
ABSTRACT
Adjuvant immunotherapies targeting CTLA4 or PD-1 recently demonstrated efficacy in the treatment of earlier stages of human cancer. We previously demonstrated using mouse spontaneous metastasis models that neoadjuvant immunotherapy and surgery was superior, compared to surgery and adjuvant immunotherapy, in eradicating the lethal metastatic disease. However, the optimal scheduling between neoadjuvant immunotherapy and surgery and how it impacts on efficacy and development of immune-related adverse events (irAEs) remains undefined. Using orthotopic 4T1.2 and E0771 mouse models of spontaneously metastatic mammary cancer, we varied the schedule and duration of neoadjuvant immunotherapies and surgery and examined how it impacted on long-term survival. In two tumor models, we demonstrated that a short duration (4–5 days) between first administration of neoadjuvant immunotherapy and resection of the primary tumor was necessary for optimal efficacy, while extending this duration (10 days) abrogated immunotherapy efficacy. However, efficacy was also lost if neoadjuvant immunotherapy was given too close to surgery (2 days). Interestingly, an additional 4 adjuvant doses of treatment following a standard 2 doses of neoadjuvant immunotherapy, did not significantly improve overall tumor-free survival regardless of the combination treatment (anti-PD-1+anti-CD137 or anti-CTLA4+anti-PD-1). Furthermore, biochemical immune-related adverse events (irAEs) increased in tumor-bearing mice that received the additional adjuvant immunotherapy. Overall, our data suggest that shorter doses of neoadjuvant immunotherapy scheduled close to the time of surgery may optimize effective anti-tumor immunity and reduce severe irAEs.
Introduction
Antibody-based therapies targeting the immune checkpoint receptors cytotoxic T-lymphocyte-associated protein 4 (CTLA4) or programmed cell-death protein 1 (PD-1) or its ligand PD-L1 alone or in combination are now approved for the treatment of various advanced cancer types including melanoma and NSCLC.Citation1 The demonstration that these cancer immunotherapies induced clinical responses and improved overall survival, even in advanced cancer has now prompted the evaluation of their efficacy in an adjuvant setting in the context of cancer surgery for patients who are at high risk of disease recurrence. Both adjuvant anti-CTLA4 and anti-PD-1 improved recurrence-free survival in the treatment of patients with resectable stage III melanoma.Citation2-Citation4 In addition, it was reported that amongst patients with stage IIIB, IIIC, or IV melanoma, the use of adjuvant anti-PD-1 compared to anti-CTLA4 resulted in a significantly longer recurrence-free survival (RFS) at 12 months and a better safety profile.Citation5
Although promising, there is scope to further improve clinical outcomes, given a significant proportion of patients who receive adjuvant anti-CTLA4 or anti-PD-1 still relapse.Citation3,Citation5 One strategy is to administer immunotherapies in a neoadjuvant setting prior to cancer surgery. Potentially, neoadjuvant treatment may offer a number of advantages, allowing one to: (i) determine therapy efficacy within the individual patient for possible additional adjuvant therapy, if needed; (ii) reduce tumor burden before surgery; and (iii) use pathological response data as surrogate outcome markers for relapse-free and overall survival.Citation6 Recently, we demonstrated the improved efficacy of neoadjuvant immunotherapy to eradicate spontaneous metastatic disease in mouse models of triple negative breast cancer (TNBC) following resection of the primary tumor.Citation7 We observed improvement in long-term survival regardless of the types of immunotherapy used, including anti-CD25 to deplete Tregs or anti-PD-1 alone or in combination with agonistic anti-CD137 to activate effector T and natural killer (NK) cells. Importantly, immediately increased tumor-localized tumor-specific CD8+ T cells and early elevated, and sustained peripheral tumor-specific CD8+ T cells after neoadjuvant immunotherapy, strongly correlated with improved survival.Citation7 Clinically, the efficacy of neoadjuvant, compared to adjuvant ipilimumab (anti-CTLA4) and nivolumab (anti-PD-1) combination immunotherapy was assessed in a small two-arm Phase 1b trial in high-risk stage IIIB/C melanoma patients (OpACIN; NCT02437279)(n = 20). Patients were randomized to either receive four cycles of ipilimumab+nivolumab every 3 weeks following tumor resection or to receive two cycles of ipilimumab+nivolumab every 3 weeks pre-surgery, followed by surgery at week 6 and another two cycles of treatment at week 12. Neoadjuvant immunotherapy was feasible as all patients underwent surgery on schedule with data suggesting a better recurrence-free benefit in the neoadjuvant compared to an adjuvant-treated group at a median follow-up of 25.6 months.Citation6 Neoadjuvant ipilimumab+nivolumab expanded more tumor-resident T cell clones compared to an adjuvant application, similar to what we observed pre-clinically.Citation7 However, due to the high toxicity rates, a median of only two courses of treatment were given in both the neoadjuvant and adjuvant arms. Currently, new trials assessing the efficacy of neoadjuvant immunotherapies alone or in combination with chemotherapy and targeted therapy against other cancer types including NSCLC are underway.Citation8
Given that mature overall survival data from current neoadjuvant immunotherapy trials are a few years away, a number of key questions still remain to be answered. In this study, we defined the optimal duration between administration of neoadjuvant immunotherapy and surgery using clinically relevant mouse tumor models. We also assessed whether additional adjuvant immunotherapy should be given after neoadjuvant immunotherapy and whether this impacted on overall survival and development of immune-related adverse events (irAEs).
Materials and methods
Mouse strains
BALB/c and C57BL/6J wild-type (WT) mice were bred-in-house or purchased from the Walter and Eliza Hall Institute for Medical Research. Female mice greater than eight weeks old were used in all experiments and were approved by the QIMR Berghofer Medical Research Institute Animal Ethics Committee.
Cell culture
The BALB/c-derived 4T1.2 or C57BL/6-derived or E0771 mammary carcinoma cell line was cultured in RPMI 1640 or DMEM containing 10% FCS, penicillin/streptomycin, and L-glutamine, respectively, as previously described.Citation7 All cell lines were routinely tested for mycoplasma but cell line authentication was not routinely performed.
Antibodies and reagents
Purified anti-mouse PD-1 mAb (RMP1-14), anti-mouse anti-CD137 (3H3), anti-mouse CTLA4 (UC10-4F10), control IgG (2A3) were obtained from BioXCell (West Lebanon). FTY720 was obtained from Sigma and administered at 25 µg/mouse i.p. as indicated. For experiments where the anti-PD-1+anti-CD137 combination was used, 100 µg/mouse each of anti-PD-1 or anti-CD137 or 200 µg/mouse of cIg were administered i.p. as indicated. For experiments where the anti-PD-1 alone or anti-PD-1+anti-CTLA4 combination was used, 250 µg/mouse each of anti-PD-1 or anti-CTLA4 and 250 or 500 µg/mouse of cIg were administered i.p as indicated.
Spontaneous tumor metastasis
For post-surgery survival experiments, the indicated dose of 4T1.2 or E0771 tumor cells were inoculated into the fourth mammary fat pad of female mice as previously describedCitation7 and survival monitored. The mean tumor size at the time of resection was ~30–80 mm2.
Flow cytometry analysis
Tumor, blood, lungs, spleen and lymph nodes were harvested from mice and processed for flow cytometry analysis as previously described.Citation7 For surface staining, single cell suspensions were stained with anti-CD45.2 FITC (104)(eBioscience), anti-TCRβ PerCP-Cy5.5 (H57-597)(eBioscience), anti-CD8α BV711 (53–6.7)(Biolegend), anti-CD4 BV605 (RM4–5)(Biolegend), anti-CD69 PE-Cy7 (H1.2F3)(eBioscience), H-2Ld tetramer to peptide SPSYVYHQF APC (MuLV env gp70 423–431)(NIH Tetramer Core facility) and live/dead dye Zombie Aqua (Biolegend) in the presence of anti-CD16/32 (2.4G2) to block FcR. To stain for Ki67 (Sol185)(eBioscience), samples were fixed and permeabilized with Foxp3 Fixation/Permeabilization kit (eBioscience). To measure intracellular cytokine staining, single-cell suspensions were incubated for 4 hrs in complete RPMI with brefeldin A (Becton Dickinson). Samples were then surface stained before being fixed/permeabilized (Becton Dickinson CytoFix/CytoPerm Kit) and stained with anti-IFNγ AF488 (XMG1.2)(Biolegend) and fluorescence minus one control for IFNγ staining. To determine absolute counts in samples, liquid counting beads (Becton Dickinson Biosciences) were added directly before samples were run on the flow cytometer. All data were collected on a Fortessa 4 (Becton Dickinson) flow cytometer and analyzed with FlowJo v10 software (Tree Star, Inc.).
gp70 tetramer-specific CD8+ T cell sorting and stimulation
Total CD8+ T cells were positively selected from the lungs of neoadjuvant-treated mice by staining cell suspensions with anti-mouse CD8 microbeads in accordance with the manufacturer’s instructions. Cell suspensions were passed through magnetized Miltenyi LS columns (Miltenyi), then removed from the magnetic field and eluted off the column. Positively selected CD8+ T cells of every two mice from the same group were pooled into one tube for cell sorting. Next, cells were further stained for with anti-TCRβ PE (H57-597)(eBioscience), anti-CD8α Brilliant Violet 711 (53–6.7)(Biolegend), H-2Ld tetramer to peptide SPSYVYHQF APC (MuLV env gp70 423–431)(kindly provided by NIH Tetramer Core facility) and 7AAD (Biolegend). Live 7AAD− CD45.2+ TCRβ+ CD8+ gp70 tetramer+ cells were sorted on the MoFlo XDP cell sorter (Beckman Coulter Life Sciences). Sorted cells were incubated for 4 h in complete RPMI media containing Cell Stimulation Cocktail (containing PMA and Ionomycin)(eBioscience) with monensin and brefeldin A (Becton Dickinson) at 37°C. Cell suspensions were then measured for intracellular IFNγ.
Histology and liver enzyme analysis
Mouse livers were perfusion fixed in 10% neutral buffered formalin overnight, processed routinely and embedded in paraffin. Four micron thick sections were cut and stained with hematoxylin and eosin (H&E). H&E stained tissue sections were imaged using Aperio Scanscope AT (Leica, Germany) and analyzed by Aperio ImageScope. The pathology of mouse liver sections was scored by referring to Sparwasser’s standards.Citation9 The liver score is the sum of individual scores for inflammatory cell infiltration in portal tracts, parenchyma, and necrosis. Serum ALT levels were measured as an indicator of liver damage. ALT levels in fresh sera were measured by Pointe (SGPT) Liquid ALT detection kit (USA).
Statistics
Statistical analysis was performed using GraphPad Prism software (San Diego, CA). Comparison of different groups was carried out using unpaired Welch’s t-test. In some experiments, to determine what immunological effects were induced after respective neoadjuvant or adjuvant immunotherapies, significance had to be performed on different days. Kaplan-Meier analyses with log-rank sum test were used for animal survival experiments. We did not adjust for all multiple comparisons between the survival curves since we were only concerned with a limited number of comparisons between groups. Data was considered to be statistically significant where the p value was equal to or less than 0.05.
Results
Proximal neoadjuvant immunotherapy and tumor resection are critical for optimal efficacy
Clinically, a concern with neoadjuvant immunotherapy is its potential to delay scheduled cancer surgery due to induction of irAEs, particularly when targeting CTLA4, as well as the potential deterioration of non-responders which may interfere with potentially curative surgery.Citation6 Therefore, we determined how varying the duration between neoadjuvant immunotherapy and resection of the primary 4T1.2 tumor from the mammary fat pad of BALB/c mice impacted on overall survival (). In our standard treatment protocol, we maintained a four or five-day window between the start of neoadjuvant immunotherapy and surgery. As we previously demonstrated, neoadjuvant anti-PD-1+anti-CD137 given on days 12 and 14 followed by surgery on day 16 significantly improved the long-term survival of 4T1.2 tumor-bearing mice (7/10) compared to similar groups of mice that received surgery on day 10 followed by adjuvant anti-PD-1+anti-CD137 on days 12 and 14 (0/10) (). However, extending this duration to 10 days, where neoadjuvant anti-PD-1+anti-CD137 therapy was given early at days 6, 8 and surgery was still performed on day 16, resulted in a complete loss of long-term survivors (). Nevertheless, this cohort still survived significantly longer compared to tumor-bearing mice that received adjuvant anti-PD-1+anti-CD137 (). To further demonstrate that a shorter duration between neoadjuvant immunotherapy and surgery was optimal for its efficacy, we setup another experiment in which neoadjuvant immunotherapy was given early on days 6 and 8, followed by surgery on day 10 or day 16. Similar to , a 10-day window between immunotherapy and surgery did not result in any long-term survivors, although their survival was prolonged (). In contrast, in similar groups of mice that received treatment on days 6 and 8, followed by surgery on day 10 (i.e maintaining a 4-day duration), all mice survived long-term (8/8) (). This enhanced survival was not unexpected given the earlier neoadjuvant immunotherapy and surgery.
Figure 1. Duration between neoadjuvant immunotherapy and removal of primary tumor impacts on long-term survival.
(a-b), Groups of BALB/c WT mice (n = 5–10/grp) were injected with 5 × 104 4T1.2 mammary carcinoma cells into the mammary fat pad. a, b, As indicated, some groups of mice were treated i.p. with anti-PD-1+anti-CD137 or cIg on days 12 and 14. (a), Additionally, some groups received neoadjuvant anti-PD-1+anti-CD137 or cIg on days 6 and 8 (early). All neoadjuvant-treated groups had their primary tumors resected on day 16. Additionally, one group of mice was resected of their tumors on day 10 and treated i.p with adjuvant anti-PD-1+anti-CD137 on days 12 and 14. (b), Additionally, some groups of mice were treated with neoadjuvant anti-PD-1+anti-CD137 or cIg on days 6 and 8 (early) with primary tumors resected either on day 10 or 16. (c) Groups of BALB/c WT mice (n = 10/grp) were injected with 2 × 104 4T1.2 mammary carcinoma cells into the mammary fat pad. As indicated in the schematic, some groups of mice were treated i.p. with either anti-PD-1+anti-CD137 or anti-PD-1 alone or cIg on days 12 and 14 while other groups received neoadjuvant anti-PD-1+anti-CD137 or anti-PD-1 alone on days 14 and 16 (close). All groups had their primary tumors resected on day 16 as indicated in the schematic. (a,b,c) The Kaplan–Meier curves for overall survival of each group are shown. Significant differences between indicated groups were determined by log-rank sum test with exact p values shown. All experiments were performed once.
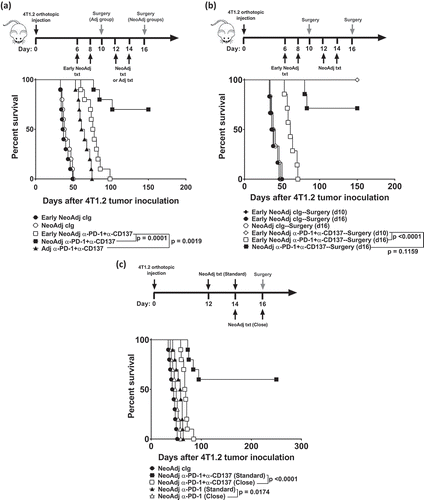
We next examined how further shortening the duration between neoadjuvant treatment and surgery impacted on overall survival (). For this experiment, two doses of neoadjuvant anti-PD-1+anti-CD137 combination immunotherapy or anti-PD-1 monotherapy were given on day 14 and on day 16 (Close), the time point when the primary tumor was resected. For the standard treatment, two doses of neoadjuvant immunotherapy were given on day 12 and 14 followed by surgery on day 16. Strikingly, this shortened window between neoadjuvant anti-PD-1+anti-CD137 and surgery resulted in the complete loss of long-term survivors (). Similarly, the duration between neoadjuvant anti-PD-1 monotherapy and surgery also significantly reduced prolongation of survival compared to the group that received standard neoadjuvant anti-PD-1 (). As expected, 60% of mice that received the standard treatment survived long-term ().
The envelope glycoprotein (gp70) is encoded by the endogenous murine leukemia virus (MuLV) and is expressed in a range of mouse cancer cell lines, including 4T1.2. Gp70 is generally silent in normal mouse tissues and can function as a dominant tumor antigen in cell lines that express MuLV.Citation7,Citation10,Citation11 We previously reported that an increase in gp70 tetramer-specific CD8+ T cells in the blood and peripheral organs occur early after neoadjuvant, but not adjuvant, immunotherapy, and this correlated with treatment efficacy.Citation7 Important to note, gp70 tetramer-specific CD8+ T cells in the lungs of adjuvant-treated mice continued to express the targets of therapy: CD137, PD-1, and CTLA4 (Supp. Figure 1A), suggesting this was not the reason why adjuvant immunotherapy was less effective. To determine if the decrease in efficacy observed with scheduling a longer duration between neoadjuvant immunotherapy and surgery impacted on the kinetics of gp70-T cell expansion, peripheral blood was obtained longitudinally from groups of mice in an experimental setup similar as ()(Supp. Figure 1B). As we previously reported, neoadjuvant immunotherapy increased peripheral gp70-T cells, with the maximum peak observed 4 days after commencement of treatment ()(Supp. Figure 1B). Interestingly, a similar gp70-T cell kinetics was observed in mice that received early neoadjuvant immunotherapy (days 6, 8) followed by surgery on day 16, suggesting that the inability to generate long-term survivors in this group was not due to changes in gp70-T cell kinetics. Next, we examined how the presence of the primary tumor impacts on the kinetics of gp70-T cells. We set up an experiment in which groups of tumor-bearing mice received neoadjuvant immunotherapy and either had their primary tumor resected or did not receive surgery ()(Supp. Figure 1C). Again, the kinetics of gp70-T cells was similar between both groups, demonstrating that the changes in proportions of gp70-T cells in the blood were induced by neoadjuvant immunotherapy and not impacted by surgery. However, the presence of the primary tumor was clearly required for the expansion of gp70-T cells as this was not observed in adjuvant immunotherapy treated mice ()(Supp. Figure 1B).
Figure 2. Duration between neoadjuvant immunotherapy and removal of primary tumor does not affect peripheral blood tumor-specific T cell expansion kinetics but attenuates its effector function.
(a), In an experimental setup similar to , peripheral blood was collected from all groups of mice (n = 5/grp) at the indicated time point and analyzed using flow cytometry. Gating on live CD45.2+ cells of lymphocyte morphology, the proportion of gp70 tetramer+ CD8+ TCRβ+ T cells are shown. Differences between neoadjuvant anti-PD-1+anti-CD137 (day 16) and early neoadjuvant anti-PD-1+anti-CD137 (day 10) (i.e. 4 days after the start of their respective therapies) were determined by unpaired Welch’s t-test with exact p-value indicated. (b), As indicated, groups of mice were treated i.p. with neoadjuvant anti-PD-1+anti-CD137 on days 11 and 13. Some groups of mice had their primary tumors resected on day 16 (surgery) compared to others that did not (No Surgery). Peripheral blood was collected from all groups of mice at the indicated time point, and the proportion of gp70 tetramer+CD8+TCRβ+ cells are shown. Differences between neoadjuvant anti-PD-1+anti-CD137 with and without surgery at day 15 were determined by unpaired Welch’s t-test with exact p-value indicated. (a-b), Data presented as mean + SEM with one naive mouse included in both experiments. Experiments were performed once. (c), In a similar experimental setup as , sorted lung gp70 tetramer+ CD8+ TCRβ+ T cells (n = 8–10 mice/group with 2 lungs pooled for each sample displayed) from all groups of mice on day 20 after tumor inoculation were re-stimulated with PMA/Io for 4 h and assessed for intracellular IFNγ+ production. Data presented as mean ± SEM. Differences between neoadjuvant anti-PD-1+anti-CD137 and early neoadjuvant anti-PD-1+anti-CD137 were determined by unpaired Welch’s t-test with exact p-value indicated. Data pooled from 2 experiments.
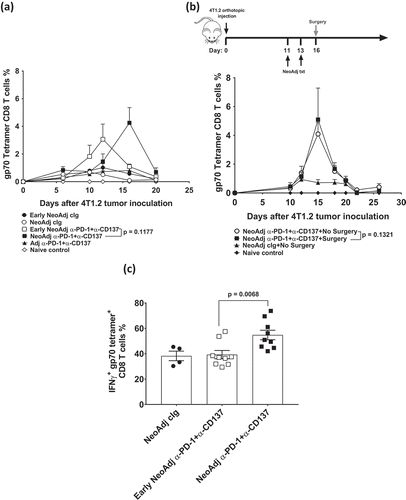
Although the expansion kinetics of gp70 tetramer-specific T cells was not different between mice that received early or standard neoadjuvant immunotherapy ()(Supp. Figure 1B), we hypothesized that the differences in their effector function may explain the difference in survival outcomes (). In an experimental setup similar to , we sorted lung gp70 tetramer-specific CD8+ T cells from early neoadjuvant or standard neoadjuvant-treated mice 20 days after tumor inoculation and restimulated them with PMA/Ionomycin and assessed their ability to produce IFNγ () (Supp. Figure 1D). We selected this time point as the level of tumor-specific T cells had decreased to a similar level between early and standard neoadjuvant-treated groups () (Supp. Figure 1B). We demonstrated that a significantly greater proportion of tumor-specific T cells from the standard compared to early neoadjuvant-treated mice produced IFNγ () (Supp. Figure 1D). Overall, our data suggest that there is an optimal window between neoadjuvant immunotherapy administration and surgery, which ultimately impacts on effector function of tumor-specific T cells.
Migration of tumor-specific T cells is critical for the efficacy of neoadjuvant immunotherapy
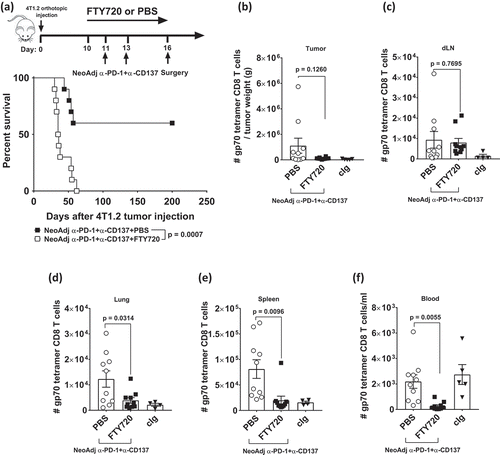
Following neoadjuvant immunotherapy, flow cytometry analysis demonstrated that gp70 tetramer-specific CD8+ T cells increased in the blood () and primary tumor as we previously described.Citation7 Using immunohistochemistry, we also observed an increased infiltration of CD8+ cells in the primary tumor of neoadjuvant compared to cIg treated mice (Supp. Figure 2A). These T cells were likely to migrate to metastatic sites to mediate their anti-tumor function. To address how their ability to circulate systemically impacts on the efficacy of neoadjuvant immunotherapy, we treated groups of mice with FTY720, a sphingosine 1 phosphate (SP1) receptor antagonist or PBS control one day before neoadjuvant anti-PD-1+anti-CD137 treatment and maintained FTY720 treatment until day 16 when the primary tumor was resected (). Strikingly, the addition of FTY720, which is thought to inhibit lymphocyte egress from lymphoid tissues, resulted in the complete loss of survival compared to the PBS-treated control group where 60% of the mice survived long-term (). In another experiment, we performed ex vivo analysis of gp70 tetramer-specific CD8+ T cells in the tumor, draining lymph node, lung, spleen, and blood of neoadjuvant-treated mice that received FTY720 or PBS on day 16, at the time point when surgery of the primary tumor would occur (-). We observed a significant decrease in tumor-specific T cells in the lung, spleen, and blood from FTY720 compared to PBS-treated groups, but not from the tumor and draining lymph node. Overall these data demonstrated the tumor and/or draining lymph node were the likely sources from which tumor-specific T cells originated.
Figure 3. The blockade of lymphocyte migration attenuates the anti-tumor efficacy of neoadjuvant immunotherapy.
Groups of BALB/c WT mice (n = 5/grp) were injected with 5 × 10Citation4 4T1.2 mammary carcinoma cells in the mammary fat pad on day 0. a, As indicated in the schematic, all mice were treated i.p. with neoadjuvant anti-PD-1+anti-CD137 mAb on days 11 and 13 with all primary tumors resected on day 16. Additionally, these groups of mice were treated i.p. with either PBS or FTY720 every day from day 10 to day 16. (a) The Kaplan–Meier curves for overall survival of each group are shown. Data pooled from 2 experiments with significant differences between indicated groups determined by log-rank sum test with exact p values shown. (b-f) In a similar experimental setup as , all mice (n = 4–5/grp) were sacrificed on day 16 and their organs were collected and single cell suspensions generated for flow cytometry. Gating on live CD45.2+ cells of lymphocyte morphology, the absolute numbers of gp70 tetramer+ CD8+ TCRβ+ cells in the tumor (b), the draining lymph node (dLN) (c), the lung (d), the spleen (e) and the blood (f) are shown. Each symbol represents a single mouse. Data presented as mean ± SEM. Data pooled from 2 experiments with significant differences determined by unpaired Welch’s t-test with exact p-value shown.
No significant benefit and greater irAEs with extended neoadjuvant-adjuvant immunotherapy
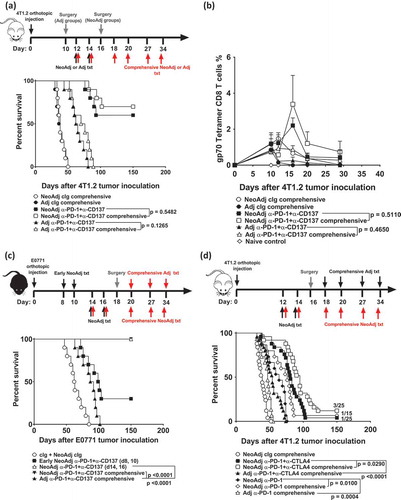
In our standard treatment protocol, two doses of neoadjuvant immunotherapy were administered followed by surgery. Clinically, some trials maintain treatment with immunotherapies after surgery.Citation8 To assess if additional doses of immunotherapies can further increase the proportion of tumor-free mice, we compared tumor-bearing mice receiving the standard two doses of neoadjuvant immunotherapy to those that received two doses of neoadjuvant immunotherapy followed by four doses of adjuvant therapy after surgery (NeoAdj comprehensive)(). Interestingly, the proportion of tumor-free mice did not significantly differ between mice that received standard or comprehensive neoadjuvant immunotherapy (). Similarly, no significant differences in survival were observed between mice receiving comprehensive compared to standard adjuvant anti-PD-1+anti-CD137 (). Furthermore, no major differences in the blood gp70-T cell kinetics of mice that received standard or comprehensive neoadjuvant immunotherapies were observed ()(Supp. Figure 3). Using E0771 as another spontaneously metastatic tumor model, we again demonstrated that standard (2 doses) or comprehensive (6 doses) of neoadjuvant anti-PD-1+anti-CD137 therapy were equally effective in generating long-term survivors (), although here the standard neoadjuvant combination immunotherapy was already completely effective. We also validated in the E0771 tumor model that the efficacy of neoadjuvant immunotherapy depended on a short duration between treatment and surgery, since early neoadjuvant immunotherapy on days 8 and 10, rather than on days 14, 16, followed by surgery on day 18, significantly reduced the number of tumor-free mice (3/10 vs 10/10)(). Given that the efficacy of anti-PD-1+anti-CTLA4 combination immunotherapy is currently being evaluated in many new neoadjuvant clinical trials, we tested this combination in 4T1.2 tumor-bearing mice (). Although comprehensive anti-PD-1+anti-CTLA4 significantly prolonged survival compared to standard neoadjuvant anti-PD-1+anti-CTLA4, the number of tumor-free survivors was not statistically different (3/25 vs 1/25)() and the kinetics of gp70-T cell expansion again did not greatly differ between these groups (Supp. Figure 4). A similar observation was seen for mice treated with comprehensive or standard anti-PD-1 monotherapy, where again the overall proportion of long-term survivors were similar (1/15 vs 0/15)(). Interestingly, standard neoadjuvant anti-PD-1 was still superior at significantly prolonging survival of treated mice compared to comprehensive adjuvant anti-PD-1 monotherapy (p = 0.0004)(). Overall our data suggest additional treatment following neoadjuvant immunotherapy and cancer surgery does not further increase the proportion of long-term survivors.
Figure 4. Comprehensive compared to standard neoadjuvant anti-PD-1+anti-CD137 are equally efficacious in impacting on long-term overall survival.
(a-b) Groups of BALB/c WT mice were injected with 5 × 10Citation4 4T1.2 mammary carcinoma cells into the mammary fat pad (n = 10/grp). As indicated, groups of mice were treated i.p. with neoadjuvant or adjuvant anti-PD-1+anti-CD137 on days 12 and 14 with all primary tumors resected on day 16 (neoadjuvant groups) or day 10 (adjuvant groups). Additionally, some groups of neoadjuvant and adjuvant-treated mice received additional anti-PD-1+anti-CD137 or cIg on days 18, 20, 27 and 34 (comprehensive). (b) In an experimental setup similar to (a), peripheral blood was collected from all groups of mice (n = 5/grp) at the indicated time point for flow cytometry. Gating on live CD45.2+ cells of lymphocyte morphology, the proportion of gp70 tetramer+ CD8+ TCRβ+ cells is shown. Data presented as mean + SEM. A naive mouse was also included in this experiment. Differences between mice that received standard or comprehensive anti-PD-1+anti-CD137 on day 20 following tumor inoculations were determined by unpaired Welch’s t-test with exact p-value indicated. c, Groups of C57BL/6J WT mice (n = 10/grp) were injected with 2 × 104 E0771 mammary carcinoma cells into the mammary fatpad. As indicated, some groups of mice were treated i.p. with neoadjuvant anti-PD-1+anti-CD137 mAb or cIg on days 8 and 10 (early), days 14 and 16 or days 14, 16, 20, 27 and 34 (comprehensive) with all primary tumors resected on day 18. One group of mice was treated with adjuvant anti-PD-1+anti-CD137 on days 20, 27 and 34 (comprehensive) with primary tumors resected on day 18. d, Groups of BALB/c WT mice (n = 5–10/grp) were injected with 2 × 10Citation4 4T1.2 mammary carcinoma cells into the mammary fat pad. As indicated, some groups of mice were treated i.p. with neoadjuvant anti-PD-1 alone, anti-PD-1+anti-CTLA4 or cIg on days 12 and 14 with all primary tumors resected on day 16. Additionally, some groups of neoadjuvant-treated mice received additional anti-PD1+anti-CTLA4 or cIg on days 18, 20, 27 and 34 (comprehensive). Other groups of mice received comprehensive adjuvant anti-PD-1 alone or anti-PD-1+anti-CTLA4 on days 18, 20, 27, 34 with primary tumors resected on day 16. (a, c, d) The Kaplan–Meier curves for overall survival of each group are shown. Significant differences between indicated groups were determined by log-rank sum test with exact p values shown. All experiments were performed once except for (d) where the data was pooled from 3 experiments. (b) is representative of two independent experiments.
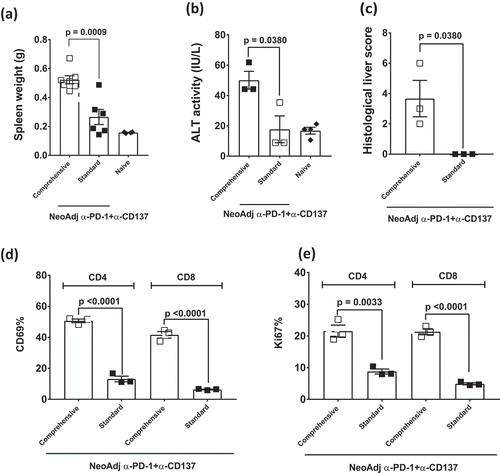
Although combination immunotherapies can increase clinical responses in a greater proportion of patients, the severity and proportion of patients who develop irAEs can also increase.Citation6,Citation12,Citation13 Therefore, we assessed whether standard or comprehensive neoadjuvant anti-PD-1+anti-CD137 therapy in 4T1.2 tumor-bearing mice affected the development of irAEs (). Long-term surviving mice were sacrificed 63 days after 4T1.2 tumor inoculation and liver, spleen and sera were collected for a series of analyses to detect biochemical irAEs, as we have previously described.Citation14 Mice that received comprehensive neoadjuvant anti-PD-1+anti-CD137 therapy (6 doses) displayed increased spleen weight compared to mice that received standard neoadjuvant anti-PD-1+anti-CD137 therapy (2 doses)(). Comprehensive, compared to standard, neoadjuvant immunotherapy-treated mice displayed increased liver damage as indicated by increased ALT levels and histological liver score (,)(Supp. Figure 5A, B). By contrast, the spleen weight and ALT levels in standard neoadjuvant immunotherapy-treated mice were similar to naive mice (,). The increased spleen weight in comprehensive neoadjuvant-treated mice also corresponded with an increase in the proportion of activated and proliferating total CD4+ and CD8+ T cells as defined by CD69 ()(Supp. Figure 5C) and Ki67 ()(Supp. Figure 5C) expression. Overall, these data suggest that extended neoadjuvant/adjuvant treatment only serves to increase systemic immune perturbation in peripheral tissues such as the liver.
Figure 5. Comprehensive compared to standard neoadjuvant anti-PD-1+anti-CD137 immunotherapy increases the severity of immune-related adverse events.
(a-e), From the same experiment as and/or another experiment in a similar setup as , long-term surviving mice (n = 3–4/grp) treated with 2 doses (Standard) or 6 doses (Comprehensive) of neoadjuvant anti-PD-1+anti-CD137 were sacrificed on day 63 after 4T1.2 tumor inoculation. Naive mice (n = 2–4/grp) were included in the experiment. Spleens were collected and single cell suspensions generated for flow cytometry. Spleen weights (a), sera ALT levels (b) and histological liver score (c) of the indicated groups of mice are shown. Gating on live CD45.2+ cells of lymphocyte morphology, the proportions of splenic CD4+ and CD8+ T cells (TCRβ+) that were (d) CD69+ or (e) Ki67+ are shown. (a-e) Data presented as mean ± SEM. Significant differences between standard and comprehensive neoadjuvant-treated groups determined by unpaired Student’s t-test with exact p-value shown. All experiments were performed once except for (a) which was pooled from two experiments.
Discussion
In this study, using two spontaneously metastatic mouse tumor models, we demonstrated that proximal (4–5 days) neoadjuvant immunotherapy and primary tumor resection was necessary for optimal efficacy, since commencing immunotherapy with delayed surgery (8 days after last treatment) was comparatively ineffective. However, further shortening the duration between neoadjuvant treatment and surgery from 4 to 5 days to 2 days also resulted in complete loss of long-term survivors, suggesting there is a window between neoadjuvant immunotherapy and surgery that is optimal for its anti-tumor efficacy. In addition, tumor-bearing mice receiving extended immunotherapy (2 neoadjuvant plus 4 adjuvant) did not display any significant increase in overall survival compared with mice simply given 2 doses of neoadjuvant immunotherapy immediately prior to surgery. This finding was observed whether mice were treated with anti-PD-1+anti-CD137 or anti-CTLA4+anti-PD-1. Furthermore, biochemical irAEs increased in tumor-bearing mice that received 6 doses, compared to 2 doses, of immunotherapy. Overall, our data suggest that a short course of neoadjuvant immunotherapy immediately prior to surgery may suffice to induce effective anti-tumor immunity while reducing the risk of developing severe irAEs.
A two-arm Phase 1b trial has assessed whether the combination of ipilimumab+nivolumab was more efficacious when administered in a neoadjuvant compared to adjuvant setting in Stage III melanoma patients who are able to receive complete resection of their primary tumor (OpACIN trial; NCT02437279).Citation6 In this study, patients were randomized to either receive 4 doses of adjuvant or a split neoadjuvant and adjuvant immunotherapy regime (2:2)(n = 10/grp). The trial met one of its co-primary endpoints, demonstrating the feasibility of the neoadjuvant regime, since all patients in the neoadjuvant arm underwent surgery at the pre-planned time point with no surgery-related adverse events attributed to immunotherapy.Citation6 However, many patients received only 2 out of the 4 planned doses of immunotherapy due to an unexpectedly high proportion of these patients developing grade 3–4 irAEs (18/20)Citation6 which appeared higher than what would be predicted from Stage IV melanoma patients treated with the same combination immunotherapy.Citation15 Despite the reduction in the planned courses of treatment, pathological complete responses (pCR) were achieved in 7/9 neoadjuvant-treated patients and none have so far relapsed, suggesting two cycles of treatment might suffice although this has to be confirmed with long-term overall survival follow-up. In our study, we observed that comprehensive neoadjuvant immunotherapy (6 doses) induced greater biochemical irAEs compared to standard neoadjuvant immunotherapy (2 doses). A possible explanation for the increased toxicities observed in the OpaCIN trial may relate to the better immune status of the melanoma patients or a lower degree of systemic immunosuppression. The former would seem less likely an explanation for the differences we observed in mice. In preliminary results from a larger follow-up phase 2 trial (n = 90)(OpACIN-neo trial, NCT02977052), patients treated with 2 cycles of ipilimumab (1 mg/kg) and nivolumab (3 mg/kg) were shown to have reduced toxicities while anti-tumor efficacy was preserved.Citation16 In our study, we demonstrated in two tumor models that additional treatment following 2 doses of neoadjuvant immunotherapy was generally not necessary as it did not significantly increase the proportion of long-term surviving mice, nor did it further expand the proportion of blood tumor-specific T cells. Going forward, with an optimal dosage now defined in the OpACIN-neo trial, it will be important to perform a randomized clinical trial powered to demonstrate whether neoadjuvant immunotherapy is superior compared to adjuvant immunotherapy in improving RFS and overall survival (OS), as we demonstrated in this and our previous study.Citation7
In published results from a phase 2 clinical trial by Amaria et al., neoadjuvant nivolumab was compared to neoadjuvant ipilimumab+nivolumab to treat patients with resectable clinical stage III or oligometastatic stage IV melanoma (NCT02519322).Citation12 Here, patients were randomized to receive up to 4 doses of neoadjuvant nivolumab every 14 days or up to 3 doses of ipilimumab+nivolumab every 21 days prior to surgery, before all patients were subsequently scheduled to receive adjuvant nivolumab. In this study, neoadjuvant ipilimumab+nivolumab or nivolumab monotherapy induced pCR rates of 45% (5/11) or 25% (3/12), respectively.Citation12 Similar to the study by Blank et al.,Citation6 grade 3/4 irAEs were observed in most of the combination treated groups (8/11) with dose delays being frequent (7/11). Interestingly, greater pCRs were observed in the Blank study compared to the Amaria study. Although there were some patients with stage IV disease in the latter, the majority of patients amongst these two studies were stage IIIb/c suggesting clinical stage differences may not fully explain the differences in response observed between these two trials. Another possibility could be the duration between treatment and surgery affected its efficacy. In the trial by Amaria, there was a 9-week window between the commencement of the three doses of neoadjuvant ipilimumab+nivolumab and surgery with most of these patients requiring dose delays between treatments due to toxicity issues.Citation12 It was unclear if this dose delay meant surgery was also delayed. In the Blank study, there was a 6-week duration between the two cycles of neoadjuvant ipilimumab+nivolumab and surgery, with all patients undergoing surgery at the pre-planned time point.Citation6 Interestingly, in another clinical trial of patients with resectable stage IIIB/C or Stage IV melanoma (n = 27), patients only received one dose of neoadjuvant anti-PD1 (pembrolizumab) before undergoing surgery 3 weeks later, followed by one year of adjuvant treatment.Citation17 In this trial, all patients had successful tumor resection and complete or near-complete pathological response was reported in 30% of patients (8/27), all of whom were reported recurrence-free.Citation17 These response rates were similar to what was described by Amaria et al.1Citation2 Clinically, the optimal time point for resection of the primary tumor post-neoadjuvant immunotherapy is not known. Our current data together with these recent neoadjuvant immunotherapy clinical trials in melanoma certainly demonstrates the importance in considering how the timing of neoadjuvant treatment and surgery impacts on its efficacy and this should be kept in mind in the design of future neoadjuvant clinical trials.
Pre-clinically, we have shown that neoadjuvant anti-PD-1 alone prolonged survival but was not curative compared to mice that received combination anti-PD-1+anti-CD137.Citation7 The clinical trial by Amaria et al., was stopped early on the basis of an early observation of disease progression, preventing surgical resection in the neoadjuvant nivolumab monotherapy-treated group (2/12).Citation12 In contrast, two cycles of neoadjuvant nivolumab monotherapy in patients with resectable early NSCLC (stage I, II, or IIIA), reported major pathological response in 9 of 20 resected tumors (45%)(NCT02259621).Citation18 All 20 patients who received nivolumab had a successful resection of their tumors 4 weeks after commencement of treatment. In contrast, a pCR of 22% (10/45) was reported in preliminary analysis of NSCLC patients receiving neoadjuvant anti-PD-L1 monotherapy.Citation19 Whether the better response of neoadjuvant anti-PD-1 monotherapy in NSCLC compared to melanoma patients was due to the former cohorts having earlier stages of disease or the presence of more tumor-reactive T cells in the former compared to the latter or the shorter duration between treatment and surgery remains to be elucidated. Ultimately, overall survival from the neoadjuvant-treated patients will validate whether neoadjuvant nivolumab monotherapy is efficacious.
In our study, the efficacy of neoadjuvant immunotherapy was critically dependent on an optimal duration between commencement of treatment and resection of the primary tumor as extending or shortening this duration abrogated the generation of long-term survivors. Although the expansion kinetics of gp70 tetramer-specific CD8+ T cells did not differ greatly between mice that received standard or early neoadjuvant immunotherapy, we observed an increased proportion of IFNγ producing lung tumor-specific T cells from the former compared to the latter group. We have previously demonstrated the requirement of IFNγ for the efficacy of neoadjuvant immunotherapy.Citation7 We propose that the timely removal of the primary tumor at the height of tumor-specific T cell expansion serves a number of purposes. First, the primary tumor represents the major source of tumor antigen and contains a significant proportion of tumor-specific gp70-T cells that are most likely to become dysfunctional/exhausted over time (expressing PD-1). We have previously demonstrated that gp70 tetramer-specific CD8+ T cells in the primary tumor represented ~50% of the tumor-infiltrating CD8+ T-cell population following neoadjuvant immunotherapyCitation7 and were likely to be reinvigorated and expand following neoadjuvant immunotherapy. As the experiment with FTY720 to block lymphocyte egress demonstrated, the tumor and draining lymph node likely represented the main source(s) of tumor-specific T cells that eventually traffic to the sites of metastases. By removing the primary tumor at the right time, we may prevent the retention of tumor-specific T cells at this site, which due to its size, may chronically stimulate tumor-specific T cells and contribute to their dysfunction. We have previously demonstrated that gp70 tetramer-specific CD8+ T cells were not effective against large primary 4T1.2 tumors as they were suppressed but not rejected.Citation7
Currently, adjuvant anti-CTLA4 and anti-PD-1 are FDA-approved for the treatment of high-risk patients with earlier stages of melanoma following surgery. However, a significant proportion of patients still relapse and our data demonstrates that neoadjuvant immunotherapy if it can be given prior to surgery, is one strategy to further improve overall survival. By understanding the key mechanisms and/or pathways activated by neoadjuvant immunotherapy, it may allow for their selective targeting which may further improve the efficacy of immunotherapies in general in the adjuvant or advanced cancer settings. Going forward, a key goal of neoadjuvant immunotherapy will be to identify immunological correlates of outcomes either from the primary tumor or in the PBMCs of these patients.
Disclosures and potential conflict of interest
M.J. Smyth declares scientific research agreements with Bristol Myers Squibb and Tizona Therapeutics and has an advisory board role for Tizona Therapeutics and Compass Therapeutics. No potential conflicts of interest were disclosed by the other authors.
Author contributions
Conception and design: J. Liu, M.J. Smyth, M.W.L. Teng
Development of methodology: J. Liu, J. S. O’Donnell, J. Madore, S. Allen
Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): J. Liu, J. S. O’Donnell, J. Yan, S. Allen, M.J. Smyth
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): J. Liu, J. S. O’Donnell, J.Madore, M.J. Smyth, M.W.L. Teng
Writing, review, and/or revision of the manuscript: J. Liu, J.S. O’Donnell, M.J. Smyth, M.W.L. Teng
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): J. Liu, J. S. O’Donnell, M.W.L. Teng
Study supervision: M.W.L. Teng
Supplemental Material
Download Zip (45.7 MB)Acknowledgments
The authors thank Liam Town and Kate Elder for mouse genotyping and maintenance during this study. The authors would also like to thank the QIMR Berghofer Medical Research Institute animal, flow cytometry and histology facilities.
Supplemental material
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
References
- Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348:56–61. doi:10.1126/science.aaa8172.
- Eggermont AM, Chiarion-Sileni V, Grob JJ, Dummer R, Wolchok JD, Schmidt H, Hamid O, Robert C, Ascierto PA, Richards JM, et al. Adjuvant ipilimumab versus placebo after complete resection of high-risk stage III melanoma (EORTC 18071): a randomised, double-blind, phase 3 trial. Lancet Oncol. 2015;16:522–530. doi:10.1016/S1470-2045(15)70122-1.
- Eggermont AM, Chiarion-Sileni V, Grob JJ, Dummer R, Wolchok JD, Schmidt H, Hamid O, Robert C, Ascierto PA, Richards JM, et al. Prolonged survival in stage III melanoma with ipilimumab adjuvant therapy. N Engl J Med. 2016;375:1845–1855. doi:10.1056/NEJMoa1611299.
- Eggermont AMM, Blank CU, Mandala M, Long GV, Atkinson V, Dalle S, Haydon A, Lichinitser M, Khattak A, Carlino MS, et al. Adjuvant pembrolizumab versus placebo in resected stage III melanoma. N Engl J Med. 2018;378:1789–1801. doi:10.1056/NEJMoa1802357.
- Weber J, Mandala M, Del Vecchio M, Gogas HJ, Arance AM, Cowey CL, Dalle S, Schenker M, Chiarion-Sileni V, Marquez-Rodas I, et al. Adjuvant nivolumab versus ipilimumab in resected stage III or IV melanoma. N Engl J Med. 2017;377:1824–1835. doi:10.1056/NEJMoa1709030.
- Blank CU, Rozeman EA, Fanchi LF, Sikorska K, van de Wiel B, Kvistborg P, Krijgsman O, van Den Braber M, Philips D, Broeks A, et al. Neoadjuvant versus adjuvant ipilimumab plus nivolumab in macroscopic stage III melanoma. Nat Med. 2018;24:1655–1661. doi:10.1038/s41591-018-0198-0.
- Liu J, Blake SJ, Yong MC, Harjunpaa H, Ngiow SF, Takeda K, Young A, O’Donnell JS, Allen S, Smyth MJ, et al. Improved efficacy of neoadjuvant compared to adjuvant immunotherapy to eradicate metastatic disease. Cancer Discov. 2016;6:1382–1399. doi:10.1158/2159-8290.CD-16-0577.
- Keung EZ, Ukponmwan EU, Cogdill AP, Wargo JA. The rationale and emerging use of neoadjuvant immune checkpoint blockade for solid malignancies. Ann Surg Oncol. 2018; 25:1814–1827.
- Mayer CT, Ghorbani P, Kuhl AA, Stuve P, Hegemann M, Berod L, Gershwin ME, Sparwasser T. Few Foxp3(+) regulatory T cells are sufficient to protect adult mice from lethal autoimmunity. Eur J Immunol. 2014;44:2990–3002. doi:10.1002/eji.201344315.
- Scrimieri F, Askew D, Corn DJ, Eid S, Bobanga ID, Bjelac JA, Tsao ML, Allen F, Othman YS, Wang SC, et al. Murine leukemia virus envelope gp70 is a shared biomarker for the high-sensitivity quantification of murine tumor burden. Oncoimmunology. 2013;2:e26889. doi:10.4161/onci.26889.
- Huang AY, Gulden PH, Woods AS, Thomas MC, Tong CD, Wang W, Engelhard VH, Pasternack G, Cotter R, Hunt D, et al. The immunodominant major histocompatibility complex class I-restricted antigen of a murine colon tumor derives from an endogenous retroviral gene product. Proc Natl Acad Sci U S A. 1996;93:9730–9735.
- Amaria RN, Reddy SM, Tawbi HA, Davies MA, Ross MI, Glitza IC, Cormier JN, Lewis C, Hwu WJ, Hanna E, et al. Neoadjuvant immune checkpoint blockade in high-risk resectable melanoma. Nat Med. 2018;24:1649–1654. doi:10.1038/s41591-018-0197-1.
- Boutros C, Tarhini A, Routier E, Lambotte O, Ladurie FL, Carbonnel F, Izzeddine H, Marabelle A, Champiat S, Berdelou A, et al. Safety profiles of anti-CTLA-4 and anti-PD-1 antibodies alone and in combination. Nat Rev Clin Oncol. 2016;13:473–486. doi:10.1038/nrclinonc.2016.58.
- Liu J, Blake SJ, Harjunpaa H, Fairfax KA, Yong MC, Allen S, Kohrt HE, Takeda K, Smyth MJ, Teng MW. Assessing immune-related adverse events of efficacious combination immunotherapies in preclinical models of cancer. Cancer Res. 2016;76:5288–5301. doi:10.1158/0008-5472.CAN-16-0194.
- Wolchok JD, Chiarion-Sileni V, Gonzalez R, Rutkowski P, Grob JJ, Cowey CL, Lao CD, Wagstaff J, Schadendorf D, Ferrucci PF, et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med. 2017;377:1345–1356. doi:10.1056/NEJMoa1709684.
- Rozeman EA MA, van de Wiel BA, Adhikari C, Sikorska K, Krijgsman O, Eriksson H, Bierman C, Grijpink-Ongering LG, Gonzalez M, Broeks A, et al. OpACIN-neo: A multicenter phase 2 study to identify the optimal neo-adjuvant combination scheme of Ipilimumab (IPI) and Nivolumab (NIVO). Ann Oncol Suppl. 2018: abstract LBA42 2018.
- Huang AC, Xu X, Orlowski RJ, George SM, Chilukuri L, Kozlov A, Carberry M, Giles L, McGettigan S, Kreider K, et al. Abstract CT181: safety, activity, and biomarkers for neoadjuvant anti-PD-1 therapy in melanoma. Cancer Res. 2018;78:CT181–CT. doi:10.1158/1538-7445.AM2018-CT181.
- Forde PM, Chaft JE, Smith KN, Anagnostou V, Cottrell TR, Hellmann MD, Zahurak M, Yang SC, Jones DR, Broderick S, et al. Neoadjuvant PD-1 blockade in resectable lung cancer. N Engl J Med. 2018;378:1976–1986. doi:10.1056/NEJMoa1716078.
- Rusch VW, Chaft JE, Johnson B, Wistuba II, Kris MG, Lee JM, Bunn PA, Kwiatkowski DJ, Reckamp KL, Finley DJ, et al. Neoadjuvant atezolizumab in resectable non-small cell lung cancer (NSCLC): initial results from a multicenter study (LCMC3). J Clin Oncol. 2018;36:8541. doi:10.1200/JCO.2018.36.15_suppl.8541.