
ABSTRACT
Multiple rodent and primate preclinical studies have advanced CAR T cells into the clinic. However, no single model accurately reflects the challenges of effective CAR T therapy in human cancer patients. To evaluate the effectiveness of next-generation CAR T cells that aim to overcome barriers to durable tumor elimination, we developed a system to evaluate CAR T cells in pet dogs with spontaneous cancer. Here we report on this system and the results of a pilot trial using CAR T cells to treat canine diffuse large B cell lymphoma (DLBCL). We designed and manufactured CD20-targeting, second-generation canine CAR T cells for functional evaluation in vitro and in vivo using lentivectors to parallel human CAR T cell manufacturing. A first-in-species trial of five dogs with DLBCL treated with CAR T was undertaken. Canine CAR T cells functioned in an antigen-specific manner and killed CD20+ targets. Circulating CAR T cells were detectable post-infusion, however, induction of canine anti-mouse antibodies (CAMA) was associated with CAR T cell loss. Specific selection pressure on CD20+ tumors was observed following CAR T cell therapy, culminating in antigen escape and emergence of CD20-disease. Patient survival times correlated with ex vivo product expansion. Altering product manufacturing improved transduction efficiency and skewed toward a memory-like phenotype of canine CAR T cells. Manufacturing of functional canine CAR T cells using a lentivector is feasible. Comparable challenges to effective CAR T cell therapy exist, indicating their relevance in informing future human clinical trial design.
Introduction
Chimeric antigen receptor (CAR) T cells have emerged as a valuable treatment option for patients with B cell neoplasia. CD19 CAR T cells have received FDA approval for relapsed/refractory precursor acute lymphoblastic leukemia (ALL) and diffuse large B cell lymphoma (DLBCL).Citation1 Complete response rates reviewed by Milone and Bhoj across 16 different trials for various B cell neoplasms ranged from 8.3-100% with a trend for higher responses rates in patients with ALL compared with chronic lymphocytic leukemia (CLL) and DLBCL.Citation2 Despite very impressive response rates, other clinical observations, including lack of uniform CAR T engraftment/expansion in patientsCitation2 alongside the emergence of CD19 escape variantsCitation1,Citation3 have become apparent as major therapeutic obstacles. Humanized mouse and primate studies have been used in preclinical CAR T cell investigation; however, the former approach is hindered by disparate results between studies and technical challenges, whereas the latter is impeded by financial and ethical concerns.Citation4 Currently, no single system accurately recapitulates the variables associated with clinical response or mechanisms of resistance, thereby mandating the requirement for new, improved cancer immunotherapy models.Citation4,Citation5
DLBCL is an aggressive and rapidly progressive disease that is commonly encountered as a spontaneous tumor in pet dogs.Citation6 Multi-agent CHOP-based chemotherapy is considered the standard of care for dogs with DLBCL and most dogs achieve remission after initial chemotherapy. However, the majority of dogs relapse with drug-resistant disease and a median progression-free survival times of 252 d are reported.Citation7 Median overall survival times for canine lymphoma typically range from 10 to 14 months.Citation8 Subsequent relapses are traditionally treated with varying rescue protocols, though cure is rare.Citation8 Canine B cell lymphomas share a number of key geneticCitation9 and immunophenotypic traits with the human form of disease.Citation10 As in humans, both malignant and normal canine B cells express CD20,Citation11 thus enabling lineage-specific targeting of this molecule. Incorporation of CD20-targeting monoclonal antibodies (rituximab) into the standard CHOP-based protocol (R-CHOP) significantly improved response rates for humans with DLBCL.Citation12 Rituximab, however, does not bind to canine CD20 and there are currently no effective canine CD20 targeting strategies for use in the canine clinic.
Beyond the specific parallels of human and canine lymphoma biology, genetics and behavior, there are numerous advantages of investigating cancer and cancer immunotherapies in canine cancer patients versus laboratory models. Canine tumors develop spontaneously in genetically outbred animals with complete and natural immune systems.Citation13 Canine cancers share histologic and genetic similarities with human neoplasms with regards to architecture and heterogeneity recognized within and between tumor classes.Citation14 Many canine malignancies follow similar clinical courses and exhibit comparable response patterns to those seen in people when treated with multiple modes of similar standard adjuvant anti-cancer therapies.Citation15 Support for this comparative approach to accelerate human translational research in the biomedical community is borne out by the formation of the preclinical cancer immunotherapy network of canine trials (PRECINCT) and U01 awards. The U01 trial site awardees are working to pioneer combination immunotherapies and identify correlative biomarkers to inform human clinical trial design as part of the Cancer Moonshot program. In line with these initiatives, we have developed canine CAR T cell therapy for evaluation in dogs with DLBCL, which we believe represents a unique translational opportunity to develop and evaluate next-generation, best-in-class, CAR T cells prior to employment in the human clinic.
Our group has previously demonstrated the ability to expand autologous T cells from canine lymphoma patients and transfect these cells using RNA-electroporation, leading to modest but transient tumor responses.Citation16 We hypothesized that permanent lentiviral transduction of canine T cells with CD20 CAR would be feasible and that this product would exert target-specific effects in vivo, resulting in anti-tumor activity. Here we report on the generation of second-generation canine (c)CD20-28-ζ and cCD20-BB-ζ CARs and their therapeutic application in a small pilot study in dogs with advanced DLBCL.
Materials and methods
Generation of lentivirus vectors
The lentivector pELNS was kindly provided by Dr. Michael Milone. pELNS is a third-generation, self-inactivating lentivector derived from pCLPS, with the EF-1α promoter in place of the CMV promoter. Modification of the multi-cloning site was undertaken to allow easier insertion and removal of scFvs resulting in the pELxPS vector. Plasmids for CAR and helper plasmids pCI VSV-G, pRSV Rev, and pMDL gag/pol.RRE (kindly provided by Dr. Michael Milone) were initially isolated using Qiagen EndoFree Maxi and Giga kits; later production of pELxPS cCD20-28-ζ, pELxPS cCD20-BB-ζ, and pCI VSV-G was performed by Nature Technology Corporation. Canine CD8α, CD28, 4-1BB, and CD3ζ components were cloned from cPBMC cDNA and synthesized using overlap extension PCR. Development of the anti-cCD20 scFv is previously described.Citation16 The cCD20-28-ζ CAR construct was generated in-house and cloned into pELxPS. The cCD20-BB-ζ construct sequence was codon-optimized for dogs, synthesized (GenScript) and cloned into pELxPS. Lentivirus was produced through transient transfection of 293T cells using Lipofectamine 2000 (Life Technologies), supernatants were concentrated by centrifugation for ≥8 h at ≥15,000g prior to titration on primary human CD4+ T cells or Jurkat cells.
Generation of anti-canine CD3/CD28 magnetic beads
Agonistic mouse anti-canine CD3 (clone CA17.2A12, BioRad) and mouse anti-canine CD28 (clone 5B8, a gift from Dr. Rainer Storb) were conjugated to magnetic tosylactivated Dynabeads (Life Technologies) as previously described.Citation16
Cell lines
K562 cells stably transduced with human FcγRII (CD32) and cloned by single-cell sorting to produce KT32 were a gift from Dr. Carl June. KT32 cells expressing cCD86 were generated to produce artificial (a)APCs as previously described.Citation16 aAPCs were cultured in K562 media containing RPMI 1640 with 2 mM L-glutamine (Mediatech), 10% fetal bovine serum (Atlanta Biologicals) 10 mM HEPES (Gibco), 1 mM sodium pyruvate (Mediatech), 100 U/ml penicillin and 100 μg/ml streptomycin (Gibco), and 30 μg/ml gentamicin (Gibco). Canine B cell lines GL1 (CD20-), CLBL-1 (CD20+) and stably transduced GL1 expressing GFP (GL1-GFP) and GL1 expressing GFP and canine CD20 (GL1-GFP-CD20) (both a gift of Dr. Avery Posey) were grown in T cell media (TCM) containing RPMI 1640 with 2 mM L-Glutamine (Mediatech), 10% heat-inactivated fetal bovine serum (Atlanta Biologicals), 10 mM HEPES (Gibco), and 100 U/ml penicillin and 100 μg/ml streptomycin (Gibco).
Animals
Peripheral Blood Monocytes (PBMCs) from healthy donor dogs were obtained following full IACUC approval (IACUC protocol numbers 806233 and 805972).
Canine PBMC isolation and T cell culture
PBMCs were isolated by centrifugation over Ficoll-Paque PLUS (GE Healthcare). Live PBMCs were enumerated by hemocytometer using trypan blue exclusion and plated on 10 cm tissue culture dishes (Falcon) at 1 × 106 cells/ml and incubated overnight at 37°C, 5% CO2 and 95% humidity. Enriched peripheral blood lymphocytes (PBLs) were pelleted on the following day using centrifugation at 218g for 5 min. aAPCs were irradiated with 10,000 rads and used at a 1:2 ratio of aAPCs: enriched PBLs to a final concentration of 2.5 × 105 aAPCs and 5 × 105 PBLs per ml with 0.5 μg/ml mouse anti-canine CD3 for T cell activation. When antibody-conjugated beads were used for T cell activation, beads were washed prior to addition to enriched PBLs at a 3:1 or 4:1 beads:PBLs. Where specified, T cells were activated with 2.5 ng/ml of concanavalin A (Sigma-Aldrich). CAR T cell stimulation was performed using 1:1 unsorted transduced T cells: irradiated CLBL-1 (10,000 rads). Cytokines were used as follows at the time of stimulation and every second day after: 30 U/ml rhIL-2 (Gibco) and 10 ng/ml rhIL-21 (eBioscience); 10 ng/mL rhIL-7 (Peprotech) and 5 ng/mL rhIL-15 (Peprotech); for Patient 429–006, 20 ng/mL of rhIL-7 and 10 ng/mL rhIL-15 were used. Cell culture supernatants at the time of harvest all tested negative for mycoplasma. All infusion products were gram stain negative. Samples of cultured CAR T cell products were taken 3, 5, and 7 d post-transduction and submitted to the University of Pennsylvania Translational and Correlative Studies Laboratory for Replication Competent Lentivirus (RCL) testing as described.Citation17
CFSE staining
Where indicated PBMCs were washed, resuspended to 1 × 107 cells/ml in DPBS, and labeled with carboxy-fluoroscein succinimidyl esterase (CFSE, 5μM, Sigma Aldrich) for 5 min at 37°C. Labeling was quenched with five volumes of TCM. Cells were washed twice prior to stimulation.
Flow cytometry and antibodies
Cells were harvested and washed in fluorescence-activated cell sorting (FACS) buffer (1% heat-inactivated fetal bovine serum in DPBS with calcium and magnesium) prior to surface staining with a combination of the following antibodies: APC- or PacificBlue-labeled rat anti-dog CD4 (clone YKIX302.9, BioRad), PE-labeled rat anti-dog CD5 (clone YKIX322.3, eBioscience), PE- FITC- or AF647-labeled rat anti-dog CD8 (clone YCATE55.9, BioRad), mouse anti-dog CD20 (clone 6C12, Invivogen) with BrilliantViolet421- or AlexaFluor488-labeled goat anti-mouse IgG secondary (clone Poly4053, Biolegend or Life Technologies), eFluor660-labeled mouse anti-dog CD25 (clone P4A10, eBioscience), PECy7-labeled mouse anti-human CD45 (clone HI30, Biolegend) and the cell viability dye 7-AAD (Biolegend). Following surface staining, cells were washed in FACS buffer and fixed in 1% paraformaldehyde (Ted Pella). Where indicated, cells were permeabilized with 0.1% saponin (Sigma Aldrich) post-fixation and stained with APC-labeled mouse anti-human CD79a antibody (clone HM57, BD Biosciences). Surface detection of the CAR was performed by labeling with a biotinylated rabbit anti-mouse IgG H + L antibody (Jackson ImmunoResearch Laboratories) followed by fluor-conjugated streptavidin secondary (BD Biosciences) before surface staining.
T cells were defined as CD5 positive, B cells were CD79a positive, and aAPCs were human CD45 positive. T and B cell enumeration from peripheral blood samples was calculated as the product of T or B cell frequency (gated on live total lymphocytes) and lymphocyte count from concurrent complete blood count (CBC) values. Flow cytometric enumerations from samples other than whole blood were performed using CountBright Beads (Life Technologies). Responder frequency and mitotic divisions were calculated from CFSE stained populations as previously described.Citation18 Acquisition was performed on a FACS Calibur, FACS Canto II, or LSR Fortessa flow cytometer (BD Biosciences) and data were analyzed using FlowJo software version X (Treestar). Flow sorting was performed on a FACS Aria (BD Biosciences).
Cytotoxicity assays
For the cCD20-28-ζ cytotoxicity assay, a fixed number of irradiated target cells were plated in 96-well plates. Co-cultures were incubated for 24 h at 37°C in a 96-well V-bottom plate prior to staining for viability (7-AAD) and for CD79a. Countbright beads (Life Technologies) were added to the samples before collection on a FACS Canto II. The number of live target cells was enumerated by back-calculating from the count of 7-AAD-, target marker positive events and Countbright bead events in each tube. Percent lysis = 100 x (1 – (Experimental/Target Alone)), percent specific lysis = 100 x (1 – (CAR Experimental/Non-transduced Experimental)). For the cCD20-BB-ζ assay, control GL1-GFP cells and target GL1-GFP-CD20 were labeled with 0.5 and 3μM Cell Trace Violet dye (Thermo Fisher Scientific), mixed together and then co-cultured with T cells for 4 h in 96 well U-bottom plate at 37°C. To quantify spontaneous lysis three wells of mixed GL1-GFP and GL1-GFP-CD20 cells were cultured in the absence of effectors. Following culture, cells were stained with 7-AAD and CD5 prior to acquisition on a FACS Canto II. Specific lysis = 100 x (1- ((target cells cultured with effector/control cells cultured with effectors)/mean (target cells cultured without effectors/control cells cultured without effectors))). For both assays, T cells were added at indicated E:T ratios in triplicate, with total T cells defining the effector.
Trial design
The purpose of the trial was to determine the safety, persistence and efficacy of lentiviral transduced, autologous T cells. Dogs with high-grade lymphoma that were refractory to multi-agent chemotherapy (or dogs for which standard-of-care multi-agent induction chemotherapy was not elected by owner) were eligible for screening following full written informed owner consent. Expression of cCD20 on malignant lymphocytes was confirmed by flow cytometry. Thorough physical examination, serum chemistry, CBC, urinalysis, thoracic radiographs and abdominal ultrasound were performed during screening to identify any co-morbidities and determine the extent of disease. Patients received intravenous CAR T cells in 0.9% PlasmaLyte A (Baxter) 13–16 d following T cell isolation and activation. Patients were eligible for multi-agent rescue chemotherapy following disease progression after CAR T therapy.
Ethical statement
Studies were approved by the University of Pennsylvania’s Institutional Animal Care and Use Committee (Protocol Number 805389) and signed informed owner consent was required for enrollment. The use of recombinant DNA was approved by the University of Pennsylvania’s Institutional Biosafety Committee (IBC#13-164).
qPCR for CAR gene quantification
Genomic DNA was isolated from fresh blood and lymph node aspirates. qPCR analysis was performed using ABI Taqman technology and a custom assay to detect the integrated (WPRE) transgene sequence in 200 ng genomic DNA. To determine copy number per unit DNA, a 9-point standard curve was generated consisting of 0.5–1 × 106 plasmid copies spiked into 200 ng non-transduced control genomic DNA. The number of copies of plasmid present in the standard curve was verified using digital qPCR with the same WPRE primer/probe set (WPRE.227.F 5ʹ-CGCAACCCCCACTGGTT-3ʹ, WPRE.289.R 5ʹ-AAAGCGAAAGTCCCGGAAA-3ʹ, WPRE.250.FAM_MGB Probe 5ʹ-TTG CCA CCA CCT GTC-3ʹ) and performed on a QuantStudioTM 3D digital PCR instrument (Life Technologies). Each data-point (sample and standard curve) was evaluated in triplicate with a positive Ct value in 3/3 replicates with % CV less than 0.95% for all quantifiable values. To control for the quantity of integrated DNA, a parallel amplification reaction was performed using 10 ng genomic DNA, and a Qualified Assay for quantification of canine gDNA consisted of primer/probe combination specific for a MC1R gene (K-9_MC1R.Fwd Primer_5ʹ-TGGTCCTCTGCCCTCAACA-3ʹ, K-9_MC1R.Rev Primer_5ʹ-TGATGAGGGTGAGGAAGAGGTT-3ʹ, K-9_MC1R.FAM Probe MGB_ 5ʹ-AGTTCTGAAAGACGCAG-3ʹ). Standards were verified by digital PCR. These amplification reactions generated a correction factor to adjust for calculated versus actual DNA input. Copies of transgene per cell were calculated according to the formula:
[Average copies of transgene (from qPCR) x gDNA input Correction Factor/Input gDNA (ng)]x 0.0063 ng gDNA/cell].
Canine anti-mouse antibodies (CAMA)
Serum samples were collected and stored at −80°C and analyzed by sandwich enzyme-linked immunosorbent assay (Biolegend) in triplicate to detect canine anti-mouse IgG. Anti-mouse immunoglobulin antibody concentrations were calculated from standard dilutions fit to a 5-parameter QC-validated curve.
Interferon-γ ELISA
1x105 T cells were co-cultured with 1 × 105 targets for 24 h in 96 well plates prior to harvesting and storage of supernatants at −80°C. Interferon-γ concentrations were quantified using a Quantikine canine-specific ELISA (R&D Systems).
Statistical analyses
Data were displayed and analyzed using GraphPad Prism 8 (GraphPad Software). Two-tailed paired t-tests were used when comparing two groups and paired one-way or two-way ANOVA with Bonferroni posttests were used to compare greater than two groups. Statistical significance was defined as P < .05 (*, P < .05; **, P ≤ 0.01; ***, P ≤ 0.001; ****, P ≤ 0.0001).
Results
Lentiviral transduction of primary canine T cells
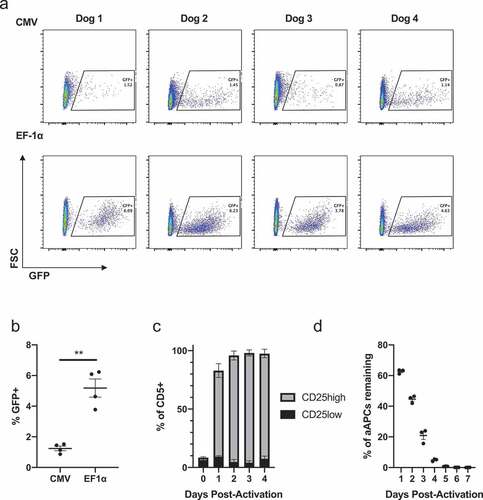
Primary canine cells were tested for their susceptibility to permanent lentiviral modification. PBMCs were isolated from four healthy dogs and activated using bead-conjugated, agonistic canine-specific anti-CD3 and anti-CD28 antibodies. Cells were then transduced with the same MOIs of lentivirus encoding GFP under the control of either CMV (pCLPS) or EF-1α (pELNS) promoters ()). Lymphocytes transduced with pELNS had a greater transduction efficiency compared to transduction with pCLPS ()); therefore, EF-1α was selected for future experiments and CAR T product manufacture. Although lentiviruses are able to infect some resting cells, quiescent T cells are known to resist gene transfer via lentivectors.Citation19 Having previously established that canine T cell stimulation with aAPCs resulted in greater T cell proliferation than anti-canine CD3/28 magnetic beads, Citation16 we further interrogated the activating properties of aAPCs to inform future T cell activation and transduction experiments. Stimulation of enriched PBLs with aAPCs in the presence of rhIL-2 and rhIL-21 resulted in T cell activation as indicated by high CD25 expression within 24 h that was maintained for at least 4 d ()). The frequency of irradiated aAPCs rapidly decreased in these co-cultures over 4 d ()). Therefore, we selected day 4 after aAPC stimulation for lentiviral transduction given clear T cell activation and absence of significant numbers of aAPCs that could act as an undesirable lentiviral vector sink at that timepoint.
Figure 1. Lentiviral transduction of primary canine T cells
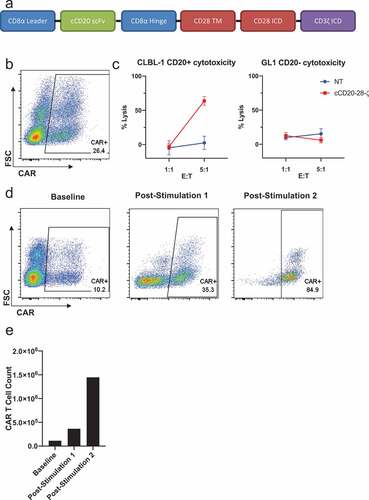
Design, manufacture and function of cCD20-28-ζ canine CAR T cells
To appraise the clinical potential of canine CAR T cells in dogs with spontaneous B cell lymphoma, a second generation, CD20-targeted canine CAR was designed using the same canine-specific anti-CD20 scFv as described previouslyCitation16 flanked by canine CD8a leader and hinge sequences and utilizing canine CD28 and canine CD3ζ signaling domains ()). This construct was cloned into pELxPS and a self-inactivating, VSV-G pseudotyped lentivirus was generated using a third-generation lentiviral packaging protocol.Citation20 Enriched PBLs from a healthy dog were transduced resulting in 26.4% transduction as measured by CAR surface expression ()). CAR T cell functionality was confirmed using an in vitro cytotoxicity assay showing antigen-specific killing of an allogeneic CD20 + B cell lymphoma cell line (CLBL-1). No significant cytotoxicity was observed against an allogeneic CD20- B cell leukemia cell line (GL1) or when using non-transduced PBLs ()). In vitro antigen-specific enrichment ()) and growth ()) of cCD20-28-ζ canine CAR T cells was documented over two rounds of re-stimulation with CLBL-1s. Together, these data demonstrate the feasibility of generating stably transduced, functional canine CAR T cells and pave the way for therapeutic evaluation in a canine patient population.
Figure 2. Design, manufacture and function of cCD20-28-ζcanine CAR T cells
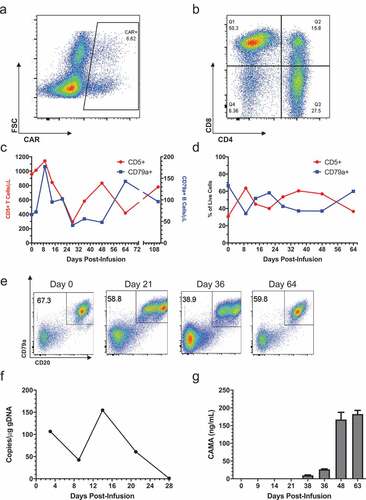
Treatment of B cell lymphoma patients with cCD20-28-ζ CAR T cells
Canine cCD20-28-ζ CAR T cells were manufactured from peripheral blood T cells isolated from four dogs with advanced DLBCL and used therapeutically in a pilot trial. In all cases, DLBCL was confirmed either by a biopsy or fine needle aspirate of an affected lymph node. Enrolled patients had either failed multi-agent chemotherapy or relapsed shortly after successful induction chemotherapy. Surface CAR expression ranged from 1.51% to 6.62% of live CD5+ T cells in products (), , Figs S1-S3) and pre-infusion products contained both CD4+ and CD8+ T cells with the CD4:CD8 ratio ranging from 1:0.35 to 1:7.8 (), , Figs S1-S3). Three patients received fresh CAR T cell products, however, patient 429–001 developed a urinary tract infection requiring intervention so the product was cryopreserved on day 14 of culture. Five days before infusion the product was thawed and re-stimulated with irradiated CLBL-1 cells. In patients 429–001, 429–003 and 429–004, CAR T cells expanded between 22 and 169 fold over 12–15 d of culture (). Limited expansion of patient 429–002’s CAR T cell product precluded infusion of a therapeutically relevant CAR T cell dose, so the CAR product was administered intranodally and the patient received an infusion of expanded autologous, non-transduced T cells. With the exception of 429–003, all dogs required rescue chemotherapy for disease progression within one month (). Tracking of 429–003’s lymph node composition revealed an initial increase in T cells accompanied by a decrease in B cell frequency ()), this inverse relationship continued throughout the disease course, with a greater frequency of T cells than B cells in the target node between weeks 4 and 7 post-infusion. Phenotypic examination of malignant nodal B cells in this patient revealed the emergence of a population of CD79a+ CD20int cells at day 21 and 36 which suggested CD20-specific immune pressure and coincided with the increased nodal T cell frequency ()). The presence of circulating CAR T cells in patient 429–003 was confirmed using qPCR, albeit at low level and could not be detected after day 28 ()). Canine anti-mouse antibodies (CAMA) were detected at day 38 and subsequently rose steadily ()). CAMA induction coincided with the intranodal loss of T cell dominance and loss of dichotomy in CD20 expression (). No adverse events ascribed to adoptive transfer of CAR T cells were documented in any patient.
Table 1. cCD20-28-ζ: clinical parameters and CAR T cell characteristics
Figure 3. Treatment of patient 429–003 with cCD20-28-ζCAR T cells
We hypothesized that higher cell doses and increased ex vivo cellular proliferation were associated with longer survival times. A significant correlation between increased overall survival and increased ex vivo population doublings was identified. In contrast, overall survival did not correlate with total T cell dose or CAR T cell dose administered (Fig S4). In summary treatment of canine DLBCL patients with permanently modified, autologous cCD20-28-ζ CAR T cells is feasible and preceded down-regulation of target antigen alongside induction of anti-CAR host immunity.
Modifying CAR manufacturing
Despite administration of low doses of CAR T cells in these four patients, we were encouraged by the early findings of feasibility of CAR T manufacture, safety, suggestion that CAR T cells exert a selective pressure on CD20+ targets, and that survival times correlated with ex vivo expansion properties. We next sought to improve the CAR manufacturing process to enhance transduction efficiencies and modify T cell phenotypes to promote CAR T persistence. First, we compared the use of aAPCs as a source of cellular stimulation in comparison with beads expressing agonistic canine CD3 and CD28 antibodies. We confirmed our previous findingCitation16 that aAPCs supported significantly greater cellular proliferation than CD3/CD28 beads (Fig S5a). However, CD3/CD28 beads induced more CD5+ T cells to divide early (day 2 post-activation) compared with aAPCs (Fig. S5b). Given the absence of aAPCs as a potential viral sink, and the fact that the majority of CD5+ T cells were highly activated by CD3/CD28 bead stimulation on day 1, we transduced CD3/CD28 activated cells with lentivirus on day 1 or day 2 and found that transduction efficiencies were significantly increased in CD3/CD28 bead stimulated cultures compared to aAPC-activated cells transduced on day 4 (Fig S5c).
Treatment of canine patients with cCD20-28-ζ CAR T cells revealed that these genetically modified T cells failed to persist for significant time periods in vivo. Although CAMA induction might partially explain this loss, we also explored whether cytokine culture conditions could be altered to maintain a less differentiated, memory T cell phenotype. We compared the phenotypes of canine T cells grown in IL-2 and IL-21 with those grown in IL-7 and IL-15. Expression of CD27 and CD62L was better maintained in both CD4+ and CD8+ subsets when cultured with IL-7 and IL-15 (Fig. S6). By changing the means of T cell activation to CD3/CD28 beads and altering the cytokines supporting T cell growth transduction efficiency of canine T cells was improved and immunophenotype was modified to one consistent with a less differentiated phenotype.
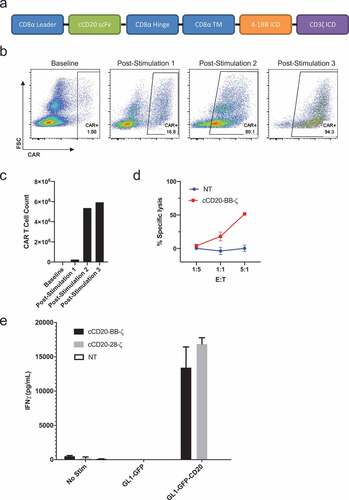
Design and function of cCD20-bb-ζ canine CAR T cells
As BB-ζ signaling confers a memory-like phenotype, greater persistence, and antitumor activity upon human CAR T cells,Citation21-Citation23 we designed a cCD20-BB-ζ CAR construct ()). Transduction of primary T cells isolated from the peripheral blood of a dog with spontaneous B cell lymphoma showed low-level expression of the cCD20-BB-ζ CAR. However, antigen-specific enrichment and expansion of CAR T cells was observed after each re-stimulation with CLBL-1s (). T cells obtained from a subsequent healthy donor blood draw were transduced with the cCD20-BB-ζ CAR, supplemented with IL-7 and IL-15, then re-stimulated with CLBL-1 prior to assessment of killing activity which revealed antigen-specific cytotoxicity ()). To further evaluate functionality of canine CAR T cells, we performed an IFNγ ELISA using both cCD20-28-ζ and cCD20-BB-ζ CAR T cells. IFNγ secretion was only observed in the presence of antigen from both cCD20-28-ζ CAR and cCD20-BB-ζ CAR T cells ()). Together these data indicate that canine primary T cells can be transduced to express a cCD20-BB-ζ CAR and these CAR T cells exhibit antigen-specific functionality.
Figure 4. Design and expansion of cCD20-BB-ζ canine CAR T cells
Treatment of a B cell lymphoma patient with cCD20-BB-ζ canine CAR T cells
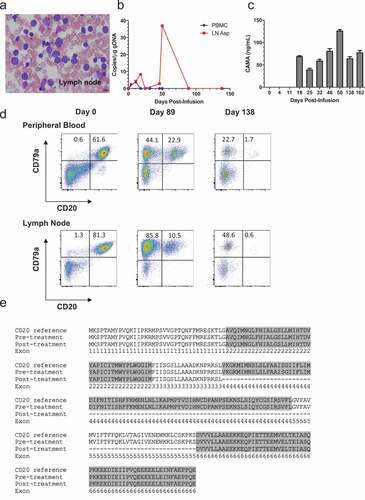
After modification of CAR T cell culture conditions, we enrolled patient 429–006, a 5 yr old male-castrated boxer with stage Va DLBCL (diagnosis confirmed by fine needle aspiration- )) for treatment with cCD20-BB-ζ CAR T cells after his owners declined to continue with multi-agent chemotherapy. As the patient was markedly leukemic at the time of blood draw (Fig S7), CD5+ T cells were selected by flow cytometric sorting, prior to activation with CD3/CD28 magnetic beads. T cells were transduced with cCD20-BB-ζ CAR at day 1 and day 2 post-stimulation and subsequently expanded with aAPCs and autologous patient PBMCs (containing 94.8% CD20+ blasts) and supplemented with IL-7 and IL-15. Cyclophosphamide (50mg/m2 PO) was given for 4 consecutive days between 2 and 6 d before CAR T cells as pre-conditioning. A total dose of 2.18 × 106 T cells/kg (comprising an estimated 1 × 105 CAR T cells/kg) was administered intravenously. No adverse effects attributable to CAR T cell administration were documented. Low levels of integrated CAR copies were documented both in the peripheral blood and lymph node aspirates within the first 11 d after infusion ()). Integrated CAR copy number peaked within the target lymph node at day 50 and then disappeared. CAMA was detected at day 18, peaking at day 50 (at the initiation of rescue chemotherapy) and remaining detectable until the patient was euthanized due to progressive disease on day 182 post-infusion ()). Flow cytometric tracking of the B cell population in the peripheral blood and a target malignant lymph node revealed the progressive loss of the target antigen CD20 on CD79a+ B cells ()). To investigate the loss of CD20 expression, peripheral blood lymphocytes were sorted into CD79a+CD20- and CD79a+CD20lo populations and subject to semi-quantitative PCR analysis using CD20 primers. In the post-treatment, CD79a+CD20- population two products were identified; the expected 891 bp sequence of CD20 and a smaller PCR product (552bp) (Fig. S8). Alignment of the pre-treatment CD20 sequence (891 bp), the smaller post-treatment sequence (552bp) and the canine CD20 reference sequence revealed the complete absence of exons 4 and 5 in the smaller post-treatment product ()). These exons encode the second extracellular loop of CD20 that is thought to contain the CD20 CAR target epitope. Together, these data suggest that immunological pressure exerted by a low dose of CD20 CAR T cells contributed to the emergence of a B cell escape variant by virtue of modified CD20.
Figure 5. Treatment of patient 429–006 with cCD20-BB-ζ CAR T cells
Discussion
We conducted a first-in-species study and demonstrated the ability to generate permanently transduced canine CD20 CAR T cells that function and kill in an antigen-specific manner. The expansion of the different patient products was variable, with increased ex vivo proliferation positively correlated with survival. In the two dogs where CAR T cells were detected by qPCR, a decrease in target antigen expression was observed. Overall, we showed that it is feasible to manufacture permanently transduced canine CAR T cells and these cells were well tolerated and detectable in dogs with DLBCL. As in human patients with B cell malignancies, development of anti-CAR immunity, poor expansion of the CAR T cell product and antigen loss emerged as three challenges for effective CAR T cell therapy in dogs with DLBCL.
Development of human anti-mouse antibodies has been detected in patients treated with CD20 CAR T cells,Citation24 and induction of cellular immunity against CD19 and CD20 CAR T cells was documented in a separate group and posited as a reason for limited CAR persistence.Citation25 Seroconversion was also implicated in limiting the activity of CARs targeting α-folate receptor in ovarian cancer patients.Citation26 Induction of CAMA in two patients emphasizes the value of using immunologically intact animals as pre-clinical models for CAR T cell therapy, and the potential role of cell-mediated immunity against the canine CAR T product is worthy of future investigation. “Caninization” of the scFv represents a potential future approach to prevent the induction of CAMA and possible cell-mediated anti-CAR immunity.
Stage V lymphoma has been associated with poor outcome in dogs with DLBCL.Citation27,Citation28 Despite a high circulating peripheral blast count in 429–006, this dog had the longest duration of survival and virtually complete loss of detectable CD20 protein from malignant lymphocytes. Ruella and coworkers demonstrated how targeting immunosuppressive components of the tumor microenvironment (TME) enabled successful treatment of Hodgkin’s lymphoma with CAR T cells.Citation29 Across multiple clinical trials of human CD19 CAR T cells, overall response rates appear to be greater for patients with ALL compared to DLBCL, Citation2 though this observation has not been formally proven. The influence of the anatomic disease location upon treatment outcomes following CAR T cell therapy for lymphoid neoplasia is uncertain, but it is certainly plausible that CAR T cells perform better outside of the TME. Future evaluation of whether CAR T cells are as functional in the potentially immunosuppressive tumor microenvironment of a neoplastic lymph node in comparison with their ability to eliminate tumor in peripheral circulation is merited.
Multiple T cell memory phenotypes exist, and product composition with defined T cell proliferative properties and differentiation status is being actively researched for cellular therapies.Citation30 Fraietta et al. reported a significant positive correlation between ex vivo product doubling and circulating CAR DNA copy number,Citation31 And increased ex vivo proliferation of T cells was associated with increased overall survival in our study. Identification of T cells with stem-like features (memory stem T cells/TSCM) was of particular significance for the field.Citation32 Fraietta et al. also showed that patients experiencing complete remission (CR) after CD19 CAR T cell therapy for CLL had significantly greater frequency of TSCM in the apheresis products used to manufacture CAR T cells.Citation31 As IL-7 and IL-15 supplementation following CD3 and CD28 stimulation are requirements for TSCM maintenanceCitation33 we changed our culture conditions accordingly. Decreasing the length of time for which CAR T cell products are cultured ex vivo prior to infusion limits T cell differentiation and results in a more active product as shown by preclinical in vitro and in vivo testing.Citation34 Similarly, using a 4-1BB-based CAR as opposed to the CD28 co-stimulatory domain favors a T central memory phenotype.Citation21,Citation23 The early data presented here support the use of IL-7 and IL-15 in culture to maintain a less differentiated, memory-like phenotype in canine T cells, as these modifications to manufacture preceded clear target antigen modulation in vivo. Work to further improve the expansion and activity of canine CAR T cells is ongoing.
Antigen loss through immunoediting has been described as a consequence of cancer immunotherapy.Citation35 In 2015, Sotillo and colleagues described the selection of preexisting CD19 splice variants following CD19 CAR T cell therapy as a mechanism of resistance in relapsed patients.Citation36 Orlando et al. also found loss of CD19 following CAR T therapy in a separate cohort of patients, however, the data there did not support the emergence of preexisting splice variants; instead, the development of de novo CD19 mutants was documented.Citation37 Irrespective of the actual mechanism of resistance, it is clear that loss of CD19 antigen is a major concern for a subset of treated patients and was the rationale for developing a CD22-specific CARCitation38 as well as a CD19/CD20 bi-specific CAR.Citation39 CD20 loss following rituximab therapy has been reported in DLBCL.Citation40 We observed evidence of selection pressure being exerted on CD20+ targets in two of our canine patients, more subtly in 429–003 as the emergence of a CD20int population and more definitively in 429–006 as the near complete absence of CD20+ cells. Our sequencing revealed loss of exons 4 and 5 in transcripts within the CD20- population, which match the domains frequently lost in human B cell malignancies targeted by anti-CD20 monoclonal antibodies.Citation41 Definitive evidence of whether this represents emergence of a splice variant or the development of a novel mutant remains to be determined, however, the loss of target antigen/epitope following CAR T cell treatment indicates that similar escape mechanisms occur in both dogs and humans and re-emphasizes the value of using immunologically competent canine patients with spontaneous disease for investigating next generation CAR T cell approaches against alternate or multiple antigens.
As the value of the dog as a pre-clinical model for cancer immunotherapy becomes appreciated, our understanding of the T cell effector/memory subsets in the dog that are relevant to our therapeutics will improve. This development alongside the increasing availability of reagents to elucidate phenotypic and functional immune cell subsets will help advance the field for both species.
Despite small number of dogs described in this first-in-species clinical trial, we observed multiple parallels with clinical CAR T cell trials and have established canine DLBCL as a preclinical entity that more faithfully represents the clinical challenges compared to rodent models. Canine DLBCL provides a unique setting to address product manufacturing issues and the induction of resistance to CAR T cell therapy. Alternative manufacturing processes and CARs against solid tumor and other hemolymphoid malignancies are currently being investigated, and evaluations of combinatorial therapies with well-characterized or novel immunomodulatory agents is planned. This will enable rapid and accurate screening of next generation CAR T cell therapies in a comprehensive, spontaneously occurring, outbred and immunocompetent model.
Author contributions
Conception and design: M.K. Panjwani, N.J. Mason
Development of methodology: M.K. Panjwani, M.J. Atherton, I. Kulikovsaya, N.J. Mason
Acquisition of data (provided animals, acquired and managed patients,
provided facilities, etc.): M.K. Panjwani, M.J. Atherton, M.A. MaloneyHuss, K.P. Haran, A. Xiong, M. Gupta, S.F. Lacey, I. Kulikovsaya N.J. Mason
Analysis and interpretation of data (e.g., statistical analysis, biostatistics,
computational analysis): M.K. Panjwani, M.J. Atherton, N.J. Mason
Writing, review, and/or revision of the manuscript: M.K. Panjwani, M.J. Atherton, I. Kulikovsaya, S.F. Lacey, N.J. Mason
Conflict of interest
M.K.P. & N.J.M. are listed as inventors on a patent pending for the development of autologous canine CAR T cells.
Supplemental Material
Download ()Acknowledgments
We would like to thank Drs. Weinstein, Walsh and Hoepp for their evaluation and interpretation of the fine needle aspirates from clinical patients, Dr. Jonathan Delong for his assistance with flow sorting and the laboratory of Dr. Michael Milone for help in generating lentivirus.
Supplementary Materials
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
References
- Zheng -P-P, Kros JM, Li J. Approved CAR T cell therapies: ice bucket challenges on glaring safety risks and long-term impacts. Drug Discov Today. 2018;23:1175–13. doi:10.1016/j.drudis.2018.02.012.
- Milone MC, Bhoj VG. The pharmacology of T cell therapies. Mol Ther Methods Clin Dev. 2018;8:210–221. doi:10.1016/j.omtm.2018.01.010.
- Majzner RG, Mackall CL. Tumor antigen escape from CAR T-cell therapy. Cancer Discov. 2018;8:1219–1226. doi:10.1158/2159-8290.CD-18-0442.
- Siegler EL, Wang P. Preclinical models in chimeric antigen receptor-engineered T-cell therapy. Hum Gene Ther. 2018;29:534–546. doi:10.1089/hum.2017.243.
- Zloza A, Karolina Palucka A, Coussens LM, Gotwals PJ, Headley MB, Jaffee EM, Lund AW, Sharpe AH, Sznol M, Wainwright DA, et al. Workshop on challenges, insights, and future directions for mouse and humanized models in cancer immunology and immunotherapy: a report from the associated programs of the 2016 annual meeting for the society for immunotherapy of cancer. J ImmunoTher Cancer. 2017;5:77. doi:10.1186/s40425-017-0278-6.
- Aresu L. Canine lymphoma, more than a morphological diagnosis: what we have learned about diffuse large B-cell lymphoma. Front Vet Sci. 2016;3:77. doi:10.3389/fvets.2016.00077.
- Childress MO, Ramos-Vara JA, Ruple A. Retrospective analysis of factors affecting clinical outcome following CHOP-based chemotherapy in dogs with primary nodal diffuse large B-cell lymphoma. Vet Comp Oncol. 2018;16:E159–68. doi:10.1111/vco.12364.
- Zandvliet M. Canine lymphoma: a review. Vet Q. 2016;36:76–104. doi:10.1080/01652176.2016.1152633.
- Richards KL, Motsinger-Reif AA, Chen H-W, Fedoriw Y, Fan C, Nielsen DM, Small GW, Thomas R, Smith C, Dave SS, et al. Gene profiling of canine B-cell lymphoma reveals germinal center and postgerminal center subtypes with different survival times, modeling human DLBCL. Cancer Res. 2013;73:5029–5039. doi:10.1158/0008-5472.CAN-12-3546.
- Rao S, Lana S, Eickhoff J, Marcus E, Avery PR, Morley PS, Avery AC. Class II major histocompatibility complex expression and cell size independently predict survival in canine B-cell lymphoma. J Vet Internal Med. 2011;25:1097–1105. doi:10.1111/j.1939-1676.2011.0767.x.
- Jubala CM, Wojcieszyn JW, Valli VEO, Getzy DM, Fosmire SP, Coffey D, Bellgrau D, Modiano JF. CD20 expression in normal canine B cells and in canine non-Hodgkin lymphoma. Vet Pathol. 2005;42:468–476. doi:10.1354/vp.42-4-468.
- Roschewski M, Staudt LM, Wilson WH. Diffuse large B-cell lymphoma-treatment approaches in the molecular era. Nat Rev Clin Oncol. 2014;11:12–23. doi:10.1038/nrclinonc.2013.197.
- Atherton MJ, Morris JS, McDermott MR, Lichty BD. Cancer immunology and canine malignant melanoma: A comparative review. Vet Immunol Immunopathol. 2016;169:15–26. doi:10.1016/j.vetimm.2015.11.003.
- Rowell JL, McCarthy DO, Alvarez CE. Dog models of naturally occurring cancer. Trends Mol Med. 2011;17:380–388. doi:10.1016/j.molmed.2011.02.004.
- LeBlanc AK, Breen M, Choyke P, Dewhirst M, Fan TM, Gustafson DL, Helman LJ, Kastan MB, Knapp DW, Levin WJ, et al. Perspectives from man’s best friend: national academy of medicine’s workshop on comparative oncology. Sci Transl Med. 2016;8:324ps5–324ps5. doi:10.1126/scitranslmed.aaf0746.
- Panjwani MK, Smith JB, Schutsky K, Gnanandarajah J, O’Connor CM, Powell DJ, Mason NJ. Feasibility and safety of RNA-transfected CD20-specific chimeric antigen receptor T cells in dogs with spontaneous B cell lymphoma. Mol Ther. 2016;24:1602–1614. doi:10.1038/mt.2016.146.
- Marcucci KT, Jadlowsky JK, Hwang W-T, Suhoski-Davis M, Gonzalez VE, Kulikovskaya I, Gupta M, Lacey SF, Plesa G, Chew A, et al. Retroviral and lentiviral safety analysis of gene-modified T cell products and infused HIV and oncology patients. Mol Ther. 2018;26:269–279. doi:10.1016/j.ymthe.2017.10.012.
- Lyons AB. Analysing cell division in vivo and in vitro using flow cytometric measurement of CFSE dye dilution. J Immunol Methods. 2000;243:147–154. doi:10.1016/s0022-1759(00)00231-3.
- Verhoeyen E, Costa C, Cosset F-L. Lentiviral vector gene transfer into human T cells. Methods Mol Biol. 2009;506:97–114. doi:10.1007/978-1-59745-409-4_8.
- Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D, Naldini L. A third-generation lentivirus vector with a conditional packaging system. J Virol. 1998;72:8463–8471.
- Salter AI, Ivey RG, Kennedy JJ, Voillet V, Rajan A, Alderman EJ, Voytovich UJ, Lin C, Sommermeyer D, Liu L, et al. Phosphoproteomic analysis of chimeric antigen receptor signaling reveals kinetic and quantitative differences that affect cell function. Sci Signal. 2018;11.
- Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, Smith JP, Walker AJ, Kohler ME, Venkateshwara VR, et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med. 2015;21:581–590. doi:10.1038/nm.3838.
- Kawalekar OU, O’Connor RS, Fraietta JA, Guo L, McGettigan SE, Posey AD, Patel PR, Guedan S, Scholler J, Keith B, et al. Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR T cells. Immunity. 2016;44:380–390. doi:10.1016/j.immuni.2016.01.021.
- Till BG, Jensen MC, Wang J, Chen EY, Wood BL, Greisman HA, Qian X, James SE, Raubitschek A, Forman SJ, et al. Adoptive immunotherapy for indolent non-Hodgkin lymphoma and mantle cell lymphoma using genetically modified autologous CD20-specific T cells. Blood. 2008;112:2261–2271. doi:10.1182/blood-2007-12-128843.
- Jensen MC, Popplewell L, Cooper LJ, DiGiusto D, Kalos M, Ostberg JR, Forman SJ. Antitransgene rejection responses contribute to attenuated persistence of adoptively transferred CD20/CD19-specific chimeric antigen receptor redirected T cells in humans. Biol Blood Marrow Transplant. 2010;16:1245–1256. doi:10.1016/j.bbmt.2010.03.014.
- Kershaw MH, Westwood JA, Parker LL, Wang G, Eshhar Z, Mavroukakis SA, White DE, Wunderlich JR, Canevari S, Rogers-Freezer L, et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res. 2006;12:6106–6115. doi:10.1158/1078-0432.CCR-06-1183.
- Teske E, van Heerde P, Rutteman GR, Kurzman ID, Moore PF, MacEwen EG. Prognostic factors for treatment of malignant lymphoma in dogs. J Am Vet Med Assoc. 1994;205:1722–1728.
- Jagielski D, Lechowski R, Hoffmann-Jagielska M, Winiarczyk S. A retrospective study of the incidence and prognostic factors of multicentric lymphoma in dogs (1998–2000). J Vet Med Ser A. 2002;49:419–424.
- Ruella M, Klichinsky M, Kenderian SS, Shestova O, Ziober A, Kraft DO, Feldman M, Wasik MA, June CH, Gill S. Overcoming the immunosuppressive tumor microenvironment of hodgkin lymphoma using chimeric antigen receptor T cells. Cancer Discov. 2017;7:1154–1167. doi:10.1158/2159-8290.CD-16-0850.
- Busch DH, Fräßle SP, Sommermeyer D, Buchholz VR, Riddell SR. Role of memory T cell subsets for adoptive immunotherapy. Semin Immunol. 2016;28:28–34. doi:10.1016/j.smim.2016.02.001.
- Fraietta JA, Lacey SF, Orlando EJ, Pruteanu-Malinici I, Gohil M, Lundh S, Boesteanu AC, Wang Y, O’Connor RS, Hwang W-T, et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat Med. 2018;24:563–571. doi:10.1038/s41591-018-0010-1.
- Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF, Almeida JR, Gostick E, Yu Z, Carpenito C, et al. A human memory T cell subset with stem cell-like properties. Nat Med. 2011;17:1290–1297. doi:10.1038/nm.2446.
- Cieri N, Camisa B, Cocchiarella F, Forcato M, Oliveira G, Provasi E, Bondanza A, Bordignon C, Peccatori J, Ciceri F, et al. IL-7 and IL-15 instruct the generation of human memory stem T cells from naive precursors. Blood. 2013;121:573–584. doi:10.1182/blood-2012-05-431718.
- Ghassemi S, Nunez-Cruz S, O’Connor RS, Fraietta JA, Patel PR, Scholler J, Barrett DM, Lundh SM, Davis MM, Bedoya F, et al. Reducing ex vivo culture improves the antileukemic activity of Chimeric Antigen Receptor (CAR) T cells. Cancer Immunol Res. 2018;6:1100–1109. doi:10.1158/2326-6066.CIR-17-0405.
- Mittal D, Gubin MM, Schreiber RD, Smyth MJ. New insights into cancer immunoediting and its three component phases–elimination, equilibrium and escape. Curr Opin Immunol. 2014;27:16–25. doi:10.1016/j.coi.2014.01.004.
- Sotillo E, Barrett DM, Black KL, Bagashev A, Oldridge D, Wu G, Sussman R, Lanauze C, Ruella M, Gazzara MR, et al. Convergence of acquired mutations and alternative splicing of CD19 enables resistance to CART-19 immunotherapy. Cancer Discov. 2015;5:1282–1295. doi:10.1158/2159-8290.CD-15-1020.
- Orlando EJ, Han X, Tribouley C, Wood PA, Leary RJ, Riester M, Levine JE, Qayed M, Grupp SA, Boyer M, et al. Genetic mechanisms of target antigen loss in CAR19 therapy of acute lymphoblastic leukemia. Nat Med. 2018;24:1504–1506. doi:10.1038/s41591-018-0146-z.
- Shah NN, Stetler-Stevenson M, Yuan CM, Shalabi H, Yates B, Delbrook C, Pico-Knijnenburg I, Simons E, Jerchel I, Wawer A, et al. Minimal residual disease negative complete remissions following anti-CD22 Chimeric Antigen Receptor (CAR) in children and young adults with relapsed/refractory Acute Lymphoblastic Leukemia (ALL). Blood. 2016;128: 650–650. doi:10.1182/blood-2016-02-701029.
- Zah E, Lin M-Y, Silva-Benedict A, Jensen MC, Chen YY. T cells expressing CD19/CD20 bispecific chimeric antigen receptors prevent antigen escape by malignant B cells. Cancer Immunol Res. 2016;4:498–508. doi:10.1158/2326-6066.CIR-15-0231.
- Duman BB, Sahin B, Ergin M, Guvenc B. Loss of CD20 antigen expression after rituximab therapy of CD20 positive B cell lymphoma (diffuse large B cell extranodal marginal zone lymphoma combination): a case report and review of the literature. Med Oncol. 2012;29:1223–1226. doi:10.1007/s12032-011-9955-3.
- Gamonet C, Bole-Richard E, Delherme A, Aubin F, Toussirot E, Garnache-Ottou F, Godet Y, Ysebaert L, Tournilhac O, Dartigeas C, et al. New CD20 alternative splice variants: molecular identification and differential expression within hematological B cell malignancies. Exp Hematol Oncol. 2015;5:7. doi:10.1186/s40164-016-0036-3.