
ABSTRACT
Immunotherapies such as adoptive cell therapy (ACT) are promising treatments for solid cancers. However, relapsing disease remains a problem and the molecular mechanisms underlying resistance are poorly defined. We postulated that the deregulated epigenetic landscape in cancer cells could underpin the acquisition of resistance to immunotherapy. To address this question, two preclinical models of ACT were employed to study transcriptional and epigenetic regulatory processes within ACT-treated cancer cells. In these models ACT consistently causes robust tumor regression, but resistance develops and tumors relapse. We identified down-regulated expression of immunogenic antigens at the mRNA level correlated with escape from immune control. To determine whether this down-regulation was under epigenetic control, we treated escaped tumor cells with DNA demethylating agents, azacytidine (AZA) and decitabine (DEC). AZA or DEC treatment restored antigen expression in a proportion of the tumor population. To explore the importance of other epigenetic modifications we isolated tumor cells refractory to DNA demethylation and screened clones against a panel of 19 different epigenetic modifying agents (EMAs). The library of EMAs included inhibitors of a range of chromosomal and transcription regulatory protein complexes, however, when tested as single agents none restored further antigen expression. These findings suggest that tumor cells employ multiple epigenetic and genetic mechanisms to evade immune control, and a combinatorial approach employing several EMAs targeting transcription and genome stability may be required to overcome tumor resistance to immunotherapy.
Introduction
T cell-based immunotherapies such as adoptive cell therapy (ACT) aim to boost tumor-specific immune responses and are promising approaches for the treatment of solid cancers such as metastatic melanoma.Citation1–Citation3 ACT involves isolating tumor-specific cytotoxic T lymphocytes (CTLs) from tumor biopsies, expanding them ex vivo, and re-infusing large numbers of these cells back into patients.Citation4 ACT studies in melanoma have demonstrated durable complete regressions in patients with promising objective response rates.Citation5–Citation7 However, many patients do not respond to treatment and overall complete response rates remain relatively low. The development of tumoral resistance to immunotherapy remains a major obstacle to successful treatment.Citation8–Citation10 Placing tumor cells under heightened immune pressure leads to immunoediting and selection of immune escape variants.Citation11,Citation12 The loss of immunogenic antigens or components of the cellular antigen processing and presentation pathways is commonly observed in escaped tumours.Citation13–Citation16 However, the molecular mechanisms underlying the evolution of resistance remain unclear.
Epigenetic modifications, such as DNA methylation or post-translational modifications to histones tails, are associated with genomic patterns of DNA accessibility, chromatin compaction and transcriptional activity. The protein machinery that adds, removes or recognizes these covalent modifications include ‘writers’ (e.g. histone acetyltransferases/methyltransferases, DNA methyltransferases), ‘erasers’ (e.g. histone deacetylases/demethylases, Ten-eleven Translocation enzymes) and ‘readers’ (e.g. bromodomain, chromodomain proteins), respectively.Citation17 Cancer cells often display aberrant epigenetic modifications promoting chromosomal instability, loss of genomic imprinting and de-regulated gene expression.Citation18 DNA hypermethylation of specific gene promotersCitation19,Citation20 can suppress the expression of tumor suppressor genesCitation21–Citation23 pro-apoptotic genesCitation24–Citation26 and antigen presentation pathways.Citation27–Citation29 Therefore, epigenetic modifications underlie important tumor resistance mechanisms making them attractive targets for cancer therapy.
The most clinically advanced epigenetic modifying agents (EMAs) are the cytosine analogues, azacytidine (AZA) and decitabine (DEC),Citation30 which are currently used for the treatment of myelodysplastic syndrome and acute myeloid leukemia.Citation31 They function as DNA methyltransferase inhibitors (DNMTis), causing hypomethylation of DNA. Treatment of tumor cells with DNMTis can re-activate hypermethylated tumor suppressor genes, increase tumor cell immunogenicity by re-activating cancer testis and differentiation antigens,Citation32–Citation36 and increase antigen presentation and MHC class I expression.Citation37 Recent studies suggest that DNMTis can induce a sustained interferon response in cancer cells by activating retroviral encoded double-stranded RNA.Citation37,Citation38 The ability of EMAs to increase tumor immunogenicity raises the possibility that the combined application of epigenetic modifiers and immunotherapies such as checkpoint blockade and ACTCitation39,Citation40 may be an effective treatment option. However, to date, clinical support for the utility of this approach remains limited.Citation41,Citation42 We hypothesize that under heightened immune pressure, tumor cells develop resistance to immunotherapy via epigenetic modification, and this resistance might be reversed by treatment with EMAs such as AZA and DEC.
We utilized two preclinical models of ACT to investigate the molecular mechanisms of resistance to ACT. Both tumor models initially respond to a single round of ACT before escaping from immune control. We analyzed the molecular features of ACT-resistant clones and screened them with a library of EMAs for antigen-reactivation. Together these studies revealed prominent transcriptional silencing of immunogenic antigens and a role for genomic instability. We conclude these are key molecular evolutionary mechanisms operative during immunotherapy, leading to the development of resistance in these models.
Results
Tumors escape from immune control after initially responding to treatment with adoptive transfer of tumor-specific CTLs
ACT using tumor-specific CTLs is a promising immunotherapy applicable to many solid tumors. To better understand the mechanisms controlling anti-tumor immunity in this setting, two preclinical models of ACT were established. B16 melanomas, engineered to express the model antigen OVA and GFP (B16.OVA), were inoculated onto C57Bl6 mice. Similarly, AB1 mesotheliomas engineered with the model antigen HA and antibiotic resistance to Geneticin (AB1.HA) were inoculated into BALB/c mice. Importantly, both tumor cell lines express their respective target antigens constitutively. ACT using in vitro activated OVA-specific (OT.I) or HA-specific (CL4) T cells was administered with total body irradiation (TBI) once tumors were well established (D8–10).
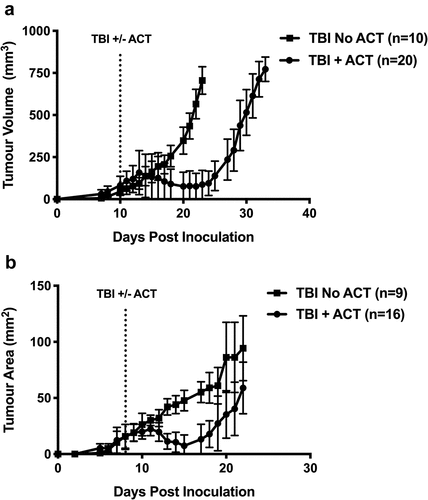
In both models, all tumors responded to ACT within 4 days ()). Although ACT reduced tumor burden considerably, no complete responses were observed. Tumour regression plateaued 5–8 days after ACT and tumor growth was held in check for a further 3–5 days ()). From this point onwards, tumors re-established growth rates similar to untreated controls. Treatment of B16.OVA tumors led to a greater delay in tumor outgrowth than was observed with AB1.HA tumors, but this did not result in increased survival. Thus, our models recapitulate the cycle of regression, remission and relapse often associated with ACT in the clinic, supporting their use to study the development of resistance mechanisms and tumor immune evasion in this setting.
Figure 1. ACT with activated CTLs induces tumor regression but fails to eliminate tumors. (a) B16.OVA.GFP tumor growth curves by tumor volume (mm3) in C57Bl6 mice with (circles) or without (squares) transfer of activated OT.I T cells. (b) Growth curves by tumor area (mm2) of AB1.HA tumors in BALB/c mice with (circles) or without (squares) transfer of activated CL4 T cells. Points represent the mean and error bars show standard deviation. Dotted lines represent treatment time points. All mice received 550 rads TBI. Data are pooled from three independent experiments for B16.OVA.GFP and AB1.HA tumors. Error bars represent the mean ± the standard deviation.
Tumor-infiltrating lymphocytes (TILs) exhibit reduced effector function in escaped B16 melanomas
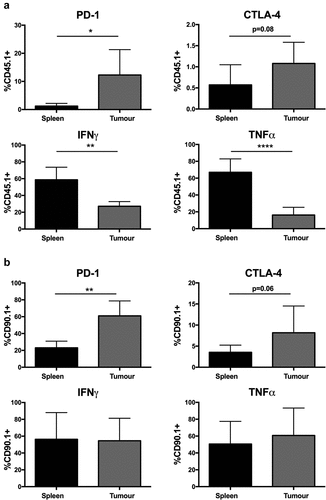
Immunosuppression of TILs within the tumor microenvironment may contribute to tumor escape from immune control. To determine whether transferred OT.I and CL4 T cells were suppressed within escaped tumors their expression of inhibitory receptors and production of effector cytokines were examined. A higher proportion of OT.I TILs in escaped tumors expressed PD-1 compared with those in the spleen (12.30% vs. 1.22%, p < .05) and fewer OT.I TILs were able to produce pro-inflammatory cytokines IFNγ (27.16% vs. 58.81%, p < .001) and TNFα (16.21% vs. 66.94%, p < .0001) upon re-stimulation ()). Similarly, an increased proportion of CL4 TILs in escaped tumors expressed PD-1 (60.98% vs. 23.03%, p < .001) ()). However, the percentages of IFNγ and TNFα producing CL4 TILs were similar to those in the spleen.
Figure 2. ACT T cells exhibit reduced effector function in escaping melanoma. The frequency of PD-1 or CTLA-4 expression on transferred T cells (CD8+CD45.1+/CD90.1+) in the spleens and tumors of C57Bl6 (a) or BALB/c (b) mice. Frequency of IFNγ and TNFα producing transferred T cells, upon ex vivo re-stimulation, in the spleens and tumors of C57Bl6 (a) or BALB/c (b) mice. All samples were analyzed 14 days after treatment with ACT. Error bars represent the mean ± SD. Data show pooled animals from two independent experiments (total animal number (a) n = 7 and (b) and n = 8). Samples from spleen and tumor were compared using a paired t-test * = p < .05, ** = p < .01, **** = p < .0001.
Immune pressure drives immunoediting of tumor cells and silencing of immunogenic antigens
To determine if the observed suppression of TILs in B16.OVA tumors was driving tumor escape, the efficacy of a second round of ACT was examined. A second transfer of freshly activated OT.I T cells was commenced once tumor escape became detectable (D24) and the impact on tumor growth was measured. B16.OVA tumors that received the second round of ACT outgrew at a similar rate to those receiving only a single treatment, indicating that activated OT.I cells were no longer effective against escaped tumors ()).
Figure 3. Tumors develop resistance to ACT by loss of immunogenic antigens. (a) B16.OVA.GFP growth curves by tumor volume (mm3) in C57Bl6 mice administered single (circles) or dual (squares) ACT. Dotted lines represent treatment time points. All mice receive treatment at the first timepoint. Data are pooled from two independent experiments and n = 10 for both groups. Error bars represent the mean ± the standard deviation. (b) GFP expression in B16.OVA tumor cells from mice with or without prior ACT was measured at D34 post inoculation. The B16.F10 parental cell line is shown as a control (grey shaded). Histograms are representative of three independent experiments (n = 3 for each experiment). (c) The viability of AB1-HA tumor explants arising from mice which received a single round of ACT were compared to explants from mice that did not receive ACT, after 2 or 7 days of culture in the presence of G418. Data pooled from two independent experiments and n = 12 for each group. ****p < .0001.
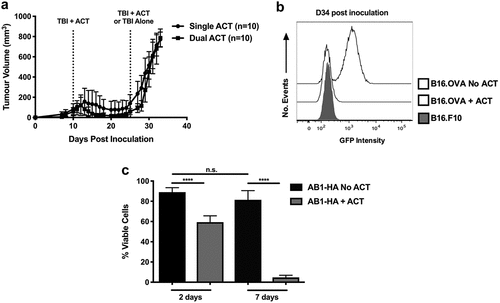
To determine whether OT.I T cells could survey escaped tumors, the expression of the target OVA antigen was analyzed in treated and untreated tumors directly ex vivo at D34 post inoculation. In B16.OVA cell lines, OVA and GFP are expressed under the control of the same promoter, allowing GFP expression to be used as a surrogate for OVA. Untreated tumors retained GFP expression at levels comparable to that of the in vitro B16.OVA cell line (MFI: 1047). After immune escape, ACT-treated tumors down-regulated GFP to a similar intensity as the GFPneg parental cell line (MFI: 199.6 vs. 246.3 p = .12) (); Supp. Figure 1). These data suggest that ACT drives the loss of OVA antigen expression by tumor cells as a mechanism of immune evasion.
In the AB1.HA model, expression of HA and the Geneticin resistance cassette is driven by the same promoter. To determine if AB1.HA tumors exhibited a similar loss of target antigen, ACT-treated and untreated tumors were harvested, and explant lines established. Tumor explants were cultured for 2 to 7 days in the presence of Geneticin and their viability was compared. After 2 days, ACT-treated explants showed significantly decreased viability compared to untreated explants (59.7% vs. 89.0% p < .0001). On day 7, this difference was further increased with less than 5% of ACT-treated tumor cells remaining viable (5.0% vs. 81.5% p < .0001). The viability of untreated explants did not decrease significantly during the culture period (89.0% vs. 81.5% p = .08). Together, these data demonstrate that loss of immunogenic antigen expression correlates with evasion of immune-mediated destruction in both models of ACT, suggesting this is an important evolutionary mechanism of resistance.
Expression of immunogenic antigens is suppressed at the level of transcription
We next investigated whether the loss of target antigens during ACT was coincident with the deletion of transgene sequences. Genomic DNA extracted from three ACT-treated B16.OVA and AB1.HA explants, each arising from different tumors, was used in PCR reactions with OVA or HA specific primers. B16.F10 or AB1 parental cell lines were used as negative controls (Lane 1). OVA or HA positive cell lines and untreated B16.OVA or AB1.HA explants were used as positive controls (Lanes 2–3). The OVA gene was detectable in all ACT-treated B16.OVA explants (Lanes 4–6) as well as the untreated explant. Interestingly, one explant (Lane 6) displayed a much lower amplification of OVA, suggesting that this tumor population had undergone some reduction in OVA gene copy number ()). Similarly, the HA gene was still detectable in all AB1.HA explants ()). However, in this case, all ACT-treated explants displayed lower amplification of HA compared to the untreated explant and positive control cell line.
Figure 4. Transcriptional silencing of immunogenic antigens in ACT resistant tumors. Escaped B16.OVA.GFP or AB1.HA tumors were harvested ~14 days after ACT and tumor explant cell lines were generated. DNA was extracted to use as a template for PCR (a, b). RNA was extracted and used to synthesize cDNA for a template in RT-PCR (c, d). DNA and RNA expression of OVA (a, c) and HA (b, d) from three tumor explants lines from ACT treated mice (ACT 1, 2, 3), alongside relevant control cell lines. All reactions were run in parallel with control reactions using the reference gene GAPDH.
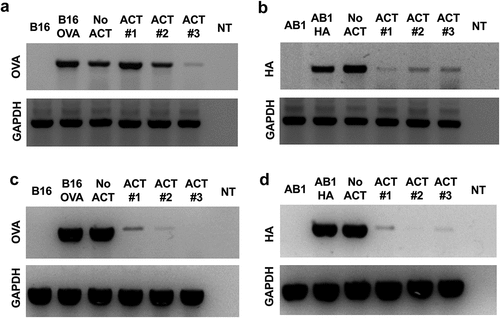
While some ACT-treated explants displayed lower OVA and HA gene copy number, no explant exhibited complete loss of either gene. Therefore, other mechanisms, besides gene deletion, must contribute to the silencing of remaining antigen-positive cells. RT-PCR analysis was used to determine if OVA and HA genes were being successfully transcribed. Reactions using cDNA generated from untreated explants and in vitro cell lines showed strong bands with either OVA or HA specific primers (Lanes 2–3). However, reactions using cDNA from ACT-treated tumor explants (Lanes 4–6) showed no or greatly reduced OVA ()) and HA ()) transcript levels. Therefore, both genetic loss and transcriptional repression appear to contribute to the down-regulation of OVA and HA expression in ACT-treated tumors.
DNA methyltransferase inhibitors azacytidine and decitabine restore antigen expression in a proportion of edited tumor cells
DNA methylation is an epigenetic modification associated with silencing of gene expression in some contexts. To determine if DNA methylation is involved in silencing of OVA antigen expression, ACT-treated tumour explants were exposed to the DNMTis, AZA and DEC. Explants were cultured in media containing either DNA hypomethylating agent at two concentrations (1 μM or 5 μM) for 72 h at which point GFP re-expression was observed in a subset of tumor cells ()). The response was dose-dependent with DEC treated cells showing greater GFP re-expression compared to AZA at the same concentration (1 μM: 6.7% vs. 0.5% p < .001; 5 μM: 8.8% vs. 4.5% p < .05) ()). Treatment with 1 μM of AZA had little effect on GFP re-expression in any of the three tumor explants, while DEC at 5 μM led to the greatest re-expression, but also affected cell viability (data not shown). Interestingly, the three ACT-treated tumor explants displayed different responsiveness to treatment with DNMTis. This observation remained consistent for both AZA and DEC across the two different concentrations.
Figure 5. Azacytidine and decitabine treatment restores antigen expression to a proportion of escaped tumor cells. (a) AB16.OVA.GFP tumor explants from ACT-treated mice were cultured in the presence of 5 uM AZA or 1 uM DEC for 72 h alongside matched untreated (vehicle) controls and GFP expression was measured by flow cytometry. Data are representative dot plots showing GFP expression in the untreated or treated tumor explant. (b) GFP expression in B16.F10 control or ACT treated (ACT1–3) explant lines after 72 h of treatment with AZA or DEC at 1 uM or 5 uM concentrations. Data are pooled from two independent experiments and n = 4 per group. Error bars represent the mean ± SD, * = p < .05, ** = p < .01, compared to untreated control cells arising from the same explant or cell line. (c) The percentage of tumor cells remaining GFP+ 2–3 weeks after treatment for 72 h with 5 uM AZA or 1 uM DEC. At least 20 individually sorted GFP+ clones giving rise to colonies were measured for each ACT-treated explant. Dots represent individual tumor colonies.
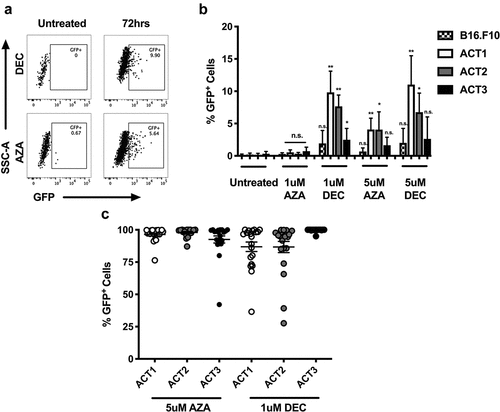
To determine if the observed re-expression of GFP after DNMTi treatment was stably maintained, GFPpos cells were single cell sorted after 72 h of treatment with AZA or DEC. Colonies arising from single GFPpos clones were expanded and their GFP expression monitored. Colonies were cultured over a period of 2–3 weeks allowing sufficient time for multiple cell divisions necessary to assess the heritability of transgene activity in daughter cells. Surprisingly, after this period >90% of sorted AZA-treated colonies and >75% of sorted DEC treated colonies remained positive for GFP (>90% of cells GFPhi), while other colonies showed a mix of GFPpos and GFPneg cells ()). These data suggest that the epigenetic effects of treatment with AZA or DEC on gene re-expression are stably maintained within tumor cells across multiple generations.
Tumor cells unresponsive to AZA or DEC are refractory to treatment with other EMAs
DNA methylation is one of the many epigenetic mechanisms that contribute to the regulation of chromosomal integrity and gene transcription. In humans, this epigenetic machinery is encoded by over 600 genes that generate a network of regulatory proteins comprising epigenetic writers, readers and erasers, some of which can be targeted by (EMAs). We therefore assessed the role of additional epigenetic mechanisms in the silencing of immunogenic antigens. Tumor cells unresponsive to treatment with AZA or DEC were sorted based on their GFPneg expression and then screened against an extensive panel of 19 EMAs that target a range of regulatory protein complexes (). Concentrations for dosing were determined from IC50 assays (Supp. Figure 2), such that cells were treated at concentrations below their IC50, up to a maximum dose of 10 μM. However, at these concentrations, none of the drugs tested as single agents successfully reinstated GFP expression in tumor cells already resistant to treatment with AZA or DEC (Supp. Figure 3).
Table 1. Tumor cells unresponsive to treatment with AZA or DEC were treated with a range of EMAs that target different epigenetic mechanisms at stated concentrations for 3, 7 and 14 days
Silencing of immunogenic antigens during ACT does not correlate with transgene deletion
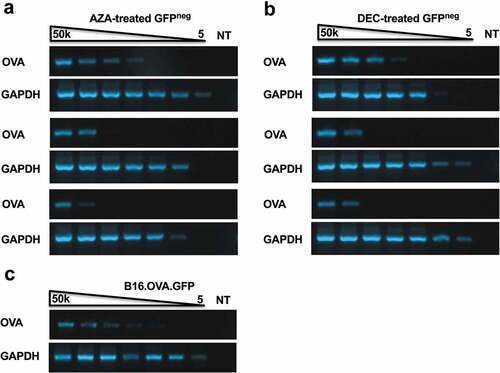
It remained possible that tumor explants refractory to further epigenetic therapy after treatment with AZA and DEC were selectively derived from OVAneg clones that had completely lost the OVA transgene. To investigate the OVA gene copy number of clonal populations, a 5-fold dilution series of genomic DNA was prepared and used as a template in PCR reactions with OVA-specific primers. The limit of detection for the OVA gene was compared between AZA or DEC resistant tumor cells (GFPneg post treatment) ()) and the in vitro B16.OVA cell line (100% GFP+) ()). Interestingly, clear differences were observed in the OVA gene copy number in the genomic DNA of certain AZA or DEC resistant tumor explants. One AZA-treated explant maintained a comparable OVA gene copy number to the control cell line while the remaining two explants displayed at least a 25-fold decrease in OVA DNA levels ()). Similarly, one DEC-treated explant showed similar levels of OVA gene copy number to the control cell line while the other two explants displayed a similar reduction in OVA copy number ()). Importantly, these results identify one explant line in each of the DNMTi treated groups with a normal level of OVA DNA, demonstrating that OVAneg tumors can maintain cells with normal transgene levels in a transcriptionally silenced state.
Figure 6. Resistant tumor cells display lower levels of OVA transgene copy number. B16.OVA tumor explants were treated with 5 uM AZA (a) or 1 uM DEC (b) for 72 h and GFPneg populations and were sorted. 1 × 106 GFPneg cells were used for genomic DNA extraction. Limit of detection for OVA or GAPDH in AZA-treated (a) or DEC treated (b) tumor explants lines from ACT treated mice (ACT 1, 2, 3) or untreated B16.OVA cells (c). Lanes 1–7 contain DNA titrations 50,000, 10,000, 2000, 400, 80, 16 and 3.2 cells/reaction. Lane 8 contains the no-template control.
Discussion
This study reports the development of acquired resistance to CTL-mediated killing in two pre-clinical models of ACT. In both models, ACT causes tumor regression; however, relapse is inevitable after a short period of remission. Additional rounds of ACT failed to control tumor outgrowth, suggesting that cell intrinsic mechanisms mediate loss of immunogenic antigens and development of resistance. The tumor models used to study the acquired resistance reported here are based on well-established models previously used to interrogate interactions between tumor antigens and immune cells.Citation43,Citation44 Both systems involve a transgenic expression of a model antigen and reporter cassette in tumor cell lines that are heterogenous with respect to genomic integration sites and transgene copy-number. In AB1.HA mesothelioma cells, antigen transgene expression is driven by the beta-actin promoter with an antibiotic resistance reporter. In contrast, in B16.OVA melanoma cancer cells, OVA transgene expression is driven by a modified Long Terminal Repeat family promoter derived from the Murine Stem Cell Virus with a GFP reporter (Supp. Figure 4). Importantly, despite using different promoter systems, these tumor models show some common molecular features associated with acquired resistance.
Immunosuppression via checkpoint signaling within solid tumors and failure of transferred T cells to persist may contribute to tumor escape. Adoptively transferred CTLs were readily detected in relapsed tumors in both models, and PD-1 expression was increased on these tumor-infiltrating cells. Upregulation of PD-1 on antigen-specific CTLs correlated with decreased ability to produce proinflammatory cytokines in the B16.OVA model, suggesting that loss of effector function had occurred. Importantly, repeated rounds of ACT targeting the same antigens had no effect on tumor growth. We conclude that T cell suppression arises during ACT; however, additional tumor intrinsic mechanisms underlie long-term ACT-resistance in these models. Furthermore, our data suggest that further treatment utilizing tumor-specific T cells will prove ineffective after the development of these immune escape mechanisms, unless resistance can be reversed, or new immunogenic targets expressed on cancer cells. We speculate similar mechanisms may play a role in the evolution of resistance to other immunotherapy modalities such as checkpoint blockade or vaccination.
Cancer cells alter their immunogenicity and evade immunosurveillance using unique mechanisms that are context-dependent.Citation33,Citation45 In ACT-resistant tumors we demonstrate that the transgene sequences encoding immunogenic epitopes are typically not completely lost, but transcription of genes coding for immunogenic antigens is decreased. Previously, it was reported that OVA down-regulation in the B16 melanoma model resulted from complete loss of the OVA gene,Citation46 while in contrast, another group demonstrated down-regulation of OVA antigen in B16 melanomas through an unknown epigenetic mechanism.Citation47 Furthermore, a recent study investigating immune escape in lung cancer patients demonstrated that early-stage untreated tumors are frequently characterized by multiple independent mechanisms of immune evasion including DNA editing, neo-antigen silencing and disruption of antigen.Citation48 Our observations are also consistent with multiple mechanisms operating during the evolution of ACT-resistance.Citation49 We find that immunosuppression of TILs may contribute to immune escape in relapsing tumor cells and this may precede the selection of clones with antigen loss. In resistant AB1.HA mesothelioma cells we observe clones with incomplete clonal loss of transgene sequences coupled with either low level or undetectable transcription. In contrast, we observe examples of resistant B16.OVA clones with normal transgene levels coupled with low-level transcription and an example with incomplete clonal loss of transgene sequences and undetectable transcription. Thus, ablating antigen expression appears to be a major molecular driver of tumor-cell intrinsic resistance arising through genetic deletions within tumor cells (in both AB1.HA and B16.OVA) or transcriptional silencing (B16.OVA). Further studies are required to track how ACT-resistance mechanisms evolve and if tumor cell intrinsic processes are selective for promoter class.
Treatment of tumor cells with DNMTis induces the expression of cancer-testis and differentiation antigens.Citation33,Citation34,Citation50 Since we noted that ACT-resistant B16.OVA tumor explants reproducibly showed normal to low OVA-copy levels but were transcriptionally silenced, we explored the potential to activate antigen expression using small molecule inhibitors of transcriptional repressor complexes and epigenetic regulatory proteins. We demonstrate stable re-expression of antigen in a proportion of ACT-resistant tumor cells after treatment with DNMTis. We took advantage of the GFP-marker to isolate cells that were activated and those that were refractory to transcriptional activation by DNMTis. We noted that restoration of antigen expression persisted long after exposure in activated cells; however, treatment of refractory cells with an extensive panel of EMAs that targeted an array of different epigenetic readers, writers and erasers did not elicit further GFP expression (). These data suggest that transient exposure to DNMTis induces stable activation of antigen gene expression persisting after cell division, however, no small molecule inhibitor, tested as a single agent, was capable of restoring antigen expression in DNMTi refractory cells. Therefore, deciphering the precise molecular processes driving genetic loss or transcriptional silencing of antigen genes may be crucial for rationalizing the combinatorial use of small molecule inhibitors that target epigenetic regulatory proteins and transcriptional regulatory complexes.
Treatment with DNMTis has been reported to synergize with the use of T cell-based immunotherapies through upregulation of MHC class I expression on tumor cells,Citation35,Citation51 which is of particular importance for tumor recognition by transferred CTLs.Citation52 EMAs have also been suggested as a complementary treatment in combination with established immunotherapies such as checkpoint blockade and ACTCitation40,Citation53 based on their ability to increase tumor visibility and immunogenicity.Citation42,Citation54,Citation55 In lung cancer patients immune escape is associated with neo-antigen silencing and promoter hypermethylation during subclone evolution.Citation48 Furthermore, EMAs induce transcriptional activation of endogenous retroviral elements which can rewire global gene expression, for example, by initiating transcription of coding genes from cryptic promoters that contain specific retroviral elements.Citation56 We present data suggesting that treatment with DNMTis may be effective in reinstating the expression of antigens silenced in tumors with acquired resistance to ACT. However, a deeper understanding of the mechanisms that modulate gene expression within tumors will be necessary in order to design appropriate therapies to support the generation of productive and lasting anti-tumor immunity.
Materials and methods
Mice
C57BL6 and BALB/c female mice were purchased from the Animal Resource Centre, Murdoch, Western Australia. OT.I mice on a C57BL/6.SJL-PtprcaPep3b/BoyJ background (CD45.1) were bred and maintained at the Telethon Kids Institute. Balb/c Clone 4 (CL4) TCR-transgenic mice were bred and maintained at the Animal Resource Centre, Murdoch, Western Australia. All animal experiments were performed in accordance with protocols approved by the Telethon Kids Institute Animal Ethics Committee or the University of Western Australia Animal Ethics Committee and conformed to the Australian National Health and Medical Research Council Australia code of practice for the care and use of animals for scientific purposes.
Cell lines and culture
The B16.F10 melanoma cell line was purchased from the ATCC. B16 lines were transduced with retroviral constructs containing the full-length membrane-bound form of OVA and eGFP and maintained in culture as previously described.Citation57 Transduction was confirmed via eGFP expression by flow cytometry. AB1 is a murine mesothelioma cell line developed by the i.p. injection of crocidolite asbestos into BALB/c mice.Citation58 AB1.HA cells are transduced to express the hemagglutinin (HA) gene of influenza virus A/PR/8/34 (H1N1) and were cultured as previously described.Citation59 HA+ cell lines were maintained under antibiotic selection in media containing 400 μg/mL Geneticin (G418). Both cell lines were passaged routinely at 70–80% confluency. Tumor explants were established from ACT-treated and untreated tumors, homogenized under sterile conditions and grown in complete media. For antibiotic resistance experiments, ACT-treated tumor explants were cultured for two or seven days in the presence of 400 μg/mL G418 and cell viability was measured by propidium iodide exclusion versus untreated explants.
T cell activation
C57Bl6 or BALB/c splenocytes were pulsed for 1 h with 1 μM OVA or HA peptide, respectively, and then washed twice and resuspended in RPMI containing no FCS. LPS was added at a final concentration of 3 μg/mL and then cells were added to an equal volume of TCR transgenic (OT.I or CL4) splenocytes and cultured in T175 flasks in a final volume of 40 mL in complete media. On day 2, cells were split 1:2 and 10 U/mL of rIL-2 (Peprotech) was added per flask. This split was repeated on day 3 and cells were collected on day 4, washed twice and resuspended in sterile PBS for adoptive transfer into tumor bearing mice.
Tumour inoculation and ACT
Mice were maintained under anesthesia using isoflurane throughout the tumor inoculation procedure. The right flank of mice was shaved and 5 × 105 tumor cells were inoculated subcutaneously in 50 μL of sterile PBS. Eight to ten days after tumor inoculation mice received 550 rads of total body irradiation (TBI) followed by i.v. transfer of up to 1 × 107 in vitro activated OT.I or CL4 T cells in 300μL sterile PBS. Tumor measurements were recorded with electronic calipers and tumor volume (mm3) or area (mm2) were calculated according to the requirements of the specific ethics applications.
Flow cytometry and sorting
Monoclonal antibodies recognising mouse CD8α APC Clone: 53–6.7, Vα2 APC-Cy7 Clone: B20.1, CD45.1 V450 Clone: A20, CD90.1 V450 Clone: OX-7, CD274 FITC Clone: TY25, CD152 PE Clone: UC10.4F10–11, IFNγ PE Clone: XMG1.2 and TNFα FITC Clone: MP6-XT22 were purchased from BD Bioscience or BioLegend. Multi-parameter analysis was performed using an LSRFortessa (BD). GFP expression in treated tumor explants was monitored using an LSRFortessa (BD) or FACSCalibur II (BD). Prior to the acquisition, cells were stained with propidium iodide (PI; Sigma) to exclude dead cells. Treated tumor explants were sorted for GFP positive and negative populations using a FACSAriaIII (BD) using single cell purity. All data were analyzed with FlowJo (TreeStar).
Intracellular cytokine staining
Tumors and spleens were harvested from mice bearing B16.OVA and AB1.HA tumors 14 days after ACT. Tissues were homogenized and single cell preparations containing OT.I or CL4 CD8+ T cells were restimulated for 1 h with OVA or CL4 peptides, respectively, before addition of Brefeldin A and incubation for a further 4 h. Cells were stained with surface antibodies to identify transgenic T cells (CD8+CD45.1+ or CD8+CD90.1+) and inhibitory surface receptors PD-1 and CTLA-4. Cells were then fixed with 1% PFA and permeabilized with 0.1% saponin before staining for intracellular effector cytokines IFNγ and TNFα.
Preparation of DNA and RNA
For DNA extraction, pelleted tumor cells were lysed in 700 μL tissue lysis buffer (Tris-HCl pH 8.0 100 mM, EDTA, 5 mM, SDS 1% (v/v), NaCl 200 mM) and incubated at 55°C overnight with addition of 0.7 μL Proteinase K. DNA was extracted using phenol-chloroform and ethanol precipitation and stored at −20°C. For RNA extraction, tumor cells were resuspended in 0.5 mL of Trizol solution (Life Technologies) immediately after sorting or directly from the culture and stored at −80°C. RNA was extracted using phenol-chloroform and the RNEasy Mini Kit (Qiagen) according to the manufacturer’s instructions. Reverse transcription was performed with 1 μg of RNA per reaction using the SuperscriptIII First Strand Synthesis System (Invitrogen) according to the manufacturer’s instructions. RNA and DNA were quantified on a Nanodrop spectrophotometer (ND-1000; Thermo Scientific).
PCR and RT-PCR
Amplification of genomic DNA was routinely performed in 25 μL reactions consisting of 12.5 μL GoTaq Green Mastermix, 1 μL each of relevant forward and reverse primers (20 μM stock), 1 μg of genomic DNA template and 9.5 μL of RNase/DNase-free water. PCR conditions were as follows: 93°C for 3 min followed by 35–40 cycles of 93°C for 30 s, 55°C for 30 s and 72°C for 45 s and a final extension at 72°C for 3 min. PCR products were run on a 1% agarose gel and visualized with ethidium bromide. RT-PCR was performed using a similar reaction mix and touchdown PCR protocol: 93°C for 3 min, followed by 15 cycles where the annealing temp reduced from 63°C by 0.5°C per cycle, followed by 20 cycles under the conditions already listed above. Primer sequences were: OVA: forward 5ʹ-CTGTGCAGATGATGTAC-3ʹ; reverse 5ʹ-TGGTTGCGATGTGCTTG-3ʹ; HA: forward 5ʹ-CAATTGGGGAAATGTAACATCGCC-3ʹ; reverse 5ʹ-AGCTTTGGGTATGAGCCCTCCTTC-3ʹ; GAPDH: forward 5ʹ- GAAGGTCGGTGTGAACGGATT-3ʹ; reverse 5ʹ-CGGAAGGGGCGGAGATGATGA-3ʹ.
Determination of IC50 values
In vitro cell viability assays were performed by seeding 1 × 103 B16 melanoma cells in flat bottom 96-well plates and adding EMAs to adherent cells at titrated concentrations. After 72-h drug exposure, alamarBlue reagent was added at a ratio of 1:10 for 4 h and cell viability determined by reading absorbance at 570 nM and 595 nM on a BioTek Synergy MX plate reader.
Schedule of epigenetic modifying agent treatment
ACT tumor explants were treated with AZA or DEC for 72 h at concentrations of 1 μM and 5 μM and GFP re-expression was measured by flow cytometry. GFPneg tumor populations were sorted after AZA and DEC treatment and then screened with a panel of EMAs at concentrations below their IC50 value (, Supp. Figure 2) for 3, 7 or 14 days. Cells were split to avoid over-confluence and drugs replenished every three days. GFP re-expression was measured by flow cytometry.
Author contributions
JW, BW and MC designed the experiments. BW, JC, CF, MB and AB performed experiments and analysed data. JW, BW, JC, MC and BF were involved in data discussion and drafting the manuscript. BW and JC wrote the manuscript. JW, MC, BF and SS edited the manuscript and supplied feedback.
Disclosure of potential conflict of interest
The authors declare no conflicts of interest.
Abbreviations
Supplemental Material
Download Zip (2.9 MB)Acknowledgments
The authors thank the Telethon Kids Institute Bioresources team for their excellent animal care and the Telethon Kids Flow Cytometry Facility for assistance in sorting experiments. We thank Dr. Peter Dallas, Scientific Writer at the Telethon Kids Institute, for helpful comments and editing assistance.
Supplemental material
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
References
- Besser MJ, Shapira-Frommer R, Itzhaki O, Treves AJ, Zippel DB, Levy D, Kubi A, Shoshani N, Zikich D, Ohayon Y, et al. Adoptive transfer of tumor-infiltrating lymphocytes in patients with metastatic melanoma: intent-to-treat analysis and efficacy after failure to prior immunotherapies. Clin Cancer Res. 2013;19(17):4792–4800. doi:10.1158/1078-0432.CCR-13-0380.
- Pilon-Thomas S, Kuhn L, Ellwanger S, Janssen W, Royster E, Marzban S, … Sarnaik AA. Efficacy of adoptive cell transfer of tumor infiltrating lymphocytes after lymphopenia induction for metastatic melanoma. J Immunother. 2012;35(8):615–620. 1997. doi:10.1097/CJI.0b013e31826e8f5f.
- Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, … Dudley ME. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res. 2011;17(13):4550–4557. doi:10.1158/1078-0432.CCR-11-0116.
- Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Sci. 2015;348(6230):62–68. doi:10.1126/science.aaa4967.
- Khammari A, Labarrière N, Vignard V, Nguyen J-M, Pandolfino M-C, Knol AC, … Dreno B. Treatment of metastatic melanoma with autologous melan-A/mart-1-specific cytotoxic T lymphocyte clones. J Invest Dermatol. 2009;129(12):2835–2842. doi:10.1038/jid.2009.144.
- Robbins PF, Kassim SH, Tran TLN, Crystal JS, Morgan RA, Feldman SA, … Rosenberg SA. A pilot trial using lymphocytes genetically engineered with an NY-ESO-1–reactive T-cell receptor: long-term follow-up and correlates with response. Clin Cancer Res. 2015;21(5):1019–1027. doi:10.1158/1078-0432.CCR-14-2708.
- Yee C, Thompson JA, Byrd D, Riddell SR, Roche P, Celis E, Greenberg PD. Adoptive T cell therapy using antigen-specific CD8+ T cell clones for the treatment of patients with metastatic melanoma: in vivo persistence, migration, and antitumor effect of transferred T cells. Proc Nat Acad Sci. 2002;99(25):16168–16173. doi:10.1073/pnas.242600099.
- Holzel M, Bovier A, Tuting T. Plasticity of tumour and immune cells: a source of heterogeneity and a cause for therapy resistance? Nat Rev Cancer. 2013;13(5):365–376. doi:10.1038/nrc3498.
- Khong HT, Restifo NP. Natural selection of tumor variants in the generation of “tumor escape” phenotypes. Nat Immunol. 2002;3(11):999–1005. doi:10.1038/ni1102-999.
- Landsberg J, Kohlmeyer J, Renn M, Bald T, Rogava M, Cron M, … Tuting T. Melanomas resist T-cell therapy through inflammation-induced reversible dedifferentiation. Nature. 2012;490(7420):412–416. doi:10.1038/nature11538.
- Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Annu Rev Immunol. 2004;22(1):329–360. doi:10.1146/annurev.immunol.22.012703.104803.
- Shankaran V, Ikeda H, Bruce AT, White JM, Swanson PE, Old LJ. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature. 2001:410. doi:10.1038/35074122.
- Jäger E, Ringhoffer M, Altmannsberger M, Arand M, Karbach J, Jäger D, … Knuth A. Immunoselection in vivo: independent loss of MHC class I and melanocyte differentiation antigen expression in metastatic melanoma. Int J Cancer. 1997;71(2):142–147. doi:10.1002/(sici)1097-0215(19970410)71:2<142::aid-ijc3>3.0.co;2-0.
- Khong HT, Wang QJ, Rosenberg SA. Identification of multiple antigens recognized by tumor-infiltrating lymphocytes from a single patient: tumor escape by antigen loss and loss of MHC expression. J Immunother. 2004;27(3):184–190. doi:10.1097/00002371-200405000-00002.
- Matsushita H, Vesely MD, Koboldt DC, Rickert CG, Uppaluri R, Magrini VJ, … Schreiber RD. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature. 2012;482(7385):400–404. doi:10.1038/nature10755.
- von Boehmer L, Mattle M, Bode P, Landshammer A, Schäfer C, Nuber N, … van Den Broek M. NY-ESO-1–specific immunological pressure and escape in a patient with metastatic melanoma. Cancer Immunity Arch. 2013;13:3. Retrieved from http://cancerimmunolres.aacrjournals.org/content/13/3/12.abstract.
- Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21(3):381–395. doi:10.1038/cr.2011.22.
- Bennett RL, Licht JD. Targeting epigenetics in cancer. Annu Rev Pharmacol Toxicol. 1992;58(1):187–207. doi: 10.1146/annurev-pharmtox-010716-105106.
- Kulis M, Esteller M. 2 - DNA methylation and cancer. In: Herceg Z, Ushijima TBT-AG, editors. Epigenetics and cancer, part A. Vol. 70. Academic Press; 2010. p. 27–56. doi: 10.1016/B978-0-12-380866-0.60002-2.
- Tellez CS, Shen L, Estécio MRH, Jelinek J, Gershenwald JE, Issa J-PJ. CpG island methylation profiling in human melanoma cell lines. Melanoma Res. 2009;19:146–155.
- Bonazzi VF, Irwin D, Hayward NK. Identification of candidate tumor suppressor genes inactivated by promoter methylation in melanoma. Genes Chromosomes Cancer. 2009;48(1):10–21. doi:10.1002/gcc.20615.
- De Smet C, Lurquin C, Lethé B, Martelange V, Boon T. DNA methylation is the primary silencing mechanism for a set of germ line- and tumor-specific genes with a CpG-rich promoter. Mol Cell Biol. 1999;19(11):7327–7335. doi:10.1128/MCB.19.11.7327.
- Kazanets A, Shorstova T, Hilmi K, Marques M, Witcher M. Epigenetic silencing of tumor suppressor genes: paradigms, puzzles, and potential. Biochimica Et Biophysica Acta (BBA) - Reviews on Cancer. 2016;1865(2):275–288. doi:10.1016/j.bbcan.2016.04.001.
- Hervouet E, Cartron P-F, Jouvenot M, Delage-Mourroux R. Epigenetic regulation of estrogen signaling in breast cancer. Epigenet. 2013;8(3):237–245. doi:10.4161/epi.23790.
- Soengas MS, Capodieci P, Polsky D, Mora J, Esteller M, Opitz-Araya X, … Lowe SW. Inactivation of the apoptosis effector Apaf-1 in malignant melanoma. Nature. 2001;409(6817):207–211. doi:10.1038/35051606.
- Zhang P, Wang J, Gao W, Yuan B-Z, Rogers J, Reed E. CHK2 kinase expression is down-regulated due to promoter methylation in non-small cell lung cancer. Mol Cancer. 2004;3:14. doi:10.1186/1476-4598-3-14.
- Chou S-D, Khan ANH, Magner WJ, Tomasi TB. Histone acetylation regulates the cell type specific CIITA promoters, MHC class II expression and antigen presentation in tumor cells. Int Immunol. 2005;17(11):1483–1494. doi:10.1093/intimm/dxh326.
- Khan ANH, Gregorie CJ, Tomasi TB. Histone deacetylase inhibitors induce TAP, LMP, tapasin genes and MHC class I antigen presentation by melanoma cells. Cancer Immunol Immunother. 2008;57(5):647–654. doi:10.1007/s00262-007-0402-4.
- Siebenkäs C, Chiappinelli KB, Guzzetta AA, Sharma A, Jeschke J, Vatapalli R, … Ahuja N. Inhibiting DNA methylation activates cancer testis antigens and expression of the antigen processing and presentation machinery in colon and ovarian cancer cells. PLoS One. 2017;12(6):e0179501. doi:10.1371/journal.pone.0179501.
- Stresemann C, Lyko F. Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. Int J Cancer. 2008;123(1):8–13. doi:10.1002/ijc.23607.
- Smith BD, Beach CL, Mahmoud D, Weber L, Henk HJ. Survival and hospitalization among patients with acute myeloid leukemia treated with azacitidine or decitabine in a large managed care population: a real-world, retrospective, claims-based, comparative analysis. Exp Hematol Oncol. 2014;3(1):10. doi:10.1186/2162-3619-3-10.
- Gang AO, Frosig TM, Brimnes MK, Lyngaa R, Treppendahl MB, Gronbak K, … Hadrup SR. 5-azacytidine treatment sensitizes tumor cells to T-cell mediated cytotoxicity and modulates NK cells in patients with myeloid malignancies. Blood Cancer J. 2014;4:e197. doi:10.1038/bcj.2014.14.
- Guo ZS, Hong JA, Irvine KR, Chen GA, Spiess PJ, Liu Y, … Schrump DS. De novo induction of a cancer/testis antigen by 5-Aza-2′-deoxycytidine augments adoptive immunotherapy in a murine tumor model. Cancer Res. 2006;66(2):1105–1113. doi:10.1158/0008-5472.CAN-05-3020.
- Klar AS, Gopinadh J, Kleber S, Wadle A, Renner C. Treatment with 5-Aza-2ʹ-deoxycytidine induces expression of NY-ESO-1 and facilitates cytotoxic T lymphocyte-mediated tumor cell killing. PLoS One. 2015;10(10):e0139221. doi:10.1371/2Fjournal.pone.0139221.
- Vo DD, Prins RM, Begley JL, Donahue TR, Morris LF, Bruhn KW, … Ribas A. Enhanced antitumor activity induced by adoptive T-cell transfer and adjunctive use of the histone deacetylase inhibitor LAQ824. Cancer Res. 2009:69. doi:10.1158/0008-5472.CAN-09-1456.
- Adair SJ, Hogan KT. Treatment of ovarian cancer cell lines with 5-aza-2′-deoxycytidine upregulates the expression of cancer-testis antigens and class I major histocompatibility complex-encoded molecules. Cancer Immunol Immunother. 2009;58(4):589–601. doi:10.1007/s00262-008-0582-6.
- Chiappinelli KB, Strissel PL, Desrichard A, Li H, Henke C, Akman B, … Strick R. Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell. 2015;162(5):974–986. doi:10.1016/j.cell.2015.07.011.
- Roulois D, Loo Yau H, Singhania R, Wang Y, Danesh A, Shen SY, … De Carvalho DD. DNA-demethylating agents target colorectal cancer cells by inducing viral mimicry by endogenous transcripts. Cell. 2015;162(5):961–973. doi:10.1016/j.cell.2015.07.056.
- Cruickshank B, Giacomantonio M, Marcato P, McFarland S, Pol J, Gujar S. Dying to be noticed: epigenetic regulation of immunogenic cell death for cancer immunotherapy. Front Immunol. 2018;9:654. doi:10.3389/fimmu.2018.00654.
- Maio M, Covre A, Fratta E, Di Giacomo AM, Taverna P, Natali PG, … Sigalotti L. Molecular pathways: at the crossroads of cancer epigenetics and immunotherapy. Clin Cancer Res. 2015;21(18):4040–4047. doi:10.1158/1078-0432.CCR-14-2914.
- Covre A, Coral S, Nicolay H, Parisi G, Fazio C, Colizzi F, … Maio M. Antitumor activity of epigenetic immunomodulation combined with CTLA-4 blockade in syngeneic mouse models. OncoImmunology. 2015;4(8):e1019978. doi:10.1080/2162402X.2015.1019978.
- Lisiero DN, Soto H, Everson RG, Liau LM, Prins RM. The histone deacetylase inhibitor, LBH589, promotes the systemic cytokine and effector responses of adoptively transferred CD8+ T cells. J ImmunoTher Cancer. 2014;2(1):8. doi:10.1186/2051-1426-2-8.
- Marzo AL, Lake RA, Robinson BWS, Scott B. T-cell receptor transgenic analysis of tumour-specific CD8 and CD4 responses in the eradication of solid tumours. Cancer Res. 1999;59:1071–1079.
- Dobrzanski MJ, Reome JB, Dutton RW. Therapeutic effects of tumour-reactive type 1 and type 2 CD8+ T cell subpopulations in established pulmonary metastases. J Immunol. 1999;162:6671–6680.
- Bai X-F, Liu J-Q, Joshi PS, Wang L, Yin L, Labanowska J, … Liu Y. Different lineages of P1A-expressing cancer cells use divergent modes of immune evasion for T-cell adoptive therapy. Cancer Res. 2006;66(16):8241–8249. doi:10.1158/0008-5472.CAN-06-0279.
- Kaluza KM, Thompson JM, Kottke TJ, Flynn Gilmer HC, Knutson DL, Vile RG. Adoptive T cell therapy promotes the emergence of genomically altered tumor escape variants. Int J Cancer. 2012;131(4):844–854. doi:10.1002/ijc.26447.
- Goldberger O, Volovitz I, Machlenkin A, Vadai E, Tzehoval E, Eisenback L. Exuberated numbers of tumour-specific T cells results in tumour escape. Cancer Res. 2008;68(9):3450–3457. doi:10.1158/0008-54772.CAN-07-5006.
- Rosenthal R, Cadieux EL, Salgado R, Al Bakir M, Moore DA, Hiley CT; The TRACERx consortium. Neoantigen-directed immune escape in lung cancer evolution. Nature. 2019;567:479–485. doi:10.1038/s41586-019-1032-7.
- Real LM, Jimenez P, Kirkin A, Serrano A, García A, Cantón J, … Ruiz-Cabello F. Multiple mechanisms of immune evasion can coexist in melanoma tumor cell lines derived from the same patient. Cancer Immunol Immunother. 2001;49(11):621–628. doi:10.1007/s002620000154.
- Sanchez-Perez L, Kottke T, Diaz RM, Ahmed A, Thompson J, Chong H, … Vile RG. Potent selection of antigen loss variants of B16 melanoma following inflammatory killing of melanocytes in vivo. Cancer Res. 2005;65(5):2009–2017. doi:10.1158/0008-5472.CAN-04-3216.
- Vlková V, Štěpánek I, Hrušková V, Šenigl F, Mayerová V, Šrámek M, … Reiniš M. Epigenetic regulations in the IFNγ signalling pathway: iFNγ-mediated MHC class I upregulation on tumour cells is associated with DNA demethylation of antigen-presenting machinery genes. Oncotarget. 2014;5(16):6923–6935. doi:10.18632/oncotarget.2222.
- Garrido F, Aptsiauri N, Doorduijn EM, Garcia Lora AM, van Hall T. The urgent need to recover MHC class I in cancers for effective immunotherapy. Curr Opin Immunol. 2016;39:44–51. doi:10.1016/j.coi.2015.12.007.
- Saleh MH, Wang L, Goldberg MS. Improving cancer immunotherapy with DNA methyltransferase inhibitors. Cancer Immunol Immunother. 2016;65(7):787–796. doi:10.1007/s00262-015-1776-3.
- Chiappinelli KB, Zahnow CA, Ahuja N, Baylin SB. Combining epigenetic and immunotherapy to combat cancer. Cancer Res. 2016;76(7):1683–1689. doi:10.1158/0008-5472.CAN-16-0584.
- Wrangle J, Wang W, Koch A, Easwaran H, Mohammad HP, Pan X, … Baylin SB. Alterations of immune response of non-small cell lung cancer with azacytidine. Oncotarget. 2013;4(11):2067–2079. doi:10.18632/oncotarget.1542.
- Brocks D, Schmidt CR, Daskalakis M, Jang HS, Shah NM, Li D, Plass C. DNMT and HDAC inhibitors induce cryptic transcription start sites encoded in long terminal repeats. Nat Genet. 2017;49(7):1052–1060. doi:10.1038/ng.3889.
- Wylie B, Seppanen E, Xiao K, Zemek R, Zanker D, Prato S, … Waithman J. Cross-presentation of cutaneous melanoma antigen by migratory XCR1+CD103− and XCR1+CD103+ dendritic cells. OncoImmunology. 2015;4(8):e1019198. doi:10.1080/2162402X.2015.1019198.
- Davis MR, Manning LS, Whitaker D, Garlepp MJ, Robinson BWS. Establishment of a murine model of malignant mesothelioma. Int J Cancer. 1992;52(6):881–886. doi:10.1002/ijc.2910520609.
- Marzo AL, Lake RA, Lo D, Sherman L, McWilliam A, Nelson D, … Scott B. Tumor antigens are constitutively presented in the draining lymph nodes. J Immunol. 1999;162:5838–5845.