
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
An effective therapy regimen for relapsed/refractory high-risk neuroblastoma (NB) includes the anti-GD2 monoclonal antibody, dinutuximab, in combination with temozolomide and irinotecan, supporting a role for chemo-immunotherapy in NB. γδ T cells are an attractive anti-tumor immunotherapy because of their direct cytotoxic activity mediated through cell surface receptors NKG2D and CD16. NKG2D facilitates the innate recognition of stress-induced ligands whereas CD16 recognizes antibody bound to tumors and activates mechanisms of antibody-dependent cellular cytotoxicity (ADCC). This study demonstrates an efficient method for expanding and storing γδ T cells from NB patient-derived apheresis products at clinically relevant amounts. The expanded patient-derived γδ T cells were cytotoxic against the K562 cell line and multiple NB cell lines. Combining γδ T cells with dinutuximab led to a 30% increase in tumor cell lysis compared to γδ T cells alone. Furthermore, low-dose temozolomide in combination with expanded γδ T cells and dinutuximab resulted in increased IFNγ secretion and increased γδ T-cell surface expression of FasL and CD107a. IMR5 NB cell line xenografts established subcutaneously in NSG mice were treated with a regimen of dinutuximab, temozolomide, and γδ T cells. This combination caused targeted killing of NB xenografts in vivo, reducing tumor burden and prolonging survival. These data support the continued preclinical testing of dinutuximab and temozolomide in conjunction with γδ T-cell immunotherapy for patients with recurrent/refractory NB.
Introduction
Neuroblastoma (NB), the most common extracranial pediatric solid tumor, is treated with multimodal therapy, including cytotoxic chemotherapy, autologous stem cell transplantation, local control with surgery and radiation, and maintenance immunotherapy. Despite increases in the intensity of therapy, high-risk NB has a 5-year event-free survival rate of <50%.Citation1,Citation2 Further dose escalation to improve survival is limited by the acute and chronic toxicities already encountered with current chemotherapy regimens.Citation3–Citation5 Cytotoxic chemotherapy has rarely been curative in high-risk NB as an individual treatment, leading to the evaluation of more targeted agents and novel modalities, most recently immunotherapy (for example, see: NCT03294954, NCT01460901, NCT01576692, NCT03242603, NCT03373097, and NCT02311621). Chemotherapy can positively impact the efficacy of immunotherapy by limiting the interference of protumorigenic immune regulatory cells systemically and within the tumor microenvironment.Citation6–Citation11 It can also be detrimental, as chemotherapy can be toxic to the therapeutic immunocompetent cells, whether innate or adaptive.Citation12,Citation13 While chemotherapy lacks specificity and generally targets proliferating cells, it can still be used as an effective method to sensitize tumors to anti-tumor cytotoxic T-cell lymphocytes.Citation14–Citation16
The use of monoclonal antibodies (mAb) as a cancer therapeutic has been clinically evaluated in an array of neoplastic disorders.Citation17 Specifically for NB, the development of antibodies to GD2 (3-F8, ch14.18 now dinutuximab, hu14.18K322A, etc.Citation18), a diasylganglioside found on a subset of NB cells, proved useful in the setting of minimal residual disease, yet showed little effect on the growth of bulky tumors in the relapsed and refractory setting.Citation19–Citation26 However, the combination of dinutuximab with chemotherapy resulted in a 47% response rate, with complete and partial responses seen in relapsed/refractory patients with bulky metastatic diseaseCitation27. Furthermore, the combination of chemotherapy and a similar monocloncal anti-GD2 antibody in newly diagnosed high-risk NB patients has shown thus far response rates as high as 80%.Citation23,Citation28,Citation29 Although increased response rates have been achieved, the effect of anti-GD2 antibody and chemotherapy combinations on cure rates in newly diagnosed high-risk NB patients is currently unknown and, therefore, actively under investigation in pilot clinical trials. Overall, the success of dinutuximab and chemotherapy combinations has established a paradigm of trial concepts and preclinical investigations to identify additional agents that will augment this effective chemo-immunotherapy backbone (NCT01576692 and NCT0379349).
The biological effectiveness of anti-GD2 antibodies, including dinutuximab, is dependent on antibody-dependent cellular cytotoxicity (ADCC) through the CD16, FcγRIII, receptor.Citation30,Citation31 We hypothesized the efficiency of monoclonal antibodies (mAbs), such as dinutuximab, could be improved by the infusion of cytotoxic cells capable of recognizing the Fc portion of mAbs. γδ T cells are innate immunocompetent cells that are an attractive candidate for immunotherapy because, unlike αβ T cells, they are not restricted by major histocompatibility complexes (MHC). Importantly, subsets of expanded γδ T cells have been shown to express high levels of CD16,Citation32 which can enhance ADCC through Fc recognition. In addition, γδ T cells have intrinsic anti-tumor activity as they also express FasL and recognize stress antigens.Citation33,Citation34 γδ T cells have the inherent ability to recognize stress antigens including MHC class I related chain A/B (MICA/B) and UL16 binding protein (ULBPs) via the NKG2D receptor (natural killer group 2, member D).Citation35,Citation36 The interaction of NKG2D with stress-inducible ligands produces rapid cell lysis and secretion of pro-inflammatory cytokines, including IFNγ.Citation34,Citation37 Studies have also shown that host γδ T- cell tumor infiltration results in an overall better prognosis.Citation38 Taken together, we hypothesize that administration of an ex vivo expanded γδ T-cell product could be an effective and novel treatment for high-risk NB.
Unfortunately, efforts aimed at expanding γδ T cells in vivo have not shown clinical benefits. For example, stimulating the production of γδ T cells in vivo with IL-2 can concurrently stimulate the production of regulatory T cells, potentially inhibiting immune surveillance of cancer cells.Citation39,Citation40 We therefore devised a novel method to successfully expand γδ T cells from peripheral blood. Our previous studies demonstrated γδ T cells from healthy donor frozen peripheral blood mononuclear cells (PBMCs) can be expanded using a serum-free expansion protocol.Citation41 Notably, newly diagnosed high-risk NB patients undergo hematopoeitic stem cell collection and storage in anticipation of autologous stem cell transplant as a standard of care, yet many of these apheresis products go unused. One goal of these investigations was to assess whether γδ T cells from NB patient apheresis products could be used as a potential source for a viable and active expansion.
NKG2D is highly expressed on healthy donor expanded γδ T cells.Citation32,Citation33,Citation42 Prior studies have shown that chemotherapy induces the expression of stress antigens such as, MHC class I chain-related protein A or protein B (MICA/B) or UL16-binding proteins (ULBPs), on the tumor cell surface, increasing tumor cell vulnerability.Citation43 By increasing susceptibility of cancer cells to recognition via the NKG2D receptor on γδ T cells, chemoimmunotherapy combinations can provide a therapeutic benefit not seen by either modality alone.Citation14,Citation15,Citation35,Citation44–Citation46 The alkylating agent, temozolomide (TMZ), is used in heavily pre-treated relapsed patients to induce tumor cell killingCitation47. TMZ is known to induce transient expression of NKG2D ligands.Citation14,Citation15,Citation35 We therefore hypothesized that dinutuximab and TMZ in combination with ex vivo expanded γδ T cells may provide a benefit to NB treatment outcomes. Herein, our data supports the ability to expand γδ T cells in serum-free conditions from apheresis hematopoietic stem cell (HSC) products collected from patients with NB and illustrates a survival benefit when combining these cells with chemotherapy and mAb therapy.
Results
Robust NB patient-derived γδ T cell expansion in serum free media
Recently, we published a good manufacturing practice (GMP)-compliant process using serum-free media to expand γδ T cells with aminobisphosphonates (e.g. zoledronic acid) combined with IL-2.Citation41 To determine whether these methods could be translated to frozen primary NB patient mobilized and apheresed PBMCs, the serum-free protocol with zoledronic acid and IL-2 supplementation was employed using cells harvested from 5 NB patients and compared to healthy controls, which were included to replicate our previoius findings. Overall, the percentage of γδ T cells from NB patient donors during 2-week cultures increased from 1.15 ± 0.90% to greater than 75% of the population (). Mean-fold expansion of NB patient-derived γδ T cells ranged from 25- to 310-fold.
Figure 1. Expansion of γδ T cells from NB patient-derived PBMCs. (a) γδ T cells were expanded using serum-free conditions from commercially available healthy donor PBMCs (n = 2, with one repeated expansion using the same donor) or NB patients (n = 6, where some patient samples were expanded multiple times). All cultures were supplemented with IL-2 on days 0 (500 IU/mL), 3 (500 IU/mL), 6 (1,000 IU/mL), and 9 (1,000 IU/mL) and zoledronic acid (5 µM) on days 0 and 3. Live cells were gated for CD3+, CD56-, and pan γδ TCR+ to determine γδ T-cell percentage. Data comparing a healthy donor versus a NB patient starting product were not significantly different by non-paired, two tail t-test on day 7 (p = 0.97) and 12 (p = 0.55). (b) An apheresis product from a single NB patient was divided into two samples and each was expanded three times. The average γδ T-cell percentage and standard deviation are shown. (c) The cellular distribution of a starting PBMC product on day 0 differs from the cellular components on day 14 of expansion, and the change is not dependent on the donor being healthy or having NB. γδ T cells comprise less than 5% of the starting cellular product and increases to 60–90% of the expanded cells.
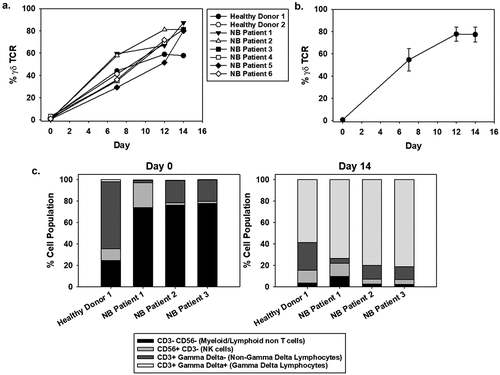
Reproducibility was tested by expanding cells from one donor in triplicate, which showed no significant variability (paired t-test) in the percentage of γδ T cells during the expansion process (). Flow cytometry analysis confirmed the resulting cell populations to be of similar composition among the NB patient and healthy PBMCs, including populations of γδ T cells (CD3+, pan-γδ+), αβ T cells (CD3+, pan-γδ−), and a low percentage of CD3− cells. Specifically, by day 14 of expansion, the myeloid/lymphoid non-T-cell population (CD3−, CD56−) comprised 2–10% of the population, NK cells (CD3−, CD56+) accounted for 4–12%, the non-γδ lymphocytes range from 4% to 26%, with the γδ lymphocytes constituting the majority of the population, at 60–82% (), Supplemental Table 1). Additional flow cytometry characterization was performed to further classify the different γδ T-cell populations, predominantly the Vδ2 subtype (Supplemental Figure 1, Supplemental Table 2). The majority of cells are CD3+ (95.57 ± 0.00) and of these, the majority are γδ T cells (85.63 ± 0.85 Vδ2 and 7.00 ± 0.07 Vδ1). Of the Vδ2 γδ T cells, we further subdivided into CD28+ CD27+ (65.90 ± 0.71), CD28- CD27+ (19.73 ± 1.56), and CD28- CD27- (12.37 ± 0.99) (Supplemental Table 2). These results therefore show that the bulk population of the γδ T cells are defined as Vδ2 γδ effector memory T cells, denoted by CD28 +CD27+ CD16+ CD45RA-CD45RO+ and CD62L- phenotype.Citation48 Based on preliminary RNAseq data (data not shown) the transcripts for perforin and granzyme are high (in the top 10% of RNA reads). Therefore, supporting that effector memory cells have high granzyme/perforin expression. Additionally, the NB patient-derived γδ T cells lack PD1/PDL1 expression, suggesting the cells can function despite a PD1/PDL1 rich tumor environment (Supplemental Table 2). As expected there is a mixed expression of CD57, as this senescence marker indicates some cells are further down the senescence pathway than others (Supplemental Table 2). Together, these data demonstrate the ability to consistently achieve similar expansion of active NB patient-derived γδ T-cell populations and the subtypes associated with these expansions.
NB patient-derived γδ T cells remain cytotoxic after freezing
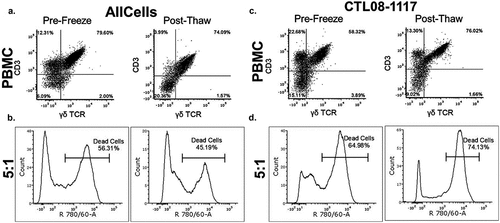
To evaluate the cytotoxicity of patient-derived γδ T cells against a standard K562, chronic myelogenous leukemia cell line, expanded γδ T cells were co-incubated at a 5:1 effector to target ratio. The γδ T cells derived from NB patients (N = 5) and healthy donors (N = 3) were used against the target cell line to determine the percentage of cells killed by effectors, which was evaluated by flow cytometry (,)). To ensure the γδ T cells would be uniform and useful as a cellular product, we evaluated γδ T-cell cytotoxicity following a serum-free freezing process. The viability of the γδ T cells post-thaw was assessed using trypan blue exclusion and was consistently greater than 70%. Eight hours after the cells were thawed, cell killing was normalized to background cell death and maintained at 51.9 ± 5.5% (N = 5), which was not significantly different from the 58.7 ± 10.3% prior to freezing (N = 5) (,)).
Figure 2. Cytotoxic activity of NB patient-derived γδ T cells Healthy donor (a, b) and NB patient (c, d) γδ T cells were expanded to day 14 and frozen at 1 × 107 cells/mL. Prior to freezing, cells were tested for cytotoxic potential against K562 cells. The left side panels show data from cells prior to freezing and the right side panels show data from cells after freezing. For the post-thaw samples, cells were thawed at 37oC and incubated in growth media for 8 hrs prior to use in the 4 hr cytotoxicity assay. (b, d) γδ T cells were used at a 5:1 effector to target ratio and incubated for 4 hr with K562 cells. Cytotoxicity was measured using flow cytometry by gating on the target cells stained with VPD450 and then analyzing the target cells for the dead stain dye, eFluor Alexa 780. There was no significant difference between pre-freeze and post-thaw γδ T-cell percentage as well as cytotoxicity (one-way ANOVA p = 0.618, N = 4). Raw representative flow cytometry data are shown.
Killing by NB patient-derived γδ T cells is enhanced when combined with dinutuximab
In the presence of mAb, the FcγRIII receptor, CD16, facilitates ADCC and this mechanism has been evaluated pre-clinically in NB models.Citation49–Citation52 The serum-free expansion and activation of γδ T cells demonstrated that patient-derived and healthy donor cells have a robust expression of CD16 (-b)). CD16 expression at the start of expansion was <20% and increased to 80% by day 6 on the total and γδ T cell populations.
Figure 3. CD16 expression of NB patient-derived γδ T cells and dinutuximab binding to NB cell lines A. γδ T cells were expanded using serum-free conditions and examined for surface expression of CD16 by flow cytometry. CD16 expression on days 0, 6, 12, and 14 of expansion is shown for the bulk culture (a) and specifically on γδ T cells (b). Multiple NB cell lines of variable genomic profiles and disease aggressiveness were evaluated for the percentage of GD2 surface expression and dinutuximab (DTX) binding (c) Biotinylated DTX (0.5 μg) was used to demonstrate the specificity of antibody binding to NB cell lines, which showed a trend between GD2 expression and DTX binding with increased surface GD2 corresponding to increased DTX binding (N = 3 per sample and the mean is shown).
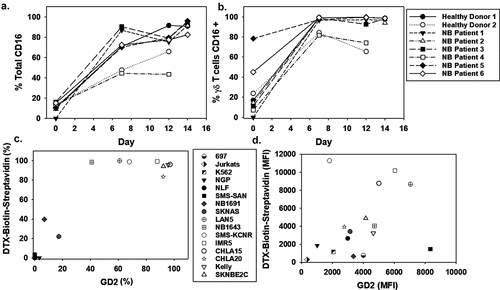
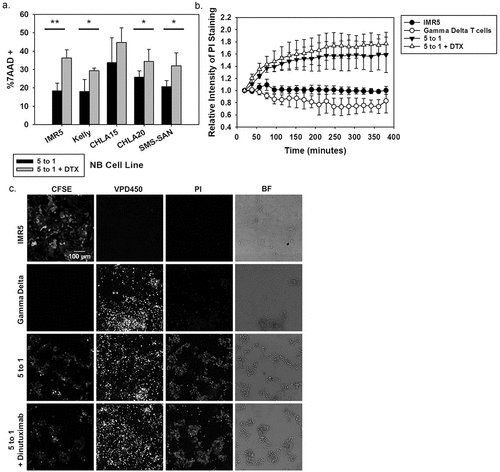
To first determine the binding potential of dinutuximab to a number of NB cell lines, dinutuximab was biotinylated and flow cytometry was used to measure its binding to NGP, NLF, SMS-SAN, NB1691, LAN5, NB1643, SMS-KCNR, IMR5, Kelly, SKNBE2C (MYCN amplified) and SKNAS, CHLA15, and CHLA20 (MYCN single copy) NB cell lines. These cell lines were derived from a variety of human NB tumors with variable GD2 expression, aggressiveness of the disease, and genomic profiles, including those with and without MYCN amplification and/or ALK mutations. High GD2 expression correlated with greater dinutuximab binding (non-linear regression second order polynomial R2 = 0.94) compared with dinutuximab binding to non-NB cell lines that lack GD2 expression such as K562, Jurkat, and 697, which were used as references for non-specific binding ()). Interestingly there is less correlation between the amount of GD2 expressed on the cell line surface and the increase in cytotoxicity with DTX. This suggests that the γδ T cells are able to recognize antibody targets on the cell surface regardless of the concentration of antibody bound. To assess the cytotoxicity potential of patient-derived γδ T cells against human-derived NB cell lines, three separate patient-derived γδ T-cell expansions were used in cytotoxicity assays with high GD2-expressing NB cell lines at 5:1 effector to target ratios, with and without dinutuximab ()). The average cell killing of IMR5, Kelly, CHLA15, CHLA20, and SMS-SAN NB cell lines increased 97.9%, 62.9%, 32.7%, 33.0%, and 54.5%, respectively, when target cells were incubated with γδ T cells and dinutuximab relative to baseline cell death of untreated cells. We performed live cell imaging over 6 hr, which confirmed increased cell killing of IMR5 cells when treated with γδ T cells and dinutuximab compared to background cell killing in target or effector cells alone (-c)). Representative still images from the live cell imaging reveals the high intensity of PI staining at 6 hrs when γδ T cells were incubated with IMR5 cells, clearly demonstrating the cytotoxic potential of patient-derived γδ T cells against NB ()).
Figure 4. Evaluating ADCC with NB patient-derived γδ T cells (a) Cytotoxicity assay using NB cell lines that showed high GD2 expression. Black bars indicate NB patient-derived γδ T cells at a 5 to 1 effector to target ratio while the grey bars represent the same assay with dinutuximab (DTX) included at 5 μg/mL. DTX alone did not show significant killing (data not shown). Expanded NB patient cells functioned as the effector cells and experiments were repeated in triplicate, mean and standard deviations (SD) are shown. (Significance determined by paired t-test * p < 0.05; **p < 0.01). (b) Live cell images at 6 hrs of IMR5 cells (stained with CFSE) co-incubated with or without γδ T cells (stained with VPD450 proliferation dye) and with or without DTX (5 μg/mL). Cell death was quantified by PI intensity (100 μM). The mean and SD (N = 4) were quantified by the relative intensity of PI staining over 6 hrs during live cell imaging. (c) Representative still images at 6 hrs: CFSE stain shows the IMR5 target cells, VPD450 shows the effector γδ T cells, PI staining identifies dead cells and bright field are captured.
Patient-derived γδ T cells in combination with dinutuximab do not affect NB growth in a xenograft murine model
To test the effectiveness of patient-derived γδ T cells in vivo, NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice were used to establish subcutaneous NB tumors with IMR5 cells. Once the tumor was palpable (125 mm3), the animals were randomized to receive γδ T cells alone or γδ T cells with 200 or 400 μg of dinutuximab, all delivered intravenously. Injections of γδ T cells were administered every 3 days over 15 days and dinutuximab was administered on days 1 and 10 ()). Mice were monitored for tumor growth ()), weight, and survival. There were no significant differences in the average tumor volume, weight, or survival between these treatment groups by one-way ANOVA on days 10 and 20.
Figure 5. In vivo effectiveness of NB patient-derived γδ T cells and dinutuximab (a) Schematic of the treatment plan for NSG mice injected subcutaneously with IMR5 cells and treated with γδ T cells, dinutuximab (DTX), or a combination of the two. (b) The mean tumor volume is shown for each group: untreated (N = 8), γδ T cells only (N = 8), γδ T cells + DTX (200 μg) (N = 6), γδ T cells + DTX (400 μg) (N = 4). The mean and standard deviation is shown for each cohort. (c) NSG mice with established IMR5 subcutaneous tumors were injected with 200 μg biotinylated DTX. Mice were sacrificed and tumors processed prior to injection (untreated) and after injection on days 1, 4, or 7. Representative flow cytometry for N = 3 is shown.
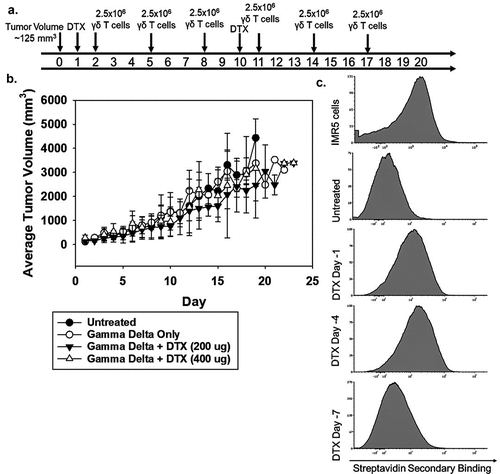
To determine the efficiency of tumor targeting by dinutuximab and γδ T cells, biotinylated dinutuximab was administered and on days 1, 4, and 7 after injection, tumors were harvested and antibody-coated tumor cells were measured by flow cytometry. The greatest binding to tumor cells was observed on day 4 with a geometric mean fluorescence intensity (MFI) of 1848. Day 1 and 7 MFI were 1459 and 1056, respectively ()). As expected, γδ T-cell homing to IMR5 cells was low, with less than 1% of the tumor being γδ T cells, which was approximately 10-fold less than the percentage of the γδ T cells found in peripheral blood (Supplemental Figure 2).Citation53 In addition, the persistence of γδ T cells in peripheral blood demonstrated a steady decline with values decreasing to near baseline by 1 week after administration (Supplemental Figure 3).
Temozolomide enhances dinutuximab and γδ T cell in vitro and in vivo killing of NB cells
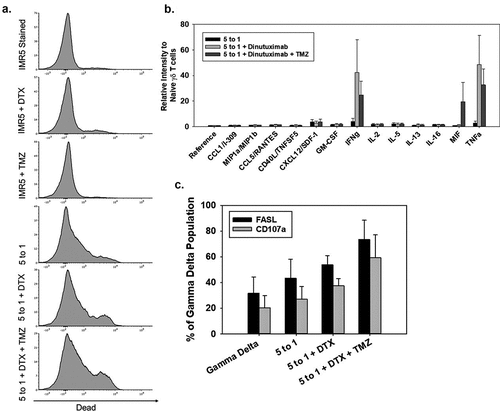
Temozolomide (TMZ) has been reported to induce stress ligands in models of glioblastoma, leading to increased γδ T cell-based killing of glioblastoma cells.Citation35 When incubating IMR5 NB cells with TMZ, there was no detected inducible increase in NKG2DL (MICA/B, ULBP1, ULBP2/5/6) with doses ranging from 100 μM to 2 mM (Supplemental Figure 4). However, when IMR5 cells were incubated at a dose of 400 μM TMZ for 1 hr prior to performing a cytotoxicity assay with γδ T cells in combination with dinutuximab, there was a 10% increase in cell death compared to IMR5 treated with only γδ T cells and dinutuximab (non-paired t-test p < 0.05) ()). To analyze the secretion of cytokines that could potentially be influencing cell death, the media from a cytotoxicity assay was collected and probed for cytokine expression ()). An increase in the secretion of IFNγ and TNFα was observed when γδ T cells/dinutuximab were cultured with IMR5 cells, but there was no significant difference when TMZ was added to this combination. When IMR5 cells were incubated with γδ T cells, dinutuximab, and TMZ there was an increase in MIF expression. In addition to measuring cytokine expression, the γδ T cells from these assays were examined for cytotoxicity markers, including FASL and CD107a, to determine if TMZ pretreatment of NB cells induces γδ T cells FAS-mediated killing. FASL and CD107a were indeed increased on γδ T cells when cultured with NB cells pretreated with dinutuximab and TMZ ()). CD112, CD15, TRAIL-R1, TRAIL-R2, and FAS were all expressed in IMR5 cells, and 400 μM TMZ did not alter the expression of these ligands on IMR5 (Supplemental Figure 5).
Figure 6. Increasing γδ T-cell function when combined with dinutuximab and TMZ (a) Representative flow cytometry of IMR5 cell killing with combinations of dinutuximab (DTX), TMZ, and γδ T cells. From top to bottom, panel A shows killing for non-treated cells or cells treated with DTX, TMZ, γδ T cells (5:1 effector:target), γδ T cells (5:1 effector:target) with DTX, or γδ T cells (5:1 effector:target) with DTX and TMZ. IMR5 background dead cell staining was 6%, which was similar to treatments with DTX (5 μg/mL; 11% dead cells) and TMZ (400 μM; 11% dead cells). Average cell death increased to approximately 30% with γδ T cells, 39% with γδ T cells and DTX, and 46% with γδ T cells, DTX, and TMZ (N = 3, p < 0.05). (b) Average and SD (N = 3) of human cytokine array quantified using ImageJ analysis. Consistent with previous findings, γδ T cells secrete proinflammatory cytokines IFNγ and TNFα mixed with NB cells. There is an increase in cytokine production when DTX and TMZ is included. All data is quantified as relative intensity to naïve γδ T cells. There is no significant difference between effector to target ratios 5 to 1 + DTX and 5 to 1 + DTX + TMZ. Data in these groups is significantly different compared to 5 to 1 effector to target ratio alone p < 0.05. (c) Flow cytometry was performed to measure FASL and CD107a on γδ T cells 4 hrs after initiation in cytotoxicity assays. Black and grey bars represent FASL and CD107a, respectively. Expression of these proteins is elevated when γδ T cells are incubated with IMR5 cells and further elevated when combined with DTX and TMZ.
To determine if the enhanced killing observed with TMZ in vitro occurs in vivo, TMZ sensitivity was tested on established IMR5 subcutaneous xenografts growing in NSG mice. Once tumors were 125mm3, TMZ was administered intraperitoneally at 125 mg/kg, 85 mg/kg, 40 mg/kg, or 20 mg/kg once a day every 3 days (for a total of six doses) (,)). The 125 mg/kg dose was lethal and the 85 mg/kg dose resulted in nearly complete tumor eradication. Doses below 85 mg/kg resulted in tumor growth, but only after completion of the six doses. Mice that received 20 mg/kg had diminished tumor growth; however, tumors progressed in every mouse in this cohort. A clear dose-response was achieved between 0 and 85 mg/kg TMZ. Combining 40 mg/kg TMZ with dinutuximab did not affect tumor growth, nor did the combination of γδ T cells and TMZ (,)). In all combination treatments using TMZ and γδ T cells, TMZ was administered 8 hrs prior to the γδ T cells. In contrast to TMZ plus γδ T cells, mice treated with the combination of patient-derived γδ T cells, dinutuximab, and 20 or 40 mg/kg TMZ dose showed a significant reduction in tumor growth compared to untreated or mice treated with any single therapy alone (p = 0.01) (–)). In addition to tumor reduction, the combination of immunotherapies (γδ T cells and dinutuximab) with TMZ resulted in significant survival benefits for mice treated with 40 mg/kg TMZ (log-rank Mantel-Cox, p = 0.0059) ()). Mice treated with a combination of γδ T cells, dinutuximab and TMZ at 20 mg/kg also showed significant survival advantage (log-rank Mantel-Cox, p < 0.05) compared to TMZ treatment alone ()). Additionally, there is a statistically significant survival advantage of γδ T cells + dinutuximab + 40 mg/kg TMZ compared to γδ T cells + dinutuximab + 20 mg/kg TMZ (log-rank Mantel-Cox, p = 0.04).
Figure 7. Enhancing NB patient-derived γδ T-cell effectiveness in vivo by combination therapy (a-b) Schematic representation of the treatment plan for NSG mice injected subcutaneously with IMR5 cells. Time T = 0 refers to when the tumor reaches a minimum of 125 mm3 and the start of treatment. NSG mice with established IMR5 subcutaneous tumors were treated over a 17-day period at varied doses of TMZ, DTX, and γδ T cells. (c) Tumor volume was measured and average tumor volume over time and standard deviation was calculated for TMZ doses of 20 mg/kg (N = 4), 40 mg/kg (N = 5), 60 mg/kg (N = 2), 85 mg/kg (N = 5), and compared to untreated controls. (d) Untreated (N = 8), 2.5 × 106 γδ Only (N = 8), γδ + dinutuximab (DTX) (400 μg) [N = 4], TMZ (40 mg/kg) [N = 5], DTX (400 μg)+TMZ (40 mg/kg) [N = 4], γδ+TMZ (40 mg/kg) [N = 5], and γδ+DTX (400 μg)+TMZ (40 mg/kg) [N = 6] were evaluated through day 30. (e) A lower dose of TMZ (20 mg/kg) [N = 4] was used alone or with various combinations of DTX and γδ T cells [minimum of N = 4 per cohort]. (f) TMZ (40 mg/kg) [N = 5] and γδ+DTX (400 μg) + TMZ (40 mg/kg) [N = 6] are compared by paired t-test over 4 weeks (*p = 0.029 at week 4). (g) Survival curves to day 50 from the start of treatment, shows a significant survival advantage among animals that received 40 mg/kg TMZ with 400 μg DTX and 2.5 × 106 γδ T cells compared to γδ T cells alone, γδ T cells + DTX, γδ T cells + TMZ, and TMZ + DTX (log-rank p < 0.001). (h) Survival curves to day 50 from the start of treatment, demonstrates significance in survival when using lower doses of TMZ (20 mg/kg) with 400 μg DTX and 2.5 × 106 γδ T cells compared to untreated animals, γδ T cells only, γδ T cells+DTX, and TMZ (20 mg/kg) only (log-rank p < 0.001).
![Figure 7. Enhancing NB patient-derived γδ T-cell effectiveness in vivo by combination therapy (a-b) Schematic representation of the treatment plan for NSG mice injected subcutaneously with IMR5 cells. Time T = 0 refers to when the tumor reaches a minimum of 125 mm3 and the start of treatment. NSG mice with established IMR5 subcutaneous tumors were treated over a 17-day period at varied doses of TMZ, DTX, and γδ T cells. (c) Tumor volume was measured and average tumor volume over time and standard deviation was calculated for TMZ doses of 20 mg/kg (N = 4), 40 mg/kg (N = 5), 60 mg/kg (N = 2), 85 mg/kg (N = 5), and compared to untreated controls. (d) Untreated (N = 8), 2.5 × 106 γδ Only (N = 8), γδ + dinutuximab (DTX) (400 μg) [N = 4], TMZ (40 mg/kg) [N = 5], DTX (400 μg)+TMZ (40 mg/kg) [N = 4], γδ+TMZ (40 mg/kg) [N = 5], and γδ+DTX (400 μg)+TMZ (40 mg/kg) [N = 6] were evaluated through day 30. (e) A lower dose of TMZ (20 mg/kg) [N = 4] was used alone or with various combinations of DTX and γδ T cells [minimum of N = 4 per cohort]. (f) TMZ (40 mg/kg) [N = 5] and γδ+DTX (400 μg) + TMZ (40 mg/kg) [N = 6] are compared by paired t-test over 4 weeks (*p = 0.029 at week 4). (g) Survival curves to day 50 from the start of treatment, shows a significant survival advantage among animals that received 40 mg/kg TMZ with 400 μg DTX and 2.5 × 106 γδ T cells compared to γδ T cells alone, γδ T cells + DTX, γδ T cells + TMZ, and TMZ + DTX (log-rank p < 0.001). (h) Survival curves to day 50 from the start of treatment, demonstrates significance in survival when using lower doses of TMZ (20 mg/kg) with 400 μg DTX and 2.5 × 106 γδ T cells compared to untreated animals, γδ T cells only, γδ T cells+DTX, and TMZ (20 mg/kg) only (log-rank p < 0.001).](/cms/asset/a67315bc-0809-4e06-a9cc-5b4d86a22dfe/koni_a_1593804_f0007_b.gif)
Discussion
The goal of these studies was to determine if a readily available cellular source material could be expanded into a cytotoxic γδ T-cell product, and if the expanded cells could be used to treat NB in a preclinical model. NB is currently treated with chemotherapy, radiation therapy, surgery, autologous stem cell transplantation, and maintenance immunotherapy containing dinutuximab.Citation1 For high-risk patients, survival outcomes remain poor.Citation3,Citation13 Since most children undergoing stem cell transplantion have additional unused apheresis products, there is potential to expand γδ T cells from these banked cells to be used as a therapeutic. Although substantial progress is being made in the field of cellular immunotherapy, NB specific advances have been limited. For example, the development and standardization of autologous chimeric antigen receptor (CAR) T-cell protocols allowed for the development of anti-GD2-based CARs.Citation52,Citation54,Citation55 However, the GD2 CAR has had some setbacks, such as severe off-tumor toxicities including fatal encephalitis in preclinical models.Citation56 In the current study, γδ T cells are presented as an alternative to αβ-based CAR T cells as γδ T cells should have specific advantages since multiple killing mechanisms are inherent to these cells, including ADCC-based mechanisms, FasL expression, and targeting of stress antigens.
Recently, we developed protocols for a serum-free expansion method for γδ T cells from normal donors and showed that cells expanded using this GMP-compliant process provided a sufficient cell source to support clinical testingCitation41. In this current study, the protocol was expanded to include cell products from children who underwent standard treatments for NB. This is the first study to utilize NB patient stem cell collection products for generating γδ T cells, which supports the expansion manufacturing process. The composition of the expanded product on day 14 is not significantly different compared to expansions using healthy donor PBMCs. In addition, the data shows that patient-derived cells retain their cytotoxic activity after storage in liquid nitrogen, which is important as it is predicted that multiple doses of the cellular product would be needed for each subject. The ability of patient-derived γδ T cells to recognize and kill tumor cells through mechanisms of ADCC and stress antigen recognition is important because it is anticipated the cells will be combined with standard of care therapy for relapsed and ultimately newly diagnosed NB, that includes both antibodies and chemotherapy that can enhance cytotoxicity by γδ T cells.Citation42,Citation57 Importantly, CD16 is upregulated during γδ T cells expansion and supports ADCC-based killing. Because the use of dinutuximab has demonstrated improved clinical outcomes in newly diagnosed and relapsed patients, the expansion and infusion of CD16+ γδ T cells in combination with dinutuximab is predicted to be beneficial, and our in vitro data shows that this combination does provide some benefit when targeting highly expressing GD2 NB cell lines. However, the tumor killing by the combination of γδ T cells and dinutuximab using an immunocompromised NSG mouse, lacking in NK, B, and T cells, is insufficient for NB tumor eradication. Furthermore, the in situ microenvironment may also affect γδ T cells/dinutuximab efficacy possibly via immune cell exhaustion of the cellular product, which may benefit from cytokine supplementation which is done clinically with anti-GD2 antibody therapy. Our data strongly supports that the lack of in vivo efficacy of the dinutuximab/γδ T cell combination alone is most likely due to insufficient trafficking of γδ T cells to the tumor, which is a well-defined issue for cellular products.Citation53,Citation58,Citation59
To enhance the effectiveness of patient-derived γδ T cells, we tested the combination of immunotherapy and chemotherapy. Combining chemotherapy and cell-based therapeutics is typically complicated by chemotherapy-induced lymphopenia. Because TMZ is rapidly metabolized to inactive products, it provides a unique opportunity for combining treatment modalities. We hypothesized that TMZ could upregulate stress antigens and would subsequently be systemically inactivated/eliminated prior to administration of the γδ T cells, thereby providing a rational means of the timing of the combination treatment. However, the in vitro data contradicts the use of TMZ as an inducer of stress-ligands in NB cells. Instead, the combination of TMZ and dinutuximab induced an increase in cytokine secretion, increased FasL expression on γδ T cells, and enhanced degranulation when γδ T cells were co-cultured with target cells. Therefore, based on these mechanisms, and apart from stress antigen expression, it was predicted the combination would be more effective in vivo than TMZ alone.
In an IMR5 in vivo murine model of NB, TMZ effectively reduced tumor growth in a dose-dependent manner. When using doses of TMZ that do not eradicate tumor growth alone, a significant benefit was observed when combining TMZ with γδ T cells and dinutuximab.
This response was not achieved with either of the single or double treatment regimens. Interestingly, the benefit of γδ T cells was achieved without co-administration of IL-2 or zoledronic acid, which have been previously used by others to support γδ T-cell survival.Citation50,Citation60 The mechanism by which TMZ enhances the effectiveness of γδ T cells and dinutuximab is not yet fully understood. One hypothesis is that TMZ does not act directly on the tumor cells. Tumor cell growth may be prevented by affecting cells within the tumor microenvironment, such as those involved in angiogenesis.Citation61 It has been shown that low dose metronomic TMZ indeed inhibits tumor angiogenesis.Citation61 Additionally, anti-angiogenic therapies show potential benefit when combined with immunotherapies for solid tumors through normalization of abnormal tumor vasculature to allow for increased infiltration of innate or adoptive immune effector cells.Citation62 Thus, TMZ may directly control tumor growth and vasculature architecture to allow for the immunotherapeutic component of treatment to be established in the tumor microenvironment,Citation63 supporting the notion that this mechanism deserves further investigation in NB. Importantly, not only can the combination reduce tumor growth, but the ability to capitalize on the non-cytotoxic anti-tumor properties of chemotherapy agents allows for lower chemotherapy dosing and can benefit high-risk NB patients by reducing short- and long-term toxicities associated with chemotherapy. For example, a strategy incorporating expanded γδ T cells from a NB patients’ apheresed-frozen PBMC product, collected during standard of care upfront therapy, potentially allows for a decrease in subsequent chemotherapeutic dosing without forgoing effectiveness to prolong survival. Additionally, there are alternative strategies to combined TMZ and cellular therapy including genetic engineering of immunocompetent cells using vectors encoding methylguanine methyltransferase,Citation35 producing drug resistance. This modification is required for protecting expanded cell therapy products after administration and during the chemotherapy challenge. However, it appears this engineering may not be necessary if the administration of the cellular therapy and chemotherapy are properly timed.
Overall, the data supports that NB patient-derived γδ T cells can be efficiently expanded, and that the expanded cells enhance the effectiveness of chemoimmunotherapy in vivo. Although effective, it remains necessary to develop methods to increase the trafficking of γδ T cells to the tumor and increase their persistence in vivo. The data shows patient-derived cells can provide benefit to the standard of care chemotherapy and dinutuximab-based immunotherapy treatments. While the majority of cellular based immunotherapies primarily rely on αβ T cells, there are limitations to the use of these cell products in solid tumor treatments. As such, alternative approaches such as the use of γδ T cells are necessary. These results support the potential for clinical use of ex vivo expanded γδ T-cell products to treat patients with highly aggressive pediatric cancers, like neuroblastoma.
Materials and methods
Expansion of γδ T cells in serum-free media
Mobilized apheresed PBMCs were obtained from consented, deceased, neuroblastoma patients at Children’s Healthcare of Atlanta (Atlanta, GA). Commercially available healthy donor frozen PBMCs were obtained from AllCells (Alameda, CA). At the time of stem cell collection, each patient had undergone two cycles of induction chemotherapy. Cells were cultured with OpTmizer (Life Technologies, Carlsbad, CA) serum-free media and supplemented with 2 mM L-glutamine and 1% penicillin/streptomycin. All cultures were stimulated with 500–1000 IU/ml of IL-2 (Peprotech, Rocky Hill, NJ) and 5 µM zoledronic acid (Sigma-Aldrich). Media changes were performed every 3 days. On days 0 and 3, cells were provided with 500 IU/mL of IL-2, whereas on day 6 and 9, 1000 IU/mL is given. Zoledronic acid was used at the start of culture and added again on day 3. Total cell numbers were monitored periodically over a 2-week period via Cellometer (Nexcelom, Lawrence, MA). Dead cells were identified by trypan blue exclusion. γδ T-cell percentage and PBMC cellular composition were monitored via flow cytometry on days 0, 7, 12, and 14.
Cell lines and cell culture
Neuroblastoma cell lines (courtesy of Children’s Oncology Group (COG) Cell Line Repository) and K562 cells were cultured in RPMI 1640 with L-glutamine (Corning cellgro, Manassass, VA) and 10% FBS and 1% Penicillin/Strep added.
Flow cytometry
Cells were washed with phosphate buffered saline (PBS) and centrifuged at 100xg. The cells were decanted and incubated with Invitrogen (San Diego, CA) eBioscience Fixable Viability Dye eFluor 780 for 30 min with shaking at room temperature. The cells were washed in 10 volumes of PBS. Supernatant was decanted and replaced with the appropriate antibody cocktail in PBS. The antibodies used from BD Biosciences (Franklin Lakes, NJ), include: BV421 Mouse Anti-Human CD3 (Clone UCHT1), PE Mouse Anti-Human TCR-1 (Clone 11F2), BUV395 Mouse Anti-Human CD56 (Clone NCAM16.2), BV711 Mouse Anti-Human CD178 (Clone NOK-1), APC Mouse Anti-Human CD107a (Clone H4A3), PE Mouse Anti-Human CD95 (Clone DX2), BV480 Mouse Anti-Human CD3 (Clone UCHT1), APC-R700 Mouse Anti-Human CD56 (Clone NCAM16.2), BV711 Mouse Anti-Human CD27 (Clone M-T271), BUV496 Mouse Anti-Human CD16 (Clone 3G8), BUV661 Mouse Anti-Human CD4 (Clone SK3), PerCP-Cy5.5 Mouse Anti-Human CD8 (Clone RPA-T8), BB515 Mouse Anti-Human CD45RA (Clone HI100), BV650 Mouse Anti-Human CD45RO (Clone UCHL1), BV421 Mouse Anti-Human CD57 (Clone NK-1), BUV563 Mouse Anti-Human CD62L (Clone DREG-56), BV786 Mouse Anti-Human PD1 (Clone EH12.1), PE-CF594 Mouse Anti-Human PDL1 (Clone MIH1), BUV737 Mouse Anti-Human FAS (Clone DX2), PE Mouse Anti-Human FASL (Clone NOK-1), and BUV395 Mouse Anti-Human CD107a (Clone H4A3). Antibodies used from BioLegend (San Diego, CA) include: APC anti-human CD314 (NKG2D) (Clone 1D11), Brilliant Violet 711 Anti-Human CD16 (Clone 3G8), BV605 Mouse Anti-Human TCR Vδ2 (Clone B6), and PE-Cy5 Mouse Anti-Human CD28 (Clone CD28.2). Antibodies obtained from R&D Systems (Minneapolis, MN) include: PE Mouse Anti-Human TRAIL-R1 (Clone 69036), APC Mouse Anti-Human TRAIL-R2 (Clone 71908), APC Mouse Anti-Human CD112 (Clone 610603), and PE Mouse Anti-Human CD155 (Clone 300907). PE-Cy7 Mouse Anti- Human TCR Vδ1 was purchased from ThermoFisher Scientific (Waltham, MA). Cells were analyzed by flow cytometry using an LSRII (BD Biosciences, Franklin Lakes, NJ) and a BD FACSymphony (BD Biosciences, Franklin Lakes, NJ).
Cytotoxicity assays
The in vitro cytotoxic potential of naïve γδ T cells against multiple malignant cell lines was assessed in flow cytometry-based cytotoxicity assays. Target cell lines included the myeloid leukemia cell line, K562 (ATCC, Manassas, VA), and the neuroblastoma cell lines, IMR5, CHLA15, Kelly, CHLA20, and SMS-SAN. Target cells were labeled with the Violet Proliferation Dye 450 (BD Biosciences, Franklin Lakes, NJ) and incubated with γδ T cells at the varied effector to target (E:T) ratios: 0:1, 1:1, 5:1, 10:1 for 4 hrs at 37°C. Target cell death was analyzed via flow cytometry using dead cell stains (eBioscience Fixable Viability Dye eFluor 780) and incubating for 30 min with shaking at room temperature and/or 7-aminoactinomycin D (7AAD) was immediately added prior to data acquisition.
Freezing/thawing γδ T cells
Cells were washed once with PBS and spun at 300xg for 5 min. Cells were resuspended in Albumin (Human) U.S.P. Albutein 5% (Grifols Therapeutics Inc.) with a 9% DMSO content at a concentration of 1 × 107 γδ T cells per mL. All reagents were kept at 4°C during the freezing process. Cells were then slowly frozen at a rate of −1°C per minute until they reached −80°C and promptly moved to liquid nitrogen storage. To thaw the cells, they were incubated in a 37°C water bath until nearly thawed and subsequently diluted in 10 times the volume of complete OpTmizer media prior to centrifugation at 300xg for 5 min. Cells were resuspended in media containing IL-2 at 1,000 IU/mL concentration.
Biotinylation of dinutuximab
Clinical grade dinutuximab was biotinylated using EZ-Link™ Sulfo-NHS-Biotin (Thermo Fisher Scientific, Waltham, MA). Five hundred micrograms of antibody were added to an Amicon Ultra filter using DPBS/Modified (GE Life Sciences, Marlborough, MA) to adjust volume. The tube was centrifuged at 300 xg for 15 min. The EZ Link Sulfo NHS-LC Biotin was reconstituted in water (10 mg/ml) and added to the concentrated antibody in the Amicon Ultra filter at 1 mg of biotin reagent per mg of protein. The reaction proceeded for 30 min on ice. Hepes Buffered Saline (HBS)/0.05% azide was used to concentrate biotinylated-dinutuximab.
Cytokine release studies
γδ T cells were incubated for 4 hrs in a cytotoxicity assay with IMR5 neuroblastoma cells in cultures of 1 mL of media per condition. Media was removed from the cells, and any floating cells were centrifuged at 500xg for 5 min. Supernatant was removed and immediately utilized in the ProteomeProfiler kit (R&D Systems, Minneapolis, MN). A Chemidoc BioRad Imager was used to acquire images and ImageJ/Fiji (NIH, Bethesda, MD) image analysis software was used for densitometry analysis. Data were normalized to basal γδ T-cell cytokine secretion culture in media after 4 hrs.
Live cell imaging
IMR5 cells were transduced with a lentiviral vector green fluorescent protein (GFP) construct under the EF1α promoter (Lentigen, Gaithersburg, MD) at a multiplicity of infection of 10. Cells were cultured and sorted using a Sony SH800 to collect the top 5% of GFP+ cells by MFI. γδ T cells were labeled using Violet Proliferation Dye 450.
IMR5 cells were plated on Lab-Tek II Chamber Coverglass 8 well chambers in the center 4 wells 24 hrs prior to imaging to allow the cells to adhere to the glass. Immediately prior to imaging, γδ T cells and 100 μM of propidium iodide (PI) (Invitrogen, Carlsbad, CA) were added to each well. Imaging was conducted over 6 hr and images were taken every 19 min. Cells were imaged using a Leica SP8 inverted confocal microscope at 10x using a 458, 488, and 514 nm argon laser. Images were analyzed using ImageJ/Fiji (NIH, Bethesda, MD) image analysis software.
In vivo mouse experiments
NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice were purchased from Jackson Laboratory (Bar Harbor, ME) and were maintained in a pathogen-free environment. Mice were cared for according to the established principles of the Institutional Animal Care and Use Committee (IACUC), and all animal protocols were approved by the IACUC. Five-week-old mice were each inoculated subcutaneously via the right flank with IMR5 cells. Mice were visually monitored, and tumor growth was measured with calipers, and treated when tumors reached approximately 125 mm3 in volume typically 30 days after inoculation.
Tumor volume was determined by the following equation.
where r1 is the length of the tumor measuring anterior to posterior and r2 is the length of the tumor dorsal to ventral. When tumors were established mice were administered dinutuximab, TMZ, or γδ T cells. Mice were injected with 200–400 μg of dinutuximab, IV every 10 days. TMZ and γδ T cells (2.5x106 cells) were injected via the tail vein every three days. This treatment plan was 17 days long. Mice were weighed and measured every other day for 4 weeks.
γδ T-cell persistence in vivo
γδ T cells were expanded from healthy donor PBMCs. On day 12 of expansion, the population was determined to be 70% pan-γδ T cells, as measured by flow cytometry. NSG mice were randomized to treatment groups, with three mice per group. Mice were injected via tail-vein with either 5x106, 10 × 106 or 15 × 106 cells. On days 1, 3, 6 and 8 following injection, blood samples from each mouse were analyzed by flow cytometry using BD LSRII Flow Cytometer (BD Biosciences, San Jose, CA). Antibodies used included FITC anti-human CD45 (BD Biosciences, San Jose, CA), APC anti-mouse CD45.1 (BD Biosciences, San Jose, CA), and PE anti-human TCR γ/δ-1 (BD Biosciences, San Jose, CA).
Stress antigen expression on NB cells and mechanism of cytotoxicity induced by TMZ
IMR5 cells were seeded at a concentration of 250,000 cells per mL in 1 mL. TMZ was added at a concentration of 100 μM to 2 mM for varying lengths of from 0 to 24 hrs. Cells were collected from the plate using 1 mL Versene (Gibco). Cells were counted, washed, and incubated with APC human ULBP-2/5/6 (R&D systems), Alexa Fluor® 488 human ULBP-1 (R&D Systems), and PE human MICA/MICB (Biolegend) for 30 min prior to FACS analysis. A similar process was used to analyze receptor status on the surface of IMR5 cells. IMR5 cells were plated at a concentration of 500,000 cells per mL. TMZ was added at a concentration of 400 μM for 8 hr. Cells were removed using Versene, washed, counted, and stained for flow cytometry analysis.
Statistical analysis
All statistical analysis and graphing were performed using Sigma Plot version 13 (Systat Software Inc,) and GraphPad Software Prism. The exact method, for example, ANOVA, T-test, or log ranked Mantel–Cox test, are described for each experiment where they are used.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Financial support
Curing Kids Cancer, Cure Childhood Cancer, NIH/NCI R21 CA223300
Supplemental Material
Download MS Word (1.5 MB)Supplementary materials
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
References
- Davidoff AM. Neuroblastoma. Semin Pediatr Surg. 2012;21:2–14. doi:10.1053/j.sempedsurg.2011.10.009.
- Bosse KR, Maris JM. Advances in the translational genomics of neuroblastoma: from improving risk stratification and revealing novel biology to identifying actionable genomic alterations. Cancer. 2016;122:20–33. doi:10.1002/cncr.29706.
- Laverdiere C, Liu Q, Yasui Y, Nathan PC, Gurney JG, Stovall M, Diller LR, Cheung N-K, Wolden S, Robison LL, et al. Long-term outcomes in survivors of neuroblastoma: a report from the Childhood Cancer Survivor Study. J Natl Cancer Inst. 2009;101:1131–1140. doi:10.1093/jnci/djp230.
- Zheng DJ, Krull KR, Chen Y, Diller L, Yasui Y, Leisenring W, Brouwers P, Howell R, Lai JS, Balsamo L, et al. Long-term psychological and educational outcomes for survivors of neuroblastoma: A report from the Childhood Cancer Survivor Study. Cancer. 2018;124:3220–3230.doi: 10.1002/cncr.31379.
- Matthay KK, Villablanca JG, Seeger RC, Stram DO, Harris RE, Ramsay NK, Swift P, Shimada H, Black CT, Brodeur GM, et al. Treatment of high-risk neuroblastoma with intensive chemotherapy, radiotherapy, autologous bone marrow transplantation, and 13-cis-retinoic acid. Children’s Cancer Group. N Engl J Med. 1999;341:1165–1173. doi:10.1056/NEJM199910143411601.
- Lutsiak ME, Semnani RT, De Pascalis R, Kashmiri SV, Schlom J, Sabzevari H. Inhibition of CD4(+)25+ T regulatory cell function implicated in enhanced immune response by low-dose cyclophosphamide. Blood. 2005;105:2862–2868. doi:10.1182/blood-2004-06-2410.
- Michaud M, Martins I, Sukkurwala AQ, Adjemian S, Ma Y, Pellegatti P, Shen S, Kepp O, Scoazec M, Mignot G, et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science. 2011;334:1573–1577. doi:10.1126/science.1208347.
- Vincent J, Mignot G, Chalmin F, Ladoire S, Bruchard M, Chevriaux A, Martin F, Apetoh L, Rébé C, Ghiringhelli F. 5-Fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res. 2010;70:3052–3061. doi:10.1158/0008-5472.CAN-09-3690.
- Ghiringhelli F, Menard C, Puig PE, Ladoire S, Roux S, Martin F, Solary E, Le Cesne A, Zitvogel L, Chauffert B. Metronomic cyclophosphamide regimen selectively depletes CD4+CD25+ regulatory T cells and restores T and NK effector functions in end stage cancer patients. Cancer Immunol Immunother. 2007;56:641–648. doi:10.1007/s00262-006-0225-8.
- Ghiringhelli F, Larmonier N, Schmitt E, Parcellier A, Cathelin D, Garrido C, Chauffert B, Solary E, Bonnotte B, Martin F. CD4+CD25+ regulatory T cells suppress tumor immunity but are sensitive to cyclophosphamide which allows immunotherapy of established tumors to be curative. Eur J Immunol. 2004;34:336–344. doi:10.1002/eji.200324181.
- Pfirschke C, Engblom C, Rickelt S, Cortez-Retamozo V, Garris C, Pucci F, Yamazaki T, Poirier-Colame V, Newton A, Redouane Y, et al. Immunogenic chemotherapy sensitizes tumors to checkpoint blockade therapy. Immunity. 2016;44:343–354. doi:10.1016/j.immuni.2015.11.024.
- Lyman GH. Impact of chemotherapy dose intensity on cancer patient outcomes. J Natl Compr Canc Netw. 2009;7:99–108.
- Matthay KK, Reynolds CP, Seeger RC, Shimada H, Adkins ES, Haas-Kogan D, Gerbing RB, London WB, Villablanca JG. Long-term results for children with high-risk neuroblastoma treated on a randomized trial of myeloablative therapy followed by 13-cis-retinoic acid: a children’s oncology group study. J Clin Oncol. 2009;27:1007–1013. doi:10.1200/JCO.2007.13.8925.
- Dasgupta A, McCarty D, Spencer HT. Engineered drug-resistant immunocompetent cells enhance tumor cell killing during a chemotherapy challenge. Biochem Biophys Res Commun. 2010;391:170–175. doi:10.1016/j.bbrc.2009.11.026.
- Dasgupta A, Shields JE, Spencer HT. Treatment of a solid tumor using engineered drug-resistant immunocompetent cells and cytotoxic chemotherapy. Hum Gene Ther. 2012;23:711–721. doi:10.1089/hum.2011.172.
- Ramakrishnan R, Assudani D, Nagaraj S, Hunter T, Cho HI, Antonia S, Altiok S, Celis E, Gabrilovich DI. Chemotherapy enhances tumor cell susceptibility to CTL-mediated killing during cancer immunotherapy in mice. J Clin Invest. 2010;120:1111–1124. doi:10.1172/JCI40269.
- Weiner LM, Surana R, Wang S. Monoclonal antibodies: versatile platforms for cancer immunotherapy. Nat Rev Immunol. 2010;10:317. doi:10.1038/nri2744.
- Ahmed M, Cheung NK. Engineering anti-GD2 monoclonal antibodies for cancer immunotherapy. FEBS Lett. 2014;588:288–297. doi:10.1016/j.febslet.2013.11.030.
- Yu AL, Uttenreuther-Fischer MM, Huang CS, Tsui CC, Gillies SD, Reisfeld RA, Kung FH. Phase I trial of a human-mouse chimeric anti-disialoganglioside monoclonal antibody ch14.18 in patients with refractory neuroblastoma and osteosarcoma. J Clin Oncol. 1998;16:2169–2180. doi:10.1200/JCO.1998.16.6.2169.
- AL BA Y, Alvarado C, Rao VJ, Castelberry RP. Usefulness of a chimeric anti- GD2 (ch14.18) and GM-CSF for refractory neuroblastoma: a POG phase II study. Proc Am Soc Clin Oncol. 1997;16:1846.
- Navid F, Sondel PM, Barfield R, Shulkin BL, Kaufman RA, Allay JA, Gan J, Hutson P, Seo S, Kim K, et al. Phase I trial of a novel anti-GD2 monoclonal antibody, Hu14.18K322A, designed to decrease toxicity in children with refractory or recurrent neuroblastoma. J Clin Oncol. 2014;32:1445–1452. doi:10.1200/JCO.2013.50.4423.
- Gilman AL, Ozkaynak MF, Matthay KK, Krailo M, Yu AL, Gan J, Sternberg A, Hank JA, Seeger R, Reaman GH, et al. Phase I study of ch14.18 with granulocyte-macrophage colony-stimulating factor and interleukin- 2 in children with neuroblastoma after autologous bone marrow transplantation or stem-cell rescue: a report from the Children’s Oncology Group. J Clin Oncol. 2009;27:85–91. doi:10.1200/JCO.2006.10.3564.
- Yu AL, Gilman AL, Ozkaynak MF, London WB, Kreissman SG, Chen HX, Smith M, Anderson B, Villablanca JG, Matthay KK, et al. Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N Engl J Med. 2010;363:1324–1334. doi:10.1056/NEJMoa0911123.
- Ozkaynak MF, Gilman AL, London WB, Naranjo A, Diccianni MB, Tenney SC, Smith M, Messer KS, Seeger R, Reynolds CP, et al. Corrigendum: a comprehensive safety trial of chimeric antibody 14.18 with GM- CSF, IL-2, and isotretinoin in high-risk neuroblastoma patients following myeloablative therapy: children’s oncology group study ANBL0931. Front Immunol. 2018;9:1641. doi:10.3389/fimmu.2018.01641.
- Bergman I, Arbit E, Rosenblum M, Larson SM, Heller G, Cheung NK. Treatment of spinal epidural neuroblastoma xenografts in rats using anti-GD2 monoclonal antibody 3F8. J Neurooncol. 1993;15:235–242.
- Cheung NK, Lazarus H, Miraldi FD, Abramowsky CR, Kallick S, Saarinen UM, Spitzer T, Strandjord SE, Coccia PF, Berger NA. Ganglioside GD2 specific monoclonal antibody 3F8: a phase I study in patients with neuroblastoma and malignant melanoma. J Clin Oncol. 1987;5:1430–1440. doi:10.1200/JCO.1987.5.9.1430.
- Rajen Mody AN, Van Ryn C, Yu AL, London WB, Shulkin BL, Parisi MT, Servaes S-E-N, Dicciani MB, Sondel PM, Maris JM, et al. Phase II randomized trial of irinotecan/temozolomide (I/T) with temsirolimus (TEM) or dinutuximab plus granulocyte colony stimulating factor (DIN/GMCSF) in children with refractory or relapsed neuroblastoma: A report from the Children’s Oncology Group (COG). ASCO Annual Meeting; Chicago, IL, 2016.
- Furman WLFS, McCarville MB, Davidoff AM, Krasin MJ, Wu J, Brennan RC, Bishop MW, Santana VM, Bahrami A, Anthony G, et al., editors. Improved clinical responses with the concomitant use of an anti- GD2 monoclonal antibody and chemotherapy in newly diagnosed children with high-risk (HR) neuroblastoma (NB): preliminary results of a phase II study. ASCO Annual Meeting; 2016; Chicago, IL. doi:10.1200/JCO.2016.34.15_suppl.10501.
- Park JR, Bagatell R, London WB, Maris JM, Cohn SL, Mattay KK, Mattay KM, Hogarty M. Children’s Oncology Group’s 2013 blueprint for research: neuroblastoma. Pediatr Blood Cancer. 2013;60:985–993. doi:10.1002/pbc.24433.
- De Maria A, Bozzano F, Cantoni C, Moretta L. Revisiting human natural killer cell subset function revealed cytolytic CD56(dim)CD16+ NK cells as rapid producers of abundant IFN-gamma on activation. Proc Natl Acad Sci USA. 2011;108:728–732. doi:10.1073/pnas.1012356108.
- Mandelboim O, Malik P, Davis DM, Jo CH, Boyson JE, Strominger JL. Human CD16 as a lysis receptor mediating direct natural killer cell cytotoxicity. Proc Natl Acad Sci USA. 1999;96:5640–5644.
- Ryan PL, Sumaria N, Holland CJ, Bradford CM, Izotova N, Grandjean CL, Jawad AS, Bergmeier LA, Pennington DJ. Heterogeneous yet stable Vdelta2(+) T-cell profiles define distinct cytotoxic effector potentials in healthy human individuals. Proc Natl Acad Sci USA. 2016;113:14378–14383. doi:10.1073/pnas.1611098113.
- Wu YL, Ding YP, Tanaka Y, Shen LW, Wei CH, Minato N, Zhang W. gammadelta T cells and their potential for immunotherapy. Int J Biol Sci. 2014;10:119–135. doi:10.7150/ijbs.7823.
- Hoeres T, Smetak M, Pretscher D, Wilhelm M. Improving the Efficiency of vgamma9vdelta2 T-cell immunotherapy in cancer. Front Immunol. 2018;9:800. doi:10.3389/fimmu.2018.00800.
- Lamb LS Jr., Bowersock J, Dasgupta A, Gillespie GY, Su Y, Johnson A, Spencer HT, Castro MG. Engineered drug resistant gammadelta T cells kill glioblastoma cell lines during a chemotherapy challenge: a strategy for combining chemo- and immunotherapy. PLoS One. 2013;8:e51805. doi:10.1371/journal.pone.0051805.
- Wu J, Groh V, Spies T. T cell antigen receptor engagement and specificity in the recognition of stress-inducible MHC class I-related chains by human epithelial gamma delta T cells. J Immunol. 2002;169:1236–1240.
- Stojanovic A, Correia MP, Cerwenka A. The NKG2D/NKG2DL axis in the crosstalk between lymphoid and myeloid cells in health and disease. Front Immunol. 2018;9:827. doi:10.3389/fimmu.2018.00827.
- Gentles AJ, Newman AM, Liu CL, Bratman SV, Feng W, Kim D, Nair VS, Xu Y, Khuong A, Hoang CD, et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med. 2015;21:938–945. doi:10.1038/nm.3909.
- Liu R, Zhou Q, La Cava A, Campagnolo DI, Van Kaer L, Shi FD. Expansion of regulatory T cells via IL-2/anti-IL-2 mAb complexes suppresses experimental myasthenia. Eur J Immunol. 2010;40:1577–1589. doi:10.1002/eji.200939792.
- Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. 2008;133:775–787. doi:10.1016/j.cell.2008.05.009.
- Sutton KS, Dasgupta A, McCarty D, Doering CB, Spencer HT. Bioengineering and serum free expansion of blood-derived gammadelta T cells. Cytotherapy. 2016;18:881–892. doi:10.1016/j.jcyt.2016.04.001.
- Vantourout P, Hayday A. Six-of-the-best: unique contributions of gammadelta T cells to immunology. Nat Rev Immunol. 2013;13:88–100. doi:10.1038/nri3384.
- Gasser S, Orsulic S, Brown EJ, Raulet DH. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature. 2005;436:1186–1190. doi:10.1038/nature03884.
- Hervieu A, Rebe C, Vegran F, Chalmin F, Bruchard M, Vabres P, Apetoh L, Ghiringhelli F, Mignot G. Dacarbazine-mediated upregulation of NKG2D ligands on tumor cells activates NK and CD8 T cells and restrains melanoma growth. J Invest Dermatol. 2013;133:499–508. doi:10.1038/jid.2012.273.
- Soriani A, Zingoni A, Cerboni C, Iannitto ML, Ricciardi MR, Di Gialleonardo V, Cippitelli M, Fionda C, Petrucci MT, Guarini A, et al. ATM-ATR-dependent up-regulation of DNAM-1 and NKG2D ligands on multiple myeloma cells by therapeutic agents results in enhanced NK-cell susceptibility and is associated with a senescent phenotype. Blood. 2009;113:3503–3511. doi:10.1182/blood-2008-08-173914.
- Berghuis D, Schilham MW, Vos HI, Santos SJ, Kloess S, Buddingh EP, Egeler RM, Hogendoorn PC, Lankester AC. Histone deacetylase inhibitors enhance expression of NKG2D ligands in Ewing sarcoma and sensitize for natural killer cell-mediated cytolysis. Clin Sarcoma Res. 2012;2:8. doi:10.1186/2045-3329-2-8.
- Middlemas DS, Stewart CF, Kirstein MN, Poquette C, Friedman HS, Houghton PJ, Brent TP. Biochemical correlates of temozolomide sensitivity in pediatric solid tumor xenograft models. Clin Cancer Res. 2000;6:998–1007.
- Pang DJ, Neves JF, Sumaria N, Pennington DJ. Understanding the complexity of gammadelta T-cell subsets in mouse and human. Immunology. 2012;136:283–290. doi:10.1111/j.1365-2567.2012.03582.x.
- Frost JD, Hank JA, Reaman GH, Frierdich S, Seeger RC, Gan J, Anderson PM, Ettinger LJ, Cairo MS, Blazar BR, et al. A phase I/IB trial of murine monoclonal anti-GD2 antibody 14.G2a plus interleukin-2 in children with refractory neuroblastoma: a report of the Children’s Cancer Group. Cancer. 1997;80:317–333.
- Fisher JP, Flutter B, Wesemann F, Frosch J, Rossig C, Gustafsson K, Anderson J. Effective combination treatment of GD2-expressing neuroblastoma and Ewing’s sarcoma using anti-GD2 ch14.18/CHO antibody with Vgamma9Vdelta2+ gammadeltaT cells. Oncoimmunology. 2016;5:e1025194. doi:10.1080/2162402X.2015.1025194.
- Mueller BM, Romerdahl CA, Gillies SD, Reisfeld RA. Enhancement of antibody- dependent cytotoxicity with a chimeric anti-GD2 antibody. J Immunol. 1990;144:1382–1386.
- Kowalczyk A, Gil M, Horwacik I, Odrowaz Z, Kozbor D, Rokita H. The GD2- specific 14G2a monoclonal antibody induces apoptosis and enhances cytotoxicity of chemotherapeutic drugs in IMR-32 human neuroblastoma cells. Cancer Lett. 2009;281:171–182. doi:10.1016/j.canlet.2009.02.040.
- Melero I, Rouzaut A, Motz GT, Coukos G. T-cell and NK-cell infiltration into solid tumors: a key limiting factor for efficacious cancer immunotherapy. Cancer Discov. 2014;4:522–526. doi:10.1158/2159-8290.CD-13-0985.
- Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G, Huls MH, Liu E, Gee AP, Mei Z, et al. Virus- specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med. 2008;14:1264–1270. doi:10.1038/nm.1882.
- Craddock JA, Lu A, Bear A, Pule M, Brenner MK, Rooney CM, Foster AE. Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. J Immunother. 2010;33:780–788. doi:10.1097/CJI.0b013e3181ee6675.
- Richman SA, Nunez-Cruz S, Moghimi B, Li LZ, Gershenson ZT, Mourelatos Z, Barrett DM, Grupp SA, Milone MC. High-affinity GD2-specific CAR T cells induce fatal encephalitis in a preclinical neuroblastoma model. Cancer Immunol Res. 2018;6:36–46. doi:10.1158/2326-6066.CIR-17-0211.
- Deniger DC, Moyes JS, Cooper LJ. Clinical applications of gamma delta T cells with multivalent immunity. Front Immunol. 2014;5:636. doi:10.3389/fimmu.2014.00636.
- Fisher DT, Chen Q, Appenheimer MM, Skitzki J, Wang WC, Odunsi K, Evans SS. Hurdles to lymphocyte trafficking in the tumor microenvironment: implications for effective immunotherapy. Immunol Invest. 2006;35:251–277. doi:10.1080/08820130600745430.
- Nolz JC, Starbeck-Miller GR, Harty JT. Naive, effector and memory CD8 T-cell trafficking: parallels and distinctions. Immunotherapy. 2011;3:1223–1233. doi:10.2217/imt.11.100.
- Pressey JG, Adams J, Harkins L, Kelly D, You Z, Lamb LS Jr. In vivo expansion and activation of gammadelta T cells as immunotherapy for refractory neuroblastoma: A phase 1 study. Medicine (Baltimore). 2016;95:e4909. doi:10.1097/MD.0000000000004864.
- Kurzen H, Schmitt S, Naher H, Mohler T. Inhibition of angiogenesis by non-toxic doses of temozolomide. Anticancer Drugs. 2003;14:515–522. doi:10.1097/01.cad.0000086842.52271.ae.
- Fukumura D, Kloepper J, Amoozgar Z, Duda DG, Jain RK. Enhancing cancer immunotherapy using antiangiogenics: opportunities and challenges. Nat Rev Clin Oncol. 2018;15:325–340. doi:10.1038/nrclinonc.2018.29.
- Roos WP, Batista LFZ, Naumann SC, Wick W, Weller M, Menck CFM, Kaina B. Apoptosis in malignant glioma cells triggered by the temozolomide-induced DNA lesion O6-methylguanine. Oncogene. 2006;26:186. doi:10.1038/sj.onc.1209785.