb) incorporated in the TCRs. (a) and (b) show flowcytometry stainings for the TCR (mCb), combined with TNFα. (c) and (d) show INFγ/TNFα stainings for TCR positive CD4+and CD8+ T cells, respectively.
Polyfunctional and cytotoxic T cell response
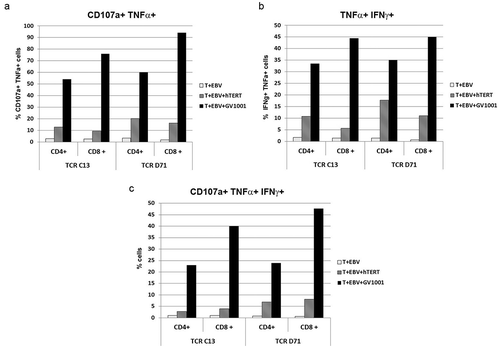
We further studied the cytotoxic capability of the redirected T cells and their polyfunctionality. The assays showed that the majority of TCR-expressing T cells acquired a polyfunctional GV1001-response, including IFNγ, TNFα and cytotoxic activity as assessed by CD107a. This applied to both TCR C13 and D71, and to both CD4+and CD8+ T cells. The CD107a-response was repeatedly higher for CD8+ than CD4+ T cells and generally higher for D71 than C13 cells. Representative data are shown in (same experiment as ). Here, 94% of CD8+and 60% of CD4+D71 cells stained double positive for TNFα/CD107a after GV1001-stimulation. For C13, the TNFα+CD107a+ proportion was 76% for CD8+ cells and 54% for CD4+cells (). A triple positive response for IFNγ, TNFαand CD107α was observed for nearly half of the CD8+ T cells (40% of C13 cells and 48% of D71+ cells; ). For CD4+ T cells, the CD107a+IFNg+TNFα+αproportion was 23% and 24% for C13 and D71 cells, respectively. The responses were antigen-specific (<1.1% in controls w/o peptide).
Figure 8. Polyfunctional and cytotoxic T cell response.
Same experiment as in and . Primary T cells were transfected with C13 or D71 and stimulated with irradiated EBV-transformed cells ± hTERT protein (2 μM) or peptide GV1001 (20 μM). The cultures were incubated overnight and analysed by flow cytometry. (a) and (b) show the proportion of cells that stained double positive for TNFα/CD107a and TNFα/IFNγ, respectively. (c) shows the proportion of cells that stained triple positive for CD107a/TNFα/IFNγ. The proportions were calculated from quartiles in dot plots, as shown in . – show representative data from three experimental runs with GV1001, of which two also included hTERT protein.
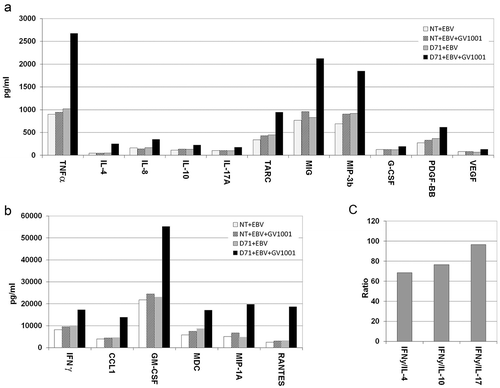
Cytokine profile of redirected T cells
The cytokine profile was investigated in multiplex assays, which showed that D71 T cells secreted a wide range of cytokines upon stimulation with peptide GV1001 (). This included the CTL and Th1-associated cytokines IFNγ and TNFα, as well as chemokines CCL-1, MDC, MIP-1A, RANTES, MIP-3β and MIG. A Th1-like cytokine profile is considered desirable for cancer eradication. We observed that the concentration of the hallmark Th1-cytokine IFNγ was >60-fold higher than the Th2-cytokine IL-4, the Th2/Treg-cytokine IL-10 and the Th17-cytokine IL-17 (). The D71-transfected T cells thus had a Th1-like cytokine profile, combined with multiple chemokines suggesting polyfunctionality.
Figure 9. Cytokine secretion by redirected T cells.
Primary T cells were transfected with TCR D71_RQR8. Transfected and non-transfected (NT) control T cells were stimulated with irradiated EBV-transformed cells ± hTERT peptide GV1001. In one of the experiments, supernatants from duplicate cultures were collected after 48 h and analysed by Bioplex cytokine assays. The cultures were kept separate from initial T cell stimulation and throughout the Bioplex assays. (a) and (b) show data from all cytokines were the stimulator index between cultures with/without peptide were > 1.5. Mean cytokine concentrations (pg/ml) from duplicate cultures are displayed. For clarity of presentation, cytokines measured at high and low levels are separated into figs. A and B, respectively. The concentration of hallmark Th1-cytokine IFNγ was compared to the concentration of three cytokines characterizing other key T cell subsets, i.e. IL-4 (Th2), IL-10 (Th2/Tr1) and IL-17 (Th17). Fig. C shows the ratio between IFNγ and these three cytokines, for D71-transfected T cells stimulated with GV1001.
Discussion
In the study reported herein, we have developed mRNA-based TCRs for transient retargeting of T cells against hTERT. We employ an in-house technology for mRNA transfection by electroporation, that yields >90% transfection efficacy in primary T cells with the applied TCR constructs C13 and D71, as measured by the marker gene RQR8. The transfection efficacy is higher than what is achievable with retrovirus or other methods for permanent retargeting of T cells. By comparison, the expression of TCR C13 and D71 in primary T cells after retroviral transduction is in our hands 30–60%.Citation25 As described, TCR D71 is consistently expressed at higher levels than C13 after mRNA transfection, even though the marker gene RQR8 is expressed at similar levels. The difference in expression between C13 and D71 thus appears to be inherent to the TCR. The mRNA transfection method was established and optimized by use of mRNA encoding GFPCitation37,Citation41 and has been further developed for TCRs.Citation42 For TCR BL71 and protein RQR8, the expression levels were in line with those obtained with GFP, which may indicate the maximum capacity of the applied method.
The use of mRNA offers both advantages and limitations. Most ACT approaches use viral vectors permanently integrating the receptor sequence into the T cell genome. The viral vectors carry the advantage of sustained anti-cancer activity after a single T cell infusion, but also represent a safety risk in the clinical setting. Since these gene-modified T cells persist and expand in the patient, adverse events may cause serious and durable complications.Citation34–Citation36 Insertional mutagenesis (risk of secondary cancer) also remains a possible concern, though this has not been reported in patients so far. Further, it is not possible to perform a controlled dose-escalation with virally retargeted cells, as the cells will proliferate after injection. After mRNA-electroporation, the TCRs are expressed and functional for about a week in the patient´s T cells, which subsequently return to their endogenous specificity. This means that the risk of adverse events is considerably lower. Further, it is possible to perform a meaningful dose-escalation, as the T cell can only expand for a short time before losing the TCR-expression, which moreover decreases upon proliferation.
The mRNA-strategy may make rapid development of cell therapy against novel targets feasible. In addition to the safety advantage, the use of mRNA eliminates the need for expensive and time consuming retrovirus production and simplifies the process for approval from regulatory authorities. The restricted time frame of the TCR-expression is, however, an efficacy limitation, and repeated injections will almost certainly be necessary to obtain clinical effects. In the setting of a first-in-man trial, we envisage two injections per week, for a period of six weeks. Further, we plan to employ a dose escalation within each patient, from week to week. Regarding the long term clinical strategy, the mRNA-based therapy may be useful in patients where a limited treatment period is desirable. It is possible that TCRs that are too toxic for long-term use, can be applied for a limited period, similar to other cancer drugs. However, if long-term activity of the redirected T cells is desired, viral vectors is desirable, and may be used after an initial documentation of safety in mRNA-based trials. The present study highlights the mRNA-approach for TCRs. Of note, the clinical efficacy of mRNA-based receptors is unproven. Interesting data have though emerged from the first mRNA-based CAR-trial.Citation43
We find that both TCR C13c and D71 confer recipient T cells with the desired GV1001-specific specificity, and that this applies both to CD4+and CD8+ T cells. Most tumour cells are HLA class II negative and will not be directly engaged by the C13 /D71- T cells, but may be killed through bystander mechanisms, including TNFα secretion. Some tumour cells upregulate HLA class II under influence by IFNγ and may thus be directly killed by the T cell.Citation44 Interestingly, the upregulation of HLA class II has been described as a mechanism for tumour escape in melanoma.Citation45 The possible implications of the redirected CD8+ T cells are interesting and not clear. The safety advantage from limited distribution of HLA class II applies even to the redirected CD8+ T cells, as these are DP4-restricted. CD8+ T cells generally have a stronger cytotoxicity potential than their CD4+counterparts. Our data show that this applies even to the HLA class II restricted TCRs, as assessed by CD107a. This suggests an increased ability to kill tumour cells and tumour-promoting stromal cells, including HLA class II positive tumour-associated macrophages, but may also lead to side effects.
The concept of Th-based ACT is largely unproven, though promising results have been obtained in a few pioneering studies.Citation46,Citation47 The natural role of these cells as “helpers” suggest that ACT with Th cells may be suitable for combination with other therapies, including checkpoint inhibitors, vaccines, oncolytic viruses or ACT with CTLs or CAR T cells.Citation5,Citation6,Citation48The cloned TCRs, C13 and D71, are widely applicable, as they recognize a universal tumour antigen (hTERT) and are DP4-restricted. These TCRs may thus be employed for exploring the Th approach across different cancers and combinatory regimes. This may be useful for clinical development and the identification of therapeutic niches for Th cells.
The limited durability of TCR expression after mRNA-transfection is a safety advantage, but also a major challenge for the efficacy. We have in previous studies with chimeric antigen receptors (CARs) observed that T cells transfected with mRNA encoding a CD19-specific CAR were functional for 8 days, even though the CAR could hardly be detected by flow cytometry beyond day fiv.Citation49 In the present TCR study, we have similarly observed peptide-specific responses in about 40% of T cells at day 6, when TCR expression is not readily detected by flow cytometry. Probably, this observation reflects that low TCR/CAR-expression, below the detection limit for flow cytometry, may be sufficient to trigger a T cell response. The RQR8 expression was detected beyond the TCR, and TCR D71 was detected longer than C13. These observations illustrate that different proteins, and different TCRs/CARs, may be retained at different levels after mRNA transfection. Such differences in expression between TCRs are not unusual. The molecular reasons for this are not entirely clear, but the differences are most likely due to intrinsic properties of the TCR, including its stability/half-life and tendency to mispairing with endogenous α/β chains. Accordingly, the functional durability of each TCR should be assessed separately. Our data suggest that the marker gene RQR8 is useful for assessing initial transfection efficiency, but not fully informative for predicting how long the T cells retain their ability to respond. A more prolonged detection of RQR8 on flow cytometry, compared to the TCRs, may be due to differences in sensitivity between the detection Abs. It may also reflect a higher expression or slower degradation of the RQR8 protein. The smaller proteins, like RQR8 (136aa), are usually expressed better than larger proteins, like the TCRs. Further, TCR expression may be affected by mispairing with endogenous α/β chains. In the clinical setting, RQR8-based measurements may be applied to monitor the fate of injected mRNA-transfected T cells. The RQR8-based measurements may over-estimate the number of TCR expressing cells. On the other hand, a higher expression and stability of RQR8 compared to the TCRs suggests that rituximab may effectively deplete T cells expressing even low TCR levels, thus supporting RQR8 as a safety strategy.
In the Bioplex cytokine assays, we observed high concentrations of the hallmark Th1 cytokine IFNγ and only low concentrations of Th2, Tr1 and Th17 cytokines. These findings indicate that the T cells have a Th1 cytokine profile, which is considered beneficial for anti-tumor responses. The flow cytometry assays analyse the cytokine profile from a different angle, capturing only a short time window and fewer cytokines, but providing information on the individual cell level. Our flow cytometry data were consistent with a polyfunctional and Th1-like response, with a large proportion of IFNγ and TNFα secreting cells, and the observation that many individual cells were polyfunctional. The cytokine profile is likely to depend on the T cell expansion protocol, the TCRs and other factors. The results reported above suggest that the applied T cell expansion and mRNA-transfection protocol yield T cells with a polyfunctional and Th1-weighted cytokine profile, at least for the TCR tested herein. These findings should be confirmed with cells produced in a full scale GMP protocol for clinical trials, which may differ in several aspects compared to an experimental set-up. As reported, we have though found that the functionality and cytokine profile of TCR C13 and D71 is robust in cells that were frozen/thawed before electroporation.
The issue of on-target, off-tumour toxicity remains a concern for hTERT. Telomerase is universally expressed across cancer forms, including stem-cell like cancer cells, but is also expressed in vital normal cell compartments like the bone marrow. Cancer vaccines targeting hTERT have not produced serious toxicity, but ACT is a more hard-hitting approach. We assume that T-helper TCRs targeting hTERT are less likely to yield intolerable toxicity, compared to CTL TCRs, as HLA-class II is not expressed by most hTERT+ cells. Further, we have chosen TCRs from T cells that circulated in long term survivors after cancer vaccination. It is conceivable, but not possible to confirm, that these cells have contributed to a favourable clinical outcome in these advanced NSCLC patients, who were treated at a time when checkpoint inhibitors were not available. Regarding safety, it is worth noting that the patients did not develop detectable side effects. However, ACT is a powerful approach where high numbers of activated T cells are infused and normal mechanisms for immune tolerance may be overwhelmed. Further, the concept of Th therapy is challenging to test in animal models, as it so heavily depends on the host immune system. Indeed, major aspects of both efficacy and toxicity may result from indirect mechanisms that are patient-specific and hard to model. This includes the mobilisation of the innate immune system as well as epitope spreading. Taken together, we consider that there is a strong rationale for using the mRNA-approach in a first-in-man trial with T-helper TCRs targeting hTERT. There will be a need for repeated injections as the TCRs are only transiently expressed. To improve safety and account for the expected inter-patient variability, we plan that each patient will receive a low-dose first injection, followed by weekly dose-escalation. Before moving into clinical testing, we need to establish full-scale GMP production of the cell product and procedures for screening patients for hTERT tumour expression and DP4-phenotype. The hTERT-specific TCRs may be applied in any tumour form, but the mRNA-strategy depends on rapid homing to tumour. As the T cells will pass through the lungs immediately after intravenous injection, patients with disease confined to the lungs may be well suited.
Materials and methods
Patients and ethical approval
Patients C and D had advanced NSCLC and were included in protocol CTN-2000,Citation27 conducted at Oslo University Hospital. The study was approved by the Norwegian Medical Agency and Regional Committee for Medical Research Ethics and performed in compliance with the World Medical Association Declaration of Helsinki. Written informed consent was obtained from the patients.
TCR cloning and in vitro mRNA synthesis
The TCR sequences from T cell clones C13 and D71 were determined as previously described.Citation50 The sequences were codon optimized and modified to express mouse TCR constant regions, in order to improve surface expression and facilitate detection of the expressed TCR on human T cells. Codon optimization used an in-house algorithm (written by M.P. and available upon request). The TCR sequences were cloned into the mRNA expression vector pCIpA102. The suicide/marker/sorter gene RQR8Citation38 was cloned upstream of the TCR α and β chains with 2A ribosome skipping sequence. All constructs were generated by in-house gene synthesis using polymerase chain reaction assembly of overlapping oligos. Phusion polymerase, quick Ligase, and NEB5α (New England Biolabs) were used for molecular cloning. Oligonucleotides were purchased from IDTDNA. The in vitro mRNA synthesis was performed essentially as previously described.Citation49 The mRNA was assessed by agarose gel electrophoresis and Nanodrop (Thermo Fisher Scientific, Waltham, MA).
Peptide GV1001 and recombinant hTERT protein
The vaccine peptide GV1001 (hTERT 611–626; EARPALLTSRLRFIPK) was supplied by Pharmexa. Recombinant hTERT protein fragment (563–735) was ordered from Genscript and initially diluted according to the manufacturer’s recommendation, in storage buffer 50 mM Tris-HCl, 10% Glycerol, pH 8.0. For use in experiments, the protein was diluted further in CellGro DC culture medium (CellGenix GmbH, Freiburg, Germany).
Primary T cells
Peripheral blood mononuclear cells (PBMCs) were isolated from healthy donor buffy coats using Lymphoprep (Axis-Shield, Oslo, Norway) density gradient centrifugation and suspended in CellGro DC culture medium (CellGenix GmbH, Freiburg, Germany) with 5% inactivated human serum (PAA Laboratories GmbH), 0.8% Mucomyst (AstraZeneca, London, UK), 50 µg/ml Gentamicin and 100 U/ml IL-2 (Proleukin, Novartis Vaccines & Diagnostics, CA) at 1.5 million cells/ml. T cells were expanded from PBMCs as previously describedCitation49 using Dynabeads ClinExVivo CD3/CD28 (Thermo Fisher, Oslo, Norway).
Electroporation of expanded T cells
Expanded T cells (day 10 post-activation) were used fresh, or after freezing/thawing, as stated. The fresh cells were washed twice and resuspended in CellGro DC medium (CellGenix GmbH). The thawed cells were resuspended in CellGro DC medium + 100 U/ml IL-2 and rested for 4 h before electroporation. For transfection, the cells (0.8ml at 5 × 106 cells/ml) were mixed with mRNA (50 ml; 100 μg/ml) and electroporated in a 4-mm gap cuvette at 500 V and 2 ms using a BTX 830 Square Wave Electroporator (BTX Technologies Inc., Hawthorne, NY, USA). Directly following transfection, cells were transferred to culture medium and cultured at 37°C in 5% CO 2 overnight to allow TCR expression.
T cell assays and flow cytometry
HLA DP04 positive EBV-transformed cells (EBV cells) generated from patient C were used as APCs. T cells were incubated with APCs, usually at ratio of 1:2, with or without (w/wo) antigen (peptide GV1001 or hTERT protein). Intracellular staining was performed using the BD Fix/ Perm reagent, according to the manufacturer´s protocol (Becton Dickinson). Briefly, the T cells were incubated w/wo antigen for 1h before addition of BD Golgiplug, BD Golgistop and CD107a-PE-Cy5 (BD Bioscience). Then, the cells were incubated overnight, before staining with mAbs for flow cytometry. The following reagents were used: QBen10-PE, QBen10-APC (R&D Systems), Fixable Viability Dye eFluor® 780, aINFγ PE-Cy7 (eBioscience), anti-mouse TCR constant beta region (a-mCb) APC, a-mCb V450, aTNFα-BV421, aCD4-BV510, aCD8-FITC, aCD4 FITC, aCD4 PE, aCD8 PE (BD). Flow cytometry acquisition was performed using Beckman-Coulter Cyan, BD LSRII or BD Fortessa instruments. The data were analysed using FlowJo software (Treestar Inc.).
Bioplex cytokine assays
T cells were incubated with EBV cells w/wo GV1001 peptide. Bioplex cytokine analyses were performed on supernatants harvested after 48 hours, according to the manufacturer’s protocol (Bio-Rad Laboratories, Hercules, Ca, USA). Supernatants were analysed in duplicates, each parallel kept separate through T cell stimulation and Bioplex assays.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors would like to thank Dr. S Sæbøe-Larssen, Dr. H Almåsbak and Dr. EM Inderberg for valuable advice, and Dr. Ines Carduso for kind help in the preparation of figures. The work was supported by the Norwegian Health Region South East and Radiumhospitalets legater. The authors thank the doctors and nurses at the Clinical Trial Unit and the staff at the Section for Immunotherapy for their excellent contributions to the vaccine trials that lead to this work. We also extend our sincere gratitude to the patients that participated in the trials.
Additional information
Funding
References
- Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, Teachey DT, Chew A, Hauck B, Wright JF, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368(16):1509–1518. doi:10.1056/NEJMoa1215134.
- Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, Bartido S, Stefanski J, Taylor C, Olszewska M, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med. 2013;5(177):177ra138. doi:10.1126/scitranslmed.3005930.
- Kochenderfer JN, Rosenberg SA. Treating B-cell cancer with T cells expressing anti-CD19 chimeric antigen receptors. Nat Rev Clin Oncol. 2013;10(5):267–276. doi:10.1038/nrclinonc.2013.46.
- Ramos CA, Heslop HE, Brenner MK. CAR-T cell therapy for lymphoma. Annu Rev Med. 2016;67::165–183. doi:10.1146/annurev-med-051914-021702.
- Abken H. Adoptive therapy with CAR redirected T cells: the challenges in targeting solid tumors. Immunotherapy. 2015;7(5):535–544. doi:10.2217/imt.15.15.
- Tanoue K, Rosewell Shaw A, Watanabe N, Porter C, Rana B, Gottschalk S, Brenner M, Suzuki M. Armed oncolytic adenovirus-expressing PD-L1 mini-body enhances antitumor effects of chimeric antigen receptor T cells in solid tumors. Cancer Res. 2017;77(8):2040–2051. doi:10.1158/0008-5472.CAN-16-1577.
- Kayser S, Bobeta C, Feucht J, Witte KE, Scheu A, Bulow HJ, Joachim S, Stevanovic S, Schumm M, Rittig SM, et al. Rapid generation of NY-ESO-1-specific CD4 T1 cells for adoptive T-cell therapy. Oncoimmunology. 2015;4(5):e1002723. doi:10.1080/2162402X.2015.1008371.
- Matsuzaki J, Tsuji T, Luescher IF, Shiku H, Mineno J, Okamoto S, Old LJ, Shrikant P, Gnjatic S, Odunsi K. Direct tumor recognition by a human CD4(+) T-cell subset potently mediates tumor growth inhibition and orchestrates anti-tumor immune responses. Sci Rep. 2015;5:14896. doi:10.1038/srep14896.
- Malandro N, Budhu S, Kuhn NF, Liu C, Murphy JT, Cortez C, Zhong H, Yang X, Rizzuto G, Altan-Bonnet G, et al. Clonal abundance of tumor-specific CD4(+) T cells potentiates efficacy and alters susceptibility to exhaustion. Immunity. 2016;44(1):179–193. doi:10.1016/j.immuni.2015.12.018.
- Church SE, Jensen SM, Antony PA, Restifo NP, Fox BA. Tumor-specific CD4+ T cells maintain effector and memory tumor-specific CD8+ T cells. Eur J Immunol. 2014;44(1):69–79. doi:10.1002/eji.201343718.
- Hadrup S, Donia M, Thor Straten P. Effector CD4 and CD8 T cells and their role in the tumor microenvironment. Cancer Microenviron. 2013;6(2):123–133. doi:10.1007/s12307-012-0127-6.
- Nitschke NJ, Bjoern J, Met O, Svane IM, Andersen MH. Therapeutic vaccination against a modified minimal survivin epitope induces functional CD4 T cells that recognize survivin-expressing cells. Scand J Immunol. 2016;84(3):191–193. doi:10.1111/sji.12456.
- Quezada SA, Simpson TR, Peggs KS, Merghoub T, Vider J, Fan X, Blasberg R, Yagita H, Muranski P, Antony PA, et al. Tumor-reactive CD4(+) T cells develop cytotoxic activity and eradicate large established melanoma after transfer into lymphopenic hosts. J Exp Med. 2010;207(3):637–650. doi:10.1084/jem.20091918.
- Nelles ME, Paige CJ. CD4(+) T cell plasticity engenders robust immunity in response to cytokine therapy. Oncoimmunology. 2015;4(3):e994370. doi:10.1080/2162402X.2015.1008371.
- Braumuller H, Wieder T, Brenner E, Assmann S, Hahn M, Alkhaled M, Schilbach K, Essmann F, Kneilling M, Griessinger C, et al. T-helper-1-cell cytokines drive cancer into senescence. Nature. 2013;494(7437):361–365. doi:10.1038/nature11824.
- Shedlock DJ, Shen H. Requirement for CD4 T cell help in generating functional CD8 T cell memory. Science. 2003;300(5617):337–339. doi:10.1126/science.1082305.
- Janssen EM, Lemmens EE, Wolfe T, Christen U, von Herrath MG, Schoenberger SP. CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes. Nature. 2003;421(6925):852–856. doi:10.1038/nature01441.
- Knutson KL, Disis ML. Tumor antigen-specific T helper cells in cancer immunity and immunotherapy. Cancer Immunol Immunother. 2005;54(8):721–728. doi:10.1007/s00262-004-0653-2.
- Kennedy R, Celis E. Multiple roles for CD4+ T cells in anti-tumor immune responses. Immunol Rev. 2008;222:129–144. doi:10.1111/j.1600-065X.2008.00616.x.
- Lurquin C, Lethe B, De Plaen E, Corbiere V, Theate I, van Baren N, Coulie PG, Boon T. Contrasting frequencies of antitumor and anti-vaccine T cells in metastases of a melanoma patient vaccinated with a MAGE tumor antigen. J Exp Med. 2005;201(2):249–257. doi:10.1084/jem.20041378.
- Inderberg-Suso EM, Trachsel S, Lislerud K, Rasmussen AM, Gaudernack G. Widespread CD4+ T-cell reactivity to novel hTERT epitopes following vaccination of cancer patients with a single hTERT peptide GV1001. Oncoimmunology. 2012;1(5):670–686. doi:10.4161/onci.20426.
- Butterfield LH, Ribas A, Dissette VB, Amarnani SN, Vu HT, Oseguera D, Wang HJ, Elashoff RM, McBride WH, Mukherji B, et al. Determinant spreading associated with clinical response in dendritic cell-based immunotherapy for malignant melanoma. Clin Cancer Res. 2003;9(3):998–1008.
- Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348(6230):69–74. doi:10.1126/science.aaa4971.
- McGranahan N, Furness AJ, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, Jamal-Hanjani M, Wilson GA, Birkbak NJ, Hiley CT, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. 2016: 1463–1469. doi:10.1126/science.aaf1490.
- Kyte JA, Gaudernack G, Faane A, Lislerud K, Inderberg EM, Brunsvig P, Aamdal S, Kvalheim G, Walchli S, Pule M. T-helper cell receptors from long-term survivors after telomerase cancer vaccination for use in adoptive cell therapy. Oncoimmunology. 2016;5(12):e1249090. doi:10.1080/2162402X.2016.1249090.
- Castelli FA, Buhot C, Sanson A, Zarour H, Pouvelle-Moratille S, Nonn C, Gahery-Segard H, Guillet JG, Menez A, Georges B, et al. HLA-DP4, the most frequent HLA II molecule, defines a new supertype of peptide-binding specificity. J Immunol. 2002;169(12):6928–6934.
- Brunsvig PF, Aamdal S, Gjertsen MK, Kvalheim G, Markowski-Grimsrud CJ, Sve I, Dyrhaug M, Trachsel S, Moller M, Eriksen JA, et al. Telomerase peptide vaccination: a phase I/II study in patients with non-small cell lung cancer. Cancer Immunol Immunother. 2006;55(12):1553–1564. doi:10.1007/s00262-006-0145-7.
- Brunsvig PF, Kyte JA, Kersten C, Sundstrom S, Moller M, Nyakas M, Hansen GL, Gaudernack G, Aamdal S. Telomerase peptide vaccination in NSCLC: A phase II trial in stage III patients vaccinated after chemoradiotherapy and an 8-year update on a phase I/II trial. Clin Cancer Res. 2011;17(21):6847–6857. doi:10.1158/1078-0432.CCR-11-1385.
- Vonderheide RH, Hahn WC, Schultze JL, Nadler LM. The telomerase catalytic subunit is a widely expressed tumor-associated antigen recognized by cytotoxic T lymphocytes. Immunity. 1999;10:673–679.
- Shay JW, Bacchetti S. A survey of telomerase activity in human cancer. Eur J Cancer. 1997;33(5):787–791. doi:10.1016/S0959-8049(97)00062-2.
- Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PL, Coviello GM, Wright WE, Weinrich SL, Shay JW. Specific association of human telomerase activity with immortal cells and cancer. Science. 1994;266:2011–2015.
- Harley CB. Telomerase and cancer therapeutics. Nat Rev Cancer. 2008;8:167–179. doi:10.1038/nrc2275.
- Linette GP, Stadtmauer EA, Maus MV, Rapoport AP, Levine BL, Emery L, Litzky L, Bagg A, Carreno BM, Cimino PJ, et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood. 2013;122(6):863–871. doi:10.1182/blood-2013-03-490565.
- Morgan RA, Chinnasamy N, Abate-Daga D, Gros A, Robbins PF, Zheng Z, Dudley ME, Feldman SA, Yang JC, Sherry RM, et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother. 2013;36(2):133–151. doi:10.1097/CJI.0b013e3182829903.
- Parkhurst MR, Yang JC, Langan RC, Dudley ME, Nathan DA, Feldman SA, Davis JL, Morgan RA, Merino MJ, Sherry RM, et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol Ther. 2011;19(3):620–626. doi:10.1038/mt.2010.272.
- Lamers CH, Sleijfer S, van Steenbergen S, van Elzakker P, van Krimpen B, Groot C, Vulto A, Den Bakker M, Oosterwijk E, Debets R, et al. Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: clinical evaluation and management of on-target toxicity. Mol Ther. 21(4): 904–912. doi:10.1038/mt.2013.17.
- Saebøe-Larssen S, Fossberg E, Gaudernack G. mRNA-based electrotransfection of human dendritic cells and induction of cytotoxic T lymphocyte responses against the telomerase catalytic subunit (hTERT). J Immunol Methods. 2002;259:191–203.
- Philip B, Kokalaki E, Mekkaoui L, Thomas S, Straathof K, Flutter B, Marin V, Marafioti T, Chakraverty R, Linch D, et al. A highly compact epitope-based marker/suicide gene for easier and safer T-cell therapy. Blood. 2014;124(8):1277–1287. doi:10.1182/blood-2014-01-545020.
- Wenandy L, Sorensen RB, Sengelov L, Svane IM. thor Straten P, Andersen MH. The immunogenicity of the hTERT540–548 peptide in cancer. Clin Cancer Res. 2008;14(1):4–7. doi:10.1158/1078-0432.CCR-07-4590.
- Structural Genomics C, China Structural Genomics C, Northeast Structural Genomics C, Graslund S, Nordlund P, Weigelt J, Hallberg BM, Bray J, Gileadi O, Knapp S, et al. Protein production and purification. Nat Methods. 2008;5(2):135–146. doi:10.1038/nmeth.f.202.
- Kyte JA, Gaudernack G. Immuno-gene therapy of cancer with tumour-mRNA transfected dendritic cells. Cancer Immunol Immunother. 2006;55(11):1432–1442. doi:10.1007/s00262-006-0161-7.
- Inderberg EM, Walchli S, Myhre MR, Trachsel S, Almasbak H, Kvalheim G, Gaudernack G. T cell therapy targeting a public neoantigen in microsatellite instable colon cancer reduces in vivo tumor growth. Oncoimmunology. 2017;6(4):e1302631. doi:10.1080/2162402X.2017.1302631.
- Beatty GL, Haas AR, Maus MV, Torigian DA, Soulen MC, Plesa G, Chew A, Zhao Y, Levine BL, Albelda SM, et al. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol Res. 2014;2(2):112–120. doi:10.1158/2326-6066.CIR-13-0170.
- Seliger B, Kloor M, Ferrone S. HLA class II antigen-processing pathway in tumors: molecular defects and clinical relevance. Oncoimmunology. 2017;6(2):e1171447. doi:10.1080/2162402X.2016.1171447.
- Donia M, Andersen R, Kjeldsen JW, Fagone P, Munir S, Nicoletti F, Andersen MH, Thor Straten P, Svane IM. Aberrant expression of MHC class II in melanoma attracts inflammatory tumor-specific CD4+ T- cells, which dampen CD8+ T-cell antitumor reactivity. Cancer Res. 2015;75(18):3747–3759. doi:10.1158/0008-5472.CAN-14-2956.
- Lu YC, Parker LL, Lu T, Zheng Z, Toomey MA, White DE, Yao X, Li YF, Robbins PF, Feldman SA, et al. Treatment of patients with metastatic cancer using a major histocompatibility complex class II-restricted T-cell receptor targeting the cancer germline antigen MAGE-A3. J Clin Oncol. 2017;35(29):3322–3329. doi:10.1200/JCO.2017.74.5463.
- Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, Dudley ME, Wunderlich JR, Somerville RP, Hogan K, Hinrichs CS, et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science. 2014;344(6184):641–645. doi:10.1126/science.1251102.
- Eriksson E, Milenova I, Wenthe J, Stahle M, Leja-Jarblad J, Ullenhag G, Dimberg A, Moreno R, Alemany R, Loskog A. Shaping the tumor stroma and sparking immune activation by CD40 and 4-1BB signaling induced by an armed oncolytic virus. Clin Cancer Res. 2017;23(19):5846–5857. doi:10.1158/1078-0432.CCR-17-0285.
- Almasbak H, Rian E, Hoel HJ, Pule M, Walchli S, Kvalheim G, Gaudernack G, Rasmussen AM. Transiently redirected T cells for adoptive transfer. Cytotherapy. 2011;13(5):629–640. doi:10.3109/14653249.2010.542461.
- Walchli S, Loset GA, Kumari S, Johansen JN, Yang W, Sandlie I, Olweus J. A practical approach to T-cell receptor cloning and expression. PLoS One. 2011;6(11):e27930. doi:10.1371/journal.pone.0027930.