
ABSTRACT
In vitro assays that evaluate CD8+ T cell-mediated cytotoxicity are important to aid in the development of novel therapeutic approaches to enhance anti-tumor immune responses. Here, we describe a novel cytotoxicity co-culture assay that circumvents the problem of highly variable allogeneic responses and obviates the constraints of HLA-restriction between effector and target cells. We show that this assay can be easily applied to a panel of tumor cell lines to provide additional insights into intrinsic drivers of sensitivity/resistance to T cell-mediated killing, and to evaluate the impact of targeted therapies on both tumor and T cell compartments.
Introduction
Immunotherapy is now established as a standard of care for many cancer types.Citation1,Citation2 Despite this rapid progress, a greater understanding of intrinsic or adaptive mechanisms of resistance to immunotherapy is required to increase the proportion of responding patients, and durability of responses. Furthermore, there is now a more urgent need to better characterize how targeted therapies, designed to specifically affect tumor cells, may impact the anti-cancer immune response to identify productive combinations and avoid potentially antagonistic drug combinations.
CD8+ cytotoxic T cells are considered as the principal cellular mediators of anti-tumor immunity due to their capacity for specific recognition of tumor-associated antigens (TAA) and intrinsic cytotoxic potential.Citation3 Therefore, in vitro assays that permit evaluation of T cell cytotoxicity against tumor cells can be used to determine mechanisms of sensitivity/resistance and to screen novel agents that may modulate the outcome of the T cell/tumor cell interaction, providing important translational insights. T cells recognize peptide antigens in the context of the major histocompatibility complex (MHC) through their T cell receptors (TCR) providing ‘signal 1ʹ. Costimulatory proteins provide ‘signal 2ʹ, via the CD28 co-receptor protein expressed on the surface of T cells. Activation of CD8+ cytotoxic T lymphocytes (CTLs) leads to target cell apoptosis through the release of lytic granules or via alternative mechanisms such as the Fas pathway, or cytokine release.Citation4 As such, most established in vitro T cell cytotoxicity assays depend on T cell antigen-specific recognition via tumor MHC-TCR engagement, and thus are either human leukocyte antigen (HLA) restricted or rely on alloreactivity.Citation5,Citation6 Autologous assays are challenging given the limitations on the numbers of HLA-matched donor T cells that can be sourced.Citation7 Moreover, allogeneic assay systems are intrinsically variable from donor to donor.Citation8 Therefore, it is difficult to assess a T cell response across a range of tumor cells with distinct genotypes, to investigate how these perturbations may impact sensitivity to T cell cytotoxicity.
We have developed a method for generating tumor cell lines that express a fragment of anti-CD3 antibody on the cell membrane.Citation9 These engineered cells provide ‘signal 1ʹ to T cells leading to T cell activation in an MHC- and antigen-independent manner, enabling T cells to be active in a redirected T cell-mediated cytotoxicity assay. This system allows for interrogation of intrinsic mechanisms of sensitivity/resistance to T cell-mediated cytotoxicity across a range of cell lines. In addition, we show that this versatile co-culture system can also be used to evaluate the activity of targeted therapies on both the tumor and T cell compartments across multiple donors.
Results
Optimizing CD8+ T cells for tumor cell cytotoxicity assays
To optimize our cytotoxicity assay, we first determined how in vitro restimulation conditions would impact the cytotoxic capacity and phenotype of CD8+ T cells. We used anti-CD3 redirected cytotoxicity against P815 mouse mastocytoma cells. These P815 cells express Fcγ-receptors, thus providing ‘signal 1ʹ and leading to CD8+T cell activation and cytotoxicity. This assessment was used to guide subsequent experiments in the anti-CD3-expressing engineered tumor cell system used. Our co-culture assay is based on two rounds of anti-CD3/CD28 stimulation of isolated primary CD8+ T cells, which are then co-cultured with DiO labeled tumor cells at various time points. After which, staining with a LIVE/DEAD Violet viability dye allows for flow cytometry analysis of live DiO+ Violet− tumor cells within the co-culture (Supplementary Fig. S1). Our data demonstrate that this second round of restimulation of CD8+ T cells following in vitro expansion with anti-CD3/CD28 dynabeads, 5 days prior to co-culture in a cytotoxicity assay, enhanced effector function (). At the highest effector: target (E: T) ratio of 10:1 versus the control 0 E: T ratio, we observed approximately 30% P815 cytotoxicity in the presence of soluble anti-CD3, when compared to almost no effect with the isotype control. Comparatively, P815 cytotoxicity was increased to 45% in the co-culture at the 10:1 E: T ratio, following restimulation of the CD8+ T cells (). This increased cytotoxicity was commensurate with an observed differentiation of the restimulated CD8+ T cells towards an effector phenotype (increased CD45RO expression, decreased CD45RA expression (), and decreased CCR7 expression () when compared to pre-stimulation).
Figure 1. Optimizing CD8+ T cells for tumor cell cytotoxicity assays.
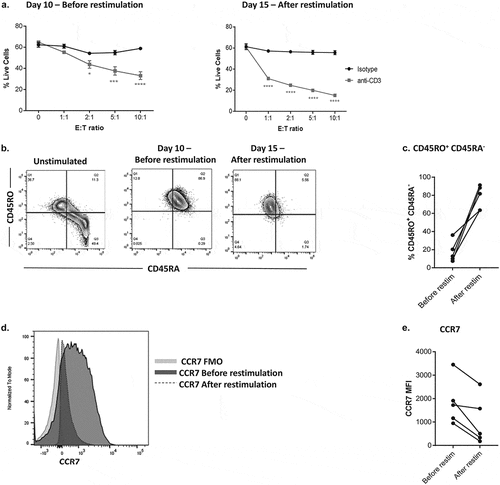
Using this expansion/reactivation protocol, we were able to generate sufficient populations of effectors T cells with robust cytotoxic activity for use in cytotoxicity assays.
Generation of anti-CD3 expressing tumor cell lines
Allogeneic T cell cytotoxicity assays are highly variable.Citation8 Therefore, TCR triggering through CD3 is an effective method for circumventing MHC/TCR alloreactivity and increasing the number of cytotoxic synapses between T cells and tumor cells. However, in contrast to P815 cells, most solid tumor cells lack Fc receptors and therefore TCR triggering at the synapse of T and tumor cells cannot be facilitated by crosslinking via addition of anti-CD3 antibody. Therefore, we generated lentiviral constructs encoding an anti-CD3 antibody fragment fused to the transmembrane domain of CD14Citation9 (). Three EGFR mutant (EGFRm) NSCLC cell lines; PC-9, NCI-H1975, and NCI-H3255, were transduced to express this scFV-CD3(OKT3)-CD14 construct to activate CD8+ T cells via their TCR and thus lead to cytotoxicity of the tumor cells (). The workflow for cell line transduction is provided in . Briefly, the anti-CD3 expressing tumor cells were transduced using viral supernatants from transfected 293T/17 cells. Multiplicity of infection (MOI) of both 1 and 10 were tested and based on sufficient transduction efficiency being achieved, cells transduced at an MOI of 1 were used for all experiments. Antibiotic selection was used to select for and maintain expression of the anti-CD3 scFv construct and transduced cells sorted by flow cytometry based on CD14 expression. Using an anti-IgG antibody (H + L), which binds to the heavy and light chains of the anti-CD3 scFV,Citation9 we examined the expression of cell surface anti-CD3 and found that all transduced cell lines expressed high levels of anti-CD3 relative to non-transduced parental controls (). Using this method, cell surface expression of anti-CD3 was routinely confirmed on the tumor cells lines.
Figure 2. Generation of anti-CD3 expressing tumor cell lines.
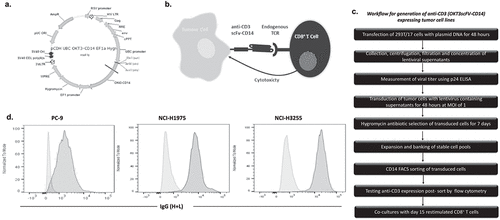
Co-culture of CD8+ T cells with tumor cells expressing anti-CD3 results in T cell-mediated cytotoxicity
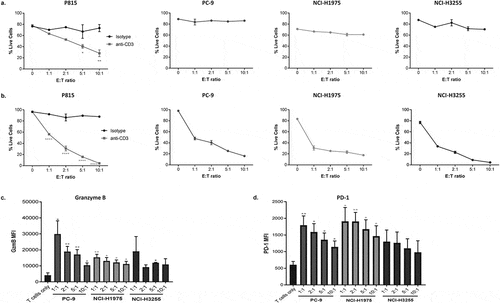
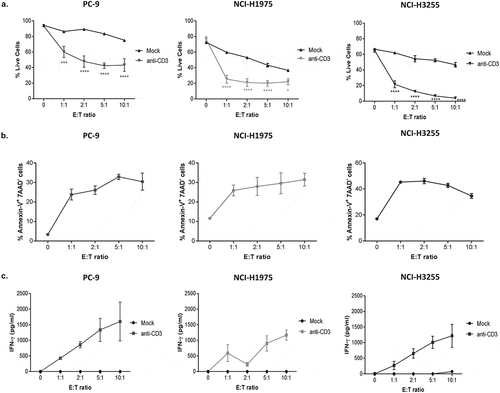
To validate the model, anti-CD3 expressing tumor cell lines were co-cultured with restimulated CD8+ effector T cells. T cell-mediated cytotoxicity against all three cell lines was evaluated at 4 and 18 h in comparison to P815 control cells, which were used as an indicator of T cell effector function due to their high susceptibility to T cell cytotoxicity as previously shown. Using our flow cytometry-based readout to analyse live tumor cells in these co-cultures, we observed that in contrast to control P815 cells (where CD3 stimulation is regulated by soluble anti-CD3 antibody), the window of T cell-mediated cytotoxicity in the anti-CD3 expressing NSCLC cell lines was small at 4 h (roughly, 3% in PC-9, 11% in NCI-H1975 and 17% in NCI-H3255) (as calculated by differences in cytotoxicity at 0 to 10:1 E: T ratio) (). However, by 18 h, the magnitude of T cell-mediated cytotoxicity was greater in both the P815 cells and in PC-9, NCI-H1975 and NCI-H3255 EGFRm cell lines (). As expected, the level of cytotoxicity was proportional to the E: T ratio. In these 18h co-cultures, the range of tumor cell death varied from 30% to 70% at the lowest E: T ratio (1:1) to 80–100% at the highest E: T ratio (10:1). We also show that the observed trends in cytotoxicity across all three EGFRm cell lines in 18h co-cultures, is replicated across multiple donors and experiments tested, with some variability depending on donor responses (Supplementary Fig. S2). Each cell line displayed slight variation in their intrinsic sensitivity to cytotoxicity in this assay. To explore this further, we also evaluated T cell function following co-culture with the different tumor cell lines, by measuring granzyme B and PD-1 expression by intracellular staining of CD8+ T cells and flow cytometry (). Granzyme B production () and PD-1 () expression were induced in CD8+ T cells by all three tumor cell lines, compared to CD8+ T cells alone, although significance was only observed in PC-9 and NCI-H1975 cells lines at all E: T ratios. However, each cell line induced differing total levels of granzyme B and PD-1, with T cells co-cultured with PC-9 tumor cells expressing the highest level of granzyme B (), while T cells co-culture with NCI-H1975 tumor cells expressed the highest level of PD-1 ().
Figure 3. Co-culture of CD8+ T cells with tumor cells expressing anti-CD3 results in target cell T cell-mediated cytotoxicity.
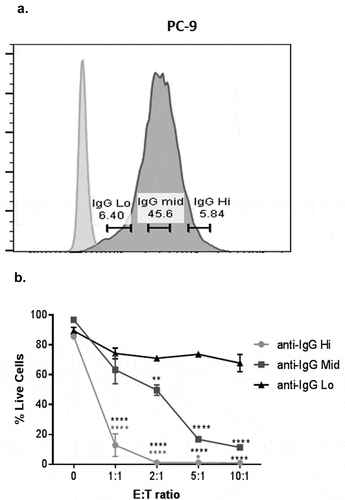
Expression levels of anti-CD3 influence the extent of tumor cell cytotoxicity by T cells
As expected, the level of anti-CD3 expression influences the magnitude of T cell-mediated cytotoxicity. Using a flow cytometric gating system, we were able to demonstrate this by gating on cells with high, medium and low expression using anti-IgG (H + L) staining (). Using, PC-9 cells (transduced with anti-CD3), we show that expression of anti-CD3 correlates with cytotoxicity, especially at higher E: T ratios (). Therefore, this gating can be applied to potentially explore the impact of TCR signal agonism on cytotoxic responses. Moreover, to circumvent this issue when comparing across cell lines with different levels of anti-CD3 expression (Supplementary Fig. S3A), this gating strategy can be specifically applied (Supplementary Fig. S3B) to select cells with similar levels of anti-CD3 expression (Supplementary Fig. S3C). This method can be used to control for variability in transduction efficiencies, thus permitting the analysis of a population of cells with highly overlapping expression levels of anti-CD3 and allowing for direct comparisons of relative cytotoxicity across multiple cell lines (Supplementary Fig. S3D).
Figure 4. Expression levels of anti-CD3 influence the extent of tumor cell cytotoxicity by T cells.
Measurements of IFN-ɣ and early tumor cell apoptotic events provide additional sensitive pharmacodynamic endpoints in this assay system
On recognition of peptide-MHC complexes on target cells, CD8+ T cells trigger programmed cell death of target cells.Citation10 Therefore, we wanted to characterize the mode of cell death in the anti-CD3-transduced tumor cells beyond evaluating overall viability in co-cultures. We examined cell death in our co-cultures using both direct DiO labelling of mock (cell lines transduced with empty vector controls) and anti-CD3 transduced tumor cells () and using annexin-V and 7-amino actinomycin D (7-AAD) staining of co-cultures with anti-CD3 expressing cells only, as a readout to evaluate apoptosis (). We found that anti-CD3 transduced tumor cells undergo significantly more T cell-mediated cytotoxicity than their empty vector mock control counterparts () and that there is an induction of early apoptotic tumor cells (annexin-V+ 7-AAD−) in the co-cultures with the anti-CD3 expressing NSCLC cell lines (). Moreover, measurement of IFN-ɣ in the co-culture supernatants of mock and anti-CD3 tumor cells also correlated with the level of tumor cell cytotoxicity and E: T ratios () and was sufficiently sensitive to be a pharmacodynamic readout of T cell activity.
Figure 5. Measurements of IFN-ɣ and early tumor cell apoptotic events provide additional sensitive pharmacodynamic endpoints in this assay system.
Investigating the impact of targeted therapies on T cell-mediated cytotoxicity using the CD3-expressing tumor cell co-culture system
Evaluating the potential impact of targeted therapies on both tumor and T cell responses is becoming increasingly important as immunotherapy becomes part of the standard of care for many cancer types. Targeted therapies may enhance or have antagonistic effects on T cell-mediated cytotoxicity. Moreover, evaluating how combinations of distinct immunotherapies may converge on cytotoxic potential against tumor cells is poorly understood.Citation11 Therefore, using the PC-9 EGFRm anti-CD3 expressing NSCLC cell line as a model, we next characterized the effects of EGFR tyrosine kinase inhibitors (TKIs) on T cell cytotoxic potential. PC-9 cells harbour an EGFR activating mutation due to a deletion in exon 19Citation12,Citation13 and are therefore sensitive to first and third generation EGFR TKIs, gefitinib and osimertinib, respectively.Citation14 Pre-treatment of PC-9 cells with gefitinib and osimertinib, at clinically relevant doses and maintenance of these concentrations in co-cultures, resulted in an increase in the amount of tumor cell death observed in co-cultures with both drugs, compared to DMSO control (). This increase in tumor cell death was mirrored in the no T cell controls (0 E: T ratio) and increased the amount of tumor cell death evaluated in combination with effector CD8+ T cells (). These observations were also confirmed using the anti-CD3 expressing NCI-H3255 EGFR mutant NSCLC cell line (Supplementary Fig. S4). These findings suggest that inhibition of EGFR signaling, whilst capable of reducing the survival of EGFRm NSCLC cells, does not negatively influence T cell-mediated cytotoxicity.
Figure 6. Investigating the impact of targeted therapies on T cell-mediated cytotoxicity using the anti-CD3-expressing tumor cell co-culture system.
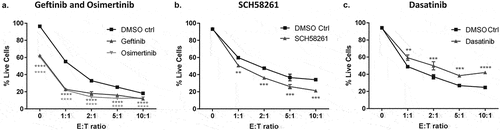
Adenosine has been shown to have suppressive effects on T cell function and is a component of some tumor microenvironments.Citation15,Citation16 The addition of SCH58261; a selective inhibitor of A2aR receptor,Citation16 was not found to directly impact tumor cell viability (0 E: T ratio), but was found to significantly enhance T cell-mediated cytotoxicity against anti-CD3 transduced PC-9 cells in the presence of AMP, when compared to DMSO control, across all E: T ratios tested (). Moreover, we show this increase in tumor cell death can be consistently observed across donors (Supplementary Fig. S5). As such, these data suggest that targeting the adenosine pathway can directly modulate CD8+ T cell cytotoxic function.
To further characterize the assay, we next investigated the effects of dasatinib, a potent inhibitor of the Src family kinases including Lck which is critical for mediating T-cell signaling downstream of the TCR.Citation17,Citation18 Our assay demonstrated a significant reduction in T cell-mediated cytotoxicity against anti-CD3 expressing PC-9 cells in the presence of 12.5 nM dasatinib compared to DMSO control and revealed that dasatinib had no effect on tumor cells alone, in the absence of T cells (0 E: T ratio) (). Furthermore, a dose-response of dasatinib from 0 to 100 nM in co-cultures with anti-CD3 expressing PC-9 cells showed that concentrations of 25, 50 and 100 nM dasatinib further significantly reduced T cell-mediated cytotoxicity of PC-9 cells (Supplementary Fig. S6). These findings confirm that modulation of TCR signaling could be measured through the magnitude T cell-mediated tumor cell cytotoxicity in the co-culture assay. Lastly, we investigated the effects of these targeted therapies on CD8+ T cell viability to exclude direct effects on T cells. We found no significant difference in the viability of CD8+ T cells when treated with gefitinib (320 nM), osimertinib (160 nM) (Supplementary Fig. S7A), SCH58261 (10 µM) (Supplementary Fig. S7B) and dasatinib (12.5 nM) (Supplementary Fig. S7C), at the same concentrations and time points used in co-culture assays.
Together these data demonstrate that this novel co-culture system can be easily applied to explore, how targeted therapies may impact both the T cell and tumor cell compartments across multiple donors, without the intrinsic variabilities normally observed with allogeneic responses.
Discussion
Understanding how CD8+ T cell anti-tumor immune responses are conditioned by specific features of tumors such as their tissue of origin and genotypes could aid in the development of successful immunotherapy treatments. Within the tumor microenvironment, this complex interaction is influenced by the presence of a number of inhibitory and costimulatory surface molecules on tumor cells and cytokines that can affect CD8+ T cell cytotoxic function.Citation19 In the system described here, we propose an in vitro approach to evaluate tumor cell sensitivity to CD8+ T cell-mediated cytotoxicity via their expression of anti-CD3 (OKT3). The ability of these engineered tumor cells to provide ‘signal 1ʹ to CD8+ T cells, thereby directly activating them, bypasses the need for TCR recognition of specific MHC-peptide complexes on tumor cells, whilst still allowing for the contributions of endogenous mechanisms of sensitivity and resistance to be assessed. Thus far, assays that allow for simultaneous assessment of CD8+ T cell-mediated cytotoxicity of different cell lines across common donors are limited due to HLA restriction and the variability associated with allogeneic responses.Citation5,Citation6 Although not antigen-specific, our assay presents a novel approach to investigating tumor escape strategies between and within various cancer types, in anti-CD3 engineered co-culture assays. In this paper, we specifically examined EGFRm NSCLC cell types and found a degree of variability in their sensitivities to T cell-mediated cytotoxicity. The determinants of these variations to T cell-mediated cytotoxicity will be investigated in future studies. Such insight could lead to the development of more personalized therapies in which the selection of drug treatments could potentially be based on tumor genetics and predicted anti-tumor immune responses, using assays such as the one described here.
Historically, the “gold standard” for measuring cytotoxicity has been assays based on chromium (Cr51) released by the lysed target cells.Citation7 This method involves the use of radioactive materials and the measurement of target cell death and has low sensitivity in assays of cultures longer than 4–8h due to the spontaneous release of Cr51.Citation20,Citation21 Alternatively, assays that measure the release of endogenous enzymesCitation22 and cytokinesCitation23 from co-culture supernatants indirectly measure cytotoxicity and may not adequately compensate for the contribution of effector cells in the readout.Citation6,Citation7 Our method relies on a flow cytometry-based readout allowing for the evaluation of direct T cell-mediated cytotoxicity by measuring tumor cell death. Moreover, the method supports the analysis of the mechanism of cell death, e.g. via measurement of annexin-V and 7AAD on tumor cells and can provide insight into T cell viability and phenotype following co-culture with target cells.
The generation of membrane-anchored anti-CD3 scFV expressing cells has been previously described to create immunosomesCitation24 and stimulator cells,Citation9 which both activate human T cells. However, this is the first description of this approach to directly engineer tumor cells to explore the cytotoxic potential of CD8+ T cells. We have shown that this protocol is amenable to a variety of adherent tumor lines and as such, can facilitate comparisons of tumor cell sensitivity across multiple tumor cells. Our data show that T cell-mediated cytotoxicity of tumor cells is comparable to that of P815 cells which express Fc receptors that allow TCR crosslinking in the presence of soluble anti-CD3 (). Whilst there remains some donor to donor variability in the degree of cytotoxicity observed, this is reduced when compared to assays that rely upon alloreactivity.Citation8,Citation25 However, it is evident that the expression of anti-CD3 on tumor cell lines leads to a larger cytotoxicity window compared to an allogeneic reaction ( – anti-CD3 vs. mock controls). This novel method provides a larger therapeutic window to evaluate pharmacodynamic responses and increases assay throughput by reducing the number of T cells required to elicit robust cytotoxicity, both of which are limitations with standard allogeneic cytotoxicity assays. Moreover, our data demonstrate that the T cell-mediated cytotoxicity observed in this assay is specifically driven via anti-CD3 expression as empty vector transduced (mock controls) cells undergo very little cell death via allorecognition in this assay and failed to induce IFN-ɣ production by CD8+ T cells when compared to co-cultures with anti-CD3 expressing tumor cells.
To optimize and standardize the methods for preparation of the CD8+ T cells used in the assay, we have characterized both their function and phenotype during pre-activation as well as during co-cultures. We have shown that a second round of stimulation of T cells with anti-CD3/CD28 leads to increased expression of CD45RO and loss of CD45RA and CCR7, resulting in increased effector function indicated by T cell-mediated cytotoxicity of P815 target cells (). Within co-cultures, CD8+ T cell cytotoxic function was evident not only by the cell death of tumor cells but also the significant production of granzyme B, in response to anti-CD3 stimulation by tumor cells. Previous studies have shown that on TCR crosslinking, stored granzyme B is released and there is de novo synthesis of lytic proteins that refills these granules, which are in turn secretedCitation26 and might therefore explain our results. PD-1 has important immunoregulatory roles in limiting effective T cell responses.Citation27 Traditionally, PD-1 has been noted as one of the hallmarks of T cell exhaustion. However, a broader view of PD-1 has highlighted its importance as firstly, being a marker of T cell activation and secondly, its expression correlating with TCR signaling strength.Citation28 As such, the induction of PD-1 expression in CD8+ T cells by anti-CD3 expressing tumor cells is expected and further shows the physiological relevance of our system.
Previously published studies demonstrate that the level of TCR engagement, e.g. by increased level of MHC/TCR clustering, can influence the relative susceptibility of a target cell to T cell-mediated cytotoxicity.Citation29,Citation30 In our assay system, we describe a simple gating strategy to enable measurement of the anti-CD3 expression levels on transduced cells in co-cultures and show that this correlates with the amount of T cell-mediated cytotoxicity observed. This is particularly useful in manipulating the amount of cytotoxicity observed in a single cell line, depending on the intended application of the assay. One caveat of this novel method is differing transduction efficiencies of tumor cells, which can result in varying levels of anti-CD3 expression across different cell lines. Therefore, this gating strategy allows cytotoxicity readouts to be normalized via anti-CD3 expression across cell lines.
Targeted and immunotherapies have been extensively investigated for their effects on cancer cells and in some cases, immune cells. However, as it relates to CD8+ T cells, most of these studies have evaluated their effects on assays such as proliferation and cytokine production in vitro and in vivo, which indirectly reflect cytotoxic function.Citation31 We have shown that our system directly allows the investigation of therapies of interest on T cell cytolytic activity. We first showed that EGFR TKIs result in an increase in target cell death in PC-9 cells, which appears to be as a consequence of their direct cytotoxic effect on the target cell and correlates with the well-reported sensitivities of this cell line to EGFR TKI based on its genotype.Citation14 These results appear to be additive in PC-9 and in a second EGFR mutant NSCLC cell line, NCI-H3255 (also characterised to be sensitive to both gefitinib and osimertinib)Citation14 at lower E: T ratios, suggesting that the effects of EGFR TKI treatment are diminished when T cell-mediated cytotoxicity is increased in the presence of higher numbers of CD8+ T cells. The reason for this is unclear but there may be a maximum threshold of cytotoxicity measurable in this assay. We also observed that SCH58261, an inhibitor of adenosine A2a receptor (A2aR), increases T cell-mediated cytotoxicity of target cells. Furthermore, this increase of cytotoxicity with SCH58261 is consistent regardless of donor variability, and overall suggests that adenosine could in fact influence T cell effector lytic function.Citation32 We also showed that the Lck inhibitor, dasatinib, decreased the amount of T cell-mediated cytotoxicity observed, showing that our assay is amenable to both enhancing and suppressive effects.
Overall, we have detailed a system that allows for engineering tumor cells to express anti-CD3 to measure their sensitivity to T cell-mediated cytotoxicity while also simultaneously phenotyping CD8+ T cells and their responses, in an in vitro co-culture assay. This system is highly adaptable to test different tumor cells, is reproducible across different donor T cells and is both robust and sensitive permitting the evaluation of novel drug combinations and interrogation of tumor intrinsic mechanisms that may influence susceptibility to T cell-mediated cytotoxicity.
Materials and methods
Generation of expression constructs encoding membrane-bound anti-CD3 single chain fragments
cDNA derived from hybridoma cells producing the OKT3-CD14 fusion protein (anti-CD3), was subjected to PCR amplification using primer pairs 5ʹ-TTCGAAGCCACCATGCCTATGGGCTC-3ʹ and 5ʹ-GGATCCTCAGGCGAAGCCTCTAG-3ʹ designed with BstB1 sites on the 3ʹ end and BamH1 5ʹ end, digested and cloned into pCDH-UbC-MCS-EF1a-Hygro to generate the lentiviral plasmid pCDH-UbC-CD14-OKT3-EF1a-Hygro.
Cell culture and generation of anti-CD3 expressing tumor cell lines – lentiviral transfection and transduction
For lentiviral transfection, 293T/17 packaging cells (ATCC) were maintained in Dulbecco‘s Modified Eagle‘s Medium (Gibco) supplemented with 10% heat-inactivated/gamma irradiated fetal bovine serum (FBS) (Invitrogen) and 1mM MEM non-essential amino acids (Gibco) at 37⁰C and 5% CO2. At 70–90% confluence, 293T/17 cells were lentivirally transfected with pCDH-UbC-CD14-OKT3-EF1a-Hygro expression plasmid or a pCDH-UBC-EF1a-Hygro empty plasmid (for mock controls) and a master mix of packaging plasmids (pVSV-G, pPACK-HI-GAG and Rev-pPACK-HI-REV) using lipofectamine and Opti-MEM (Gibco). Supernatant containing lentivirus was collected 48 h later, filtered through a 0.2 µm filter (EMD Millipore) and aliquots stored at −80⁰C for transduction of tumor cells. A Lenti-X™ p24 Rapid Titer Kit (Clontech) was used to determine lentiviral titers of collected supernatants. All tumor cells were maintained in RPMI Medium 1640 – GlutaMAX™-I (Gibco) supplemented with 10% FBS, at 37⁰C and 5% CO2. PC-9, NCI-H1975 and NCI-H3255 EGFRm tumor cells were grown to 80% confluence and were retrovirally transduced using lentiviral supernatants at a multiplicity of infection (MOI) of 1 and 10, based on p24 ELISA results, in the presence of polybrene for 48 h. Positively expressing cells were selected for using hygromycin B (Invitrogen) for 1 week, cells were then maintained in hygromycin (30 µg/ml for PC-9 and NCI-H3255; 60 µg/ml for NCI-H1975), passaged and bulked up for all experiments. Anti-CD3 transgene expression was confirmed in cells transduced at a MOI of 1 and 10 by flow cytometry staining and all future experiments were conducted with cells transduced with an MOI of 1.
Sorting of anti-CD3 expressing tumor cell lines
Expression of CD14 membrane-bound anti-CD3 OKT3 scFV on transduced cells was detected using either an Alexa Fluor® 647 AffiniPure Fab Fragment Goat anti-Mouse IgG (H + L) antibody (Jackson Immunoresearch) or human CD14 PE-conjugated antibody (R&D Systems). For sorting of transduced cells, PC-9 NCI-H1975 and NCI-H3255 anti-CD3-expressing cell lines were stained with human CD14 PE-conjugated antibody (R&D Systems) and parental (non-anti-CD3 expressing) cells were used as negative controls. PC-9, NCI-H3255 and NCI-H1975 high expressing CD14 cells, with overlapping expression levels, were gated for and sorted on a FACS Aria sorter. Sorted cells were cultured in the presence of hygromycin as before, passaged and bulked up for all experiments.
CD8+ T cell isolation, activation, and expansion
PBMCs were isolated from blood from healthy volunteer donors (National Health Service (NHS) blood and transplant unit at Addenbrooke’s hospital) using density centrifugation with Ficoll-Paque Plus (GE Healthcare). CD8+ T cells were purified from PBMC using the EasySep™ Human CD8+ T Cell Enrichment Kit (Stem Cell Technologies) and a RoboSep (Stem Cell Technologies), according to the manufacturer’s instructions. Isolated CD8+ T cells were activated with CD3/CD28 dynabeads (Thermo Scientific) at a 1:1 ratio, cultured in complete culture assay medium (DMEM- GlutaMAX™-I, 10% FBS) supplemented with 1% penicillin-streptomycin and 100 U/ml IL-2 (Peprotech) and maintained at a density of 1 × 106 cells/ml, with medium changed every 2–3 days. On day 10, CD8+ T cells were re-stimulated with CD3/CD28 dynabeads as above with medium changes every 2–3 days.
CD8+ T cell cytotoxic assay
One day prior to co-culture assays (day 14 of CD8+ T cell activation), P815, PC-9, NCI-H1975, and NCI-H3255 tumor cells were labeled with Vybrant DiO Cell-Labelling Solution (Thermo Fisher Scientific) at a 1:200 dilution for 8 min, according to the manufacturer’s instructions. Cells were washed, resuspended at 2 × 105 cells/ml in culture assay medium and 50 µl/well (10,000 cells) plated in 96 well-U-bottom plates. Plates were centrifuged and incubated overnight at 37°C and 5% CO2. On day 15 of T-cell activation, CD8+ T cells were harvested, dynabeads removed magnetically and cells were resuspended at 1 × 106 cells/ml in culture assay medium. For all co-cultures, CD8+ T cells were added to respective wells of the U-bottom plate containing DiO-labelled tumor cells at 1:1, 2:1, 5:1 and 10:1 effector: target (E: T) ratios. Plates were centrifuged and incubated for either 4 or 18 h. After incubation, supernatants were removed and stored at −80⁰C, wells were washed with PBS and cells were detached with accutase and returned to the incubator for 10 min. Cells were then transferred to a 96-well V-bottom plate and centrifuged. Harvested cells were stained with a LIVE/DEAD Fixable Violet Dead Cell Stain Kit (Thermo Scientific) at a dilution of 1:2500 in PBS for 10 min at room temperature. Cells were then washed, fixed in 3.7% formaldehyde and data acquisition carried out using a BD FACS Canto II or BD LSR Fortessa to analyze Violet+ DiO – (live tumor cells). In some experiments, P815 mouse mastocytoma cells were used as controls to control for T cell-mediated cytotoxicity capacity. P815 cells were DiO labeled and plated as was done for tumor cells and were pre-treated with 0.5 µg/ml anti-CD3 (OKT3 clone) for 1 min (eBioscience) or an isotype-matched control, before the addition of CD8+ T cells at their respective ratios. Where indicated, staining with an Alexa Fluor® 647 AffiniPure Fab Fragment Goat Anti-Mouse IgG (H + L) antibody was also done in experiments to gate on cells with similar anti-CD3 (OKT3) scFV expression levelsCitation9 (Jackson ImmunoResearch). FITC Annexin-V Apoptosis Detection Kit I (BD Biosciences) was used in experiments to analyze apoptotic cell death in co-cultures. For co-culture experiments in the presence of EGFR tyrosine kinase inhibitors (TKIs), anti-CD3 tumor cells were pre-treated with 320 nM Gefitinib and 160 nM Osimertinib after being DiO-labelled, 24 h before the addition of CD8+ T cells for 18 h. SCH58261 was added at a final concentration of 10 µM at the same time as the addition of the CD8+ T cells, in the presence of 100 µM adenosine monophosphate (AMP) (Sigma Aldrich). Experiments using dasatinib (AstraZeneca in-house supply) were similarly carried out using a 12.5 nM concentration (as described byCitation17) added at the same time as the CD8+ T cells. Dasatinib dose-response experiments were similarly repeated using concentrations indicated. All drug concentrations were maintained throughout the 18h co-cultures. Flow cytometric analysis was performed using a BD Fortessa. Data were analyzed using FlowJo (Tree Star) as shown in Figure S1B.
T cell phenotyping
T cells were phenotyped by flow cytometry staining before (Day 0 – naïve and Day 10–10 days after 1st round of stimulation) and after restimulation (Day 15), as well as in co-cultures with tumor cells. Briefly, CD8+ T cells were stained with LIVE/DEAD™ Fixable Blue Dead Cell Stain Kit (Thermo Scientific), followed by surface staining at 4 ⁰C with: – anti-CD8 AF647, anti-CD8 PerCPCy5.5, anti-CD45RO Alexa Fluor 488, anti-CD45RA BV605, anti-CCR7-PE, anti-PD-1 PerCpCy5.5 (all from Biolegend) antibodies. Staining of CCR7 was performed at 37 ⁰C for 30 min. Intracellular granzyme B staining was performed with anti-Granzyme B-AF647 antibody (Biolegend), after permeabilization with Fixation/Permeabilization Solution Kit (BD Biosciences). Flow cytometric analysis was performed using a BD Fortessa. Data were analyzed using FlowJo (Tree Star).
IFN-ɣ measurement
Supernatants from co-culture assays were stored at −80⁰C, prior to analysis for IFN-ɣ production using the Human IFN-ɣ DuoSet ELISA (R&D systems), according to the manufacturer’s instructions. Plates were washed using a BioTek EL406 microplate washer dispenser and read using an Envision L plate reader (PerkinElmer).
Statistics
Two-way ANOVA and t-test were used to assess significance using PRISM software. The P values are represented as ****P < 0.0001 ***P < 0.001, **P < 0.01, *P < 0.05. The data are presented as mean ± standard error of the mean (SEM) or mean ± standard (SD) where indicated.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplemental Material
Download MS Word (574.7 KB)SUPPLEMENTAL DATA
Supplemental data for this article can be accessed at publisher’s website.
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
References
- Yang Y. Cancer immunotherapy: harnessing the immune system to battle cancer. J Clin Invest. 2015;125:3335–3337. doi:10.1172/JCI83871.
- Kvistborg P, Yewdell JW. Enhancing responses to cancer immunotherapy. Science. 2018;359:516–517. doi:10.1126/science.aar6574.
- Apetoh L, Smyth MJ, Drake CG, Abastado JP, Apte RN, Ayyoub M, Starmann J, Tjwa M, Plate KH, Sültmann H, et al. Consensus nomenclature for CD8(+) T cell phenotypes in cancer. Oncoimmunology. 2015;4:e998538. doi:10.1080/2162402X.2015.1008371.
- Barry M, Bleackley RC. Cytotoxic T lymphocytes: all roads lead to death. Nat Rev Immunol. 2002;2:401–409. doi:10.1038/nri819.
- Al Qudaihi G, Lehe C, Negash M, Al-Alwan M, Ghebeh H, Mohamed SY, Saleh A-JM, Al-Humaidan H, Tbakhi A, Dickinson A, et al. Enhancement of lytic activity of leukemic cells by CD8+ cytotoxic T lymphocytes generated against a WT1 peptide analogue. Leuk Lymphoma. 2009;50:260–269. doi:10.1080/10428190802578478.
- Gourdain P, Boucau J, Kourjian G, Lai NY, Duong E, Le Gall S. A real-time killing assay to follow viral epitope presentation to CD8 T cells. J Immunol Methods. 2013;398–399:60–67. doi:10.1016/j.jim.2013.09.009.
- Zaritskaya L, Shurin MR, Sayers TJ, Malyguine AM. New flow cytometric assays for monitoring cell-mediated cytotoxicity. Expert Rev Vaccines. 2010;9:601–616. doi:10.1586/erv.10.49.
- Emerson RO, Mathew JM, Konieczna IM, Robins HS, Leventhal JR. Defining the alloreactive T cell repertoire using high-throughput sequencing of mixed lymphocyte reaction culture. PLoS One. 2014;9:e111943. doi:10.1371/journal.pone.0111943.
- Leitner J, Kuschei W, Grabmeier-Pfistershammer K, Woitek R, Kriehuber E, Majdic O, Zlabinger G, Pickl WF, Steinberger P. T cell stimulator cells, an efficient and versatile cellular system to assess the role of costimulatory ligands in the activation of human T cells. J Immunol Methods. 2010;362:131–141. doi:10.1016/j.jim.2010.09.020.
- Martinez-Lostao L, Anel A, Pardo J. How do cytotoxic lymphocytes kill cancer cells? Clin Cancer Res. 2015;21:5047–5056. doi:10.1158/1078-0432.CCR-15-0685.
- Karachaliou N, Gonzalez-Cao M, Sosa A, Berenguer J, Bracht JWP, Ito M, Rosell R. The combination of checkpoint immunotherapy and targeted therapy in cancer. Ann Transl Med. 2017;5:388. doi:10.21037/atm.
- Park MY, Jung MH, Eo EY, Kim S, Lee SH, Lee YJ, Park JS, Cho YJ, Chung JH, Kim CH, et al. Generation of lung cancer cell lines harboring EGFR T790M mutation by CRISPR/Cas9-mediated genome editing. Oncotarget. 2017;8:36331–36338. doi:10.18632/oncotarget.16752.
- Hamamoto J, Yasuda H, Aizawa K, Nishino M, Nukaga S, Hirano T, Kawada I, Naoki K, Betsuyaku T, Soejima K. Non-small cell lung cancer PC-9 cells exhibit increased sensitivity to gemcitabine and vinorelbine upon acquiring resistance to EGFR-tyrosine kinase inhibitors. Oncol Lett. 2017;14:3559–3565. doi:10.3892/ol.2017.6591.
- Cross DA, Ashton SE, Ghiorghiu S, Eberlein C, Nebhan CA, Spitzler PJ, Orme JP, Finlay MRV, Ward RA, Mellor MJ, et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 2014;4:1046–1061. doi:10.1158/2159-8290.CD-14-0337.
- Arab S, Kheshtchin N, Ajami M, Ashurpoor M, Safvati A, Namdar A, Mirzaei R, Mousavi Niri N, Jadidi-Niaragh F, Ghahremani MH, et al. Increased efficacy of a dendritic cell-based therapeutic cancer vaccine with adenosine receptor antagonist and CD73 inhibitor. Tumour Biol. 2017;39:1010428317695021. doi:10.1177/1010428317695021.
- Leone RD, Lo YC, Powell JD. A2aR antagonists: next generation checkpoint blockade for cancer immunotherapy. Comput Struct Biotechnol J. 2015;13:265–272. doi:10.1016/j.csbj.2015.03.008.
- Lee KC, Ouwehand I, Giannini AL, Thomas NS, Dibb NJ, Bijlmakers MJ. Lck is a key target of imatinib and dasatinib in T-cell activation. Leukemia. 2010;24:896–900. doi:10.1038/leu.2010.11.
- Das J, Chen P, Norris D, Padmanabha R, Lin J, Moquin RV, Shen Z, Cook LS, Doweyko AM, Pitt S, et al. 2-aminothiazole as a novel kinase inhibitor template. Structure-activity relationship studies toward the discovery of N-(2-chloro-6-methylphenyl)-2-[[6-[4-(2-hydroxyethyl)-1- piperazinyl)]-2-methyl-4-pyrimidinyl]amino)]-1,3-thiazole-5-carboxamide (dasatinib, BMS-354825) as a potent pan-Src kinase inhibitor. J Med Chem. 2006;49:6819–6832. doi:10.1021/jm060727j.
- Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol. 2013;13:227–242. doi:10.1038/nri3405.
- Jedema I, van der Werff NM, Barge RMY, Willemze R, Falkenburg JH. New CFSE-based assay to determine susceptibility to lysis by cytotoxic T cells of leukemic precursor cells within a heterogeneous target cell population. Blood. 2004;103:2677–2682. doi:10.1182/blood-2003-06-2070.
- Brunner KT, Mauel J, Cerottini JC, Chapuis B. Quantitative assay of the lytic action of immune lymphoid cells on 51-Cr-labelled allogeneic target cells in vitro; inhibition by isoantibody and by drugs. Immunology. 1968;14:181–196.
- Batchelor RH, Zhou M. Use of cellular glucose-6-phosphate dehydrogenase for cell quantitation: applications in cytotoxicity and apoptosis assays. Anal Biochem. 2004;329:35–42. doi:10.1016/j.ab.2004.02.007.
- Miyahira Y, Murata K, Rodriguez D, Rodriguez JR, Esteban M, Rodrigues MM, Zavala F. Quantification of antigen specific CD8+ T cells using an ELISPOT assay. J Immunol Methods. 1995;181:45–54.
- Derdak SV, Kueng HJ, Leb VM, Neunkirchner A, Schmetterer KG, Bielek E, Majdic O, Knapp W, Seed B, Pickl WF. Direct stimulation of T lymphocytes by immunosomes: virus-like particles decorated with T cell receptor/CD3 ligands plus costimulatory molecules. Proc Natl Acad Sci U S A. 2006;103:13144–13149. doi:10.1073/pnas.0602283103.
- Ekong UD, Miller SD, O‘Gorman MR. In vitro assays of allosensitization. Pediatr Transplant. 2009;13:25–34. doi:10.1111/j.1399-3046.2008.01042.x.
- Isaaz S, Baetz K, Olsen K, Podack E, Griffiths GM. Serial killing by cytotoxic T lymphocytes: T cell receptor triggers degranulation, re-filling of the lytic granules and secretion of lytic proteins via a non-granule pathway. Eur J Immunol. 1995;25:1071–1079. doi:10.1002/eji.1830250432.
- Jin HT, Ahmed R, Okazaki T. Role of PD-1 in regulating T-cell immunity. Curr Top Microbiol Immunol. 2011;350:17–37. doi:10.1007/82_2010_116.
- Simon S, Labarriere N. PD-1 expression on tumor-specific T cells: friend or foe for immunotherapy? Oncoimmunology. 2017;7:e1364828. doi:10.1080/2162402X.2017.1364828.
- Huang J, Zarnitsyna VI, Liu B, Edwards LJ, Jiang N, Evavold BD, Zhu C. The kinetics of two-dimensional TCR and pMHC interactions determine T-cell responsiveness. Nature. 2010;464(7290):932–936. doi:10.1038/nature08944.
- Fooksman DR, Grönvall GK, Tang Q, Edidin M. Clustering class I MHC modulates sensitivity of T cell recognition. J Immunol. 2006;176:6673–6680.
- Vanneman M, Dranoff G. Combining immunotherapy and targeted therapies in cancer treatment. Nat Rev Cancer. 2012;12:237–251. doi:10.1038/nrc3237.
- Ohta A, Ohta A, Madasu M, Kini R, Subramanian M, Goel N, Sitkovsky M. A2A adenosine receptor may allow expansion of T cells lacking effector functions in extracellular adenosine-rich microenvironments. J Immunol. 2009;183:5487–5493. doi:10.4049/jimmunol.0901247.