
ABSTRACT
The application of chimeric antigen receptor (CAR)-T cell therapy in patients with advanced solid tumors remains a significant challenge. Simultaneously targeting antigen and the solid tumor microenvironment are two major factors that greatly impact CAR-T cell therapy outcomes. In this study, we engineered CAR-T cells to specifically target B7-H3, a protein commonly found in solid human tumors, using a single-chain variable fragment (scFv) derived from an anti-B7-H3 monoclonal antibody. We tested the antitumor activity of B7-H3 CAR-T cells in mouse models with solid human tumors and determined that B7-H3 CAR-T cells exhibited potent antitumor activity against B7-H3+ tumor cells in vitro and in vivo. In addition, PD-1 decoy receptors were engineered to include extracellular PD-1 fused to the intracellular stimulatory domain of either CD28 or IL-7 receptor, respectively, which were then introduced into B7-H3 CAR-T cells. As a result, these newly modified, superior CAR-T cells exhibited more persistent antitumor activity in B7-H3+/B7-H1+ tumors in vivo. Our findings indicate that B7-H3 specific CAR-T cells have the potential to treat multiple types of advanced solid tumors.
Introduction
Chimeric antigen receptor (CAR) composed of antibody binding domain connected to T cell-activated signaling domain with one or two co-stimulatory domains, can recognize tumor antigen with high specificity and trigger T cell activation in a non-MHC restricting manner.Citation1−Citation3 CAR-T cells have demonstrated remarkable efficacy in patients with several types of hematologic malignancies.Citation4,Citation5 However, the treatment of solid tumor, responsible for >95% of human cancer, with this technology has been difficult. In addition to inappropriate T cell trafficking and insufficient infiltration into solid tumors,Citation6 a suppressive tumor microenvironment (TME)Citation7 and a lack of tumor-specific or tumor selective targets on solid tumorsCitation8 may contribute to the limited functionality of this method.Citation9 Therefore, engineering CAR-T cells that selectivity recognize tumor antigens on solid tumors and that are resistance to the suppressive TME may improve CAR T cell efficacy in the clinic.Citation10
Eighteen years ago, B7-H3 (CD276) was molecularly cloned and identified as a member of the B7/CD28 family with immune modulatory functions.Citation11 While it appears that B7-H3 mRNA is ubiquitously expressed in various mouse and human tissues under normal conditions, immunohistochemical (IHC) analysis demonstrates that the expression of its protein is limited to a select subset of tissues.Citation12,Citation13 Interestingly, B7-H3 protein is frequently upregulated in the majority of solid human tumors such as prostate cancer, non-small-cell lung cancer, pancreatic cancer, breast cancer, ovarian cancer and colorectal cancerCitation14-Citation24 and within the tumor vasculature as well.Citation25 Several studies have also shown that B7-H3 overexpression is associated with tumor progression, metastatic potential and a poor prognosis.Citation26-Citation29 The immune-modulatory functions of B7-H3 remain controversial, with data supporting both stimulatory and inhibitory roles in T cell immunity in different model systems.Citation30 A monoclonal antibody (mAb) recognizing B7-H3 is being used to treat refractory pediatric brain cancers and thus far the clinical trial has yielded safe and promising results.Citation31 Therefore, B7-H3 may represent a potential target for CAR-T cell directed therapy.
Tumor-infiltrating T lymphocytes (TILs) play an important role in the control of tumor progression albeit their activity is largely impaired in the TME of patients with advanced cancers. One of the major mechanisms of TIL dysfunction is mediated by the PD-1/B7-H1 interaction, which attenuates the response of effector T-cells upon antigen encounter.Citation32 Cell surface B7-H1 protein is upregulated in the TME, largely due to interferon-gamma secreted by T cells, and serves as a major mechanism of immune escape.Citation33,Citation34 Hence, the PD-1/B7-H1 axis may contribute to limiting the efficacy of CAR-T cells in the TME. In this study, we constructed a new chimeric antigen receptor binding domain using an scFv derived from a newly generated B7-H3 mAb. Because activated CAR-T cells express high levels of PD-1, to conquer this immune escape mechanism, we also genetically engineered CAR-T cells to co-express a PD-1 decoy receptor that replaces the PD-1 transmembrane and intracellular signaling domains with a co-stimulating signaling domain of CD28 or a constitutively active IL-7 receptor, so as to convert or compete possible inhibitory signal to improve T cell function. Here we show that B7-H3 directed CAR-T cells display efficient cytotoxic ability against solid tumors both in vitro and in humanized mouse tumor models in vivo.
Materials and methods
Mice
NCG mice (NOD-Prkdcem26Cd52Il2rgem26Cd22/Nju) and Balb/c mice were purchased from Model Animal Resource Information Platform of Nanjing University. Six to eight weeks of age female mice were maintained under specific pathogen-free facilities at Sun Yat-sen University and Fujian Medical University. All animal experiments were conducted in accordance with NIH guidelines and were approved by the Institutional Animal Care and Use Committees of the respective universities.
Tissue and blood samples
Formalin-fixed paraffin-embedded or surgical tissue samples were obtained from Department of Pathology, Union Hospital of Fujian Medical University. Informed consent was obtained before sample collection. Buffy coat collected from healthy adult volunteers was obtained from Fujian Blood Center under consent, and was used for isolation of PBMCs. The study was approved by the Committee for the Ethical Review of Research, Fujian Medical University (No. 2016–33).
Cell lines and culture conditions
The human melanoma (624Mel), lung cancer (PG, A549), liver cancer (Huh7, HepG2), breast cancer (MDA-MB-231), ovarian cancer (SKOV3), cervical cancer (HeLa), squamous carcinoma (SCC-47), colon cancer (HT-29, SW620), immortal epithelial (HLB100) cell lines were purchased from ATCC. Lenti-X 293T cells were purchased from TaKara. MDA-MB-231-H3KO cell line was generated from MDA-MB-231 cells by genetically knocking out B7-H3 gene using pLentiCRISPR-E vector (Addgene, 78852). PG cell line were transduced with lentiviral vector encoding the luciferase reporter gene luc2P (Photinus pyralis) or the human 4Ig-B7-H3 cDNA (pLVX vector, Clontech) to generate PG-luc cell line and PG-hB7-H3 cell line, respectively. All cells were maintained in RPMI-1640 supplemented with 10% fetal bovine serum and 2 mmol/L L-glutamine (Invitrogen).
Generation of monoclonal antibody
Female Balb/c mice at the age of 8–10 weeks were immunized subcutaneously (s.c.) at multiple sites with 200 µL of emulsion comprising 100 μg of human 4Ig-B7-H3-mFc fusion protein and complete Freund’s adjuvant (CFA) (Sigma-Aldrich). Three weeks later mice were immunized with 50–100 μg of the protein with incomplete Freund’s adjuvant (IFA) (Sigma-Aldrich) by s.c., three times altogether. Mice were bled 2 weeks after each immunization for serum titer testing. When the titer was sufficient, mice were boosted with a 60 μg/mouse booster of protein in PBS via intraperitoneal injection (i.p.). Hybridomas were obtained by fusing immune mouse spleen cells and SP2/0-Ag14 myeloma cell line (ATCC). Boosted mice were sacrificed by carbon dioxide and spleens were harvested aseptically. Whole spleen was dissociated into single-cell suspensions and red blood cells were lysed by ACK buffer. SP2/0-Ag14 myeloma and spleen cells were mixed at 1:1 ratio in a 50-mL conical centrifuge tube. After centrifugation, the supernatant was discarded and cell fusion was performed with 50% polyethylene glycol (PEG) (Roche). The fused cells were cultured for 8–10 days in hypoxanthine-aminopterin-thymidine (HAT) selection medium (Sigma-Aldrich), and the antibody-producing hybridomas that could bind to hB7-H3 expressing cells were screened by ELISA and confirmed by flow cytometry analysis. Briefly, 96-well half area high binding plates were precoated with the hB7-H3-hFc at 2µg/mL and kept at 4°C overnight. After washing and blocking, culture supernatants of the hybridoma cells were added into the wells. The plate was then incubated for 2 h at room temperature. After washing, each well was added with 30 µL of 1:100,000 diluted anti-mouse-Ig-HRP antibody, followed by incubation at room temperature for 1 h. The plate was then washed 7 times with PBS/Tween before adding 50 µL of TMB solution to each well and incubated for 15 min, followed by addition of stop solution. Absorbance was read at 450 nm by an automated ELISA reader. To confirm the supernatants of hybridomas for binding hB7-H3, hB7-H3 expressing CHO cells (CHO/hB7-H3) were incubated with the supernatants, and the cells were then washed and further incubated with anti-mIgG-APC (eBiosciences). Flow cytometry analysis was performed using FACSCalibur or FACSVerse (BD Biosciences). The subcloning of positive hybridoma was performed using the limiting dilution technique for 3–5 times to achieve a monoclonal culture.
Construction of the B7-H3 targeted CAR vectors
The variable region sequence of the mAb-7E12 was cloned from hybridoma using 5ʹRACE PCR according to manufacturer’s instructions, and then a scFv specific for B7-H3 was engineered. To generate B7-H3 targeted CARs, H3 CAR (second generation) was constructed by linking the B7-H3 specific scFv to a hinge and transmembrane domain of CD8, followed by an intracellular domain of 4-1BB and an intracellular domain of CD3ζ. Similarly, H3/DS CAR (third generation) was constructed by incorporating another co-stimulatory intracellular domain of CD28 to the H3 CAR. CAR linked with a truncated form of the CD3ζ intracellular domain lacking signal transduction was designed as a control. Control, H3 and H3/DS CARs were further linked to the reporter enhanced green fluorescence protein (EGFP) with a T2A oligopeptide, respectively. An extracellular domain of PD1 was fused with the transmembrane and cytoplasmic domains of CD28Citation35 or of mutant IL-7R (with a TTGTCCCAC insertion at transmembrane region)Citation36 to generate a chimeric decoy receptor PD28 or PDmut7R. The chimeric decoy receptors were respectively linked to H3 CAR with a T2A oligopeptide to produce H3/PD28 CAR and H3/PDmut7R CAR. All the CAR cassettes were subsequently subcloned to the lentiviral vector (pLVX-EF1alpha-IRES-ZsGreen1, TaKara).
Production of lentivirus and T cell transduction
Lentivirus particles were produced by transient transfection of Lenti-X 293T cells with corresponding CAR-expressing plasmids and two packaging plasmids psPAX2 and pMD.2G on a 3:2:1 ratio using Polyethylenimine (Polysciences, 23966–2). Lentivirus-containing culture medium were collected at 48 and 72-h post-transfection and centrifuged at 1000 g for 10 min to remove cell debris. The supernatants were further filtered through 0.45 μm PVDF low protein-binding filters and concentrated by ultra-centrifuging at 100,000 g for 2 h. Subsequently, the lentivirus particles were re-suspended in appropriate volume using RPMI-1640 medium at 4°C overnight. Final viral samples were titrated and stored at −80°C in single-use aliquots.
Human peripheral blood mononuclear cells (PBMCs) from healthy donors were negatively purified using a pan T-cell isolation kit (Invitrogen,11344D), and then activated by culture with anti-CD3/CD28 beads (Thermo Fisher Scientific,11131D) according to the manufacturer’s protocol. Two days post-activation, PBMCs were transduced with indicated lentivirus particles coated on the non-tissue culture plates with 20μg/mL RetroNectin (Takara, T100B). Spinoculation was performed for 2 h at 900 g, 32°C, followed by incubation for 48 h. After that, cells were pelleted and resuspended in the fresh media containing 20 ng/mL rhIL-7 (Peprotech, 200–07) and 10ng/mL rhIL-15 (Peprotech, 200–15). Cells were counted and fed every 2 days. Transduction efficiencies were analyzed by flow cytometry to confirm the generation of CAR-T cells. Five days after transduction, CAR-T cells were cultured in the medium with low FBS and without cytokines for 2 days prior to being used for functional assays in vitro.
T cell functional assays
The cytotoxicity of T cells was determined by incubating corresponding T cells with 0.5µM CellTrace FarRed (Invitrogen, C34564)-labeled target cells (PG, SKOV3, MDA-MB-231 or MDA-MB-231-H3KO) at the indicated effector:target (E:T) ratios for 12 h. Then, 0.01mg/mL DAPI (Sigma-Aldrich, D8417) were added to each reaction, and the cells were immediately analyzed by flow cytometry within 10 s. The % cell lysis was calculated as follows: [(FarRed+ DAPI+ cells – spontaneous apoptosis)/total FarRed+ cells] × 100%.
Cytokine-releasing assays were performed by co-culturing 1 × 106 T cells with 5 × 105 irradiated (100 Gy) target cells at a 2:1 ratio. The supernatants from each coculture reaction were collected 24 h later. Cytokine levels of IL-2, IL-4, IL-6, IL-10, IFN-gamma, and TNF-α in the supernatants were determined using the Human Th1/Th2 Cytokine Kit II (BD Biosciences) in accordance with the manufacturer’s instructions.
Flow cytometry and antibodies
Chinese hamster ovary (CHO) cells stably transfected with hB7-H3, hB7-H1, hB7-H4, or mB7-H3 were used to assess the binding activity of the B7-H3 mAb and its scFv-Fc antibody by flow cytometry. The CHO cells were incubated with respective mAbs on ice for 30 min, followed by washed and further incubated with anti-mIgG-APC (eBioscience). The expression levels of cell-surface B7-H3 or B7-H1 were detected on human tumor cell lines by using anti-human B7-H3 mAb (7E12) and anti-human B7-H1 mAb (5H1). Transduction efficiency of CAR-T cells were analyzed using 4Ig-hB7-H3-mFc fusion protein, anti-human PD-1 mAb (BD Biosciences) or EGFP. T cell phenotypes were determined with fluorescent-labeled antibodies against CD3, CD4, CD8, CD45RA, CD127, CD45RO, CD127, CD44, CD62L, CCR7, PD-1 (BD Biosciences, eBioscience, or BioLegend). Flow cytometry analysis was performed using FACSCalibur or FACSVerse (BD Biosciences).
Immunohistochemistry
Mouse tumor tissues and human tissue sections were analyzed for B7-H3 expression. Human tissue microarray sections were purchased from US Biomax or Zhuoli Biotech (Shanghai). Pathology samples of human tumors and surgical liver tissues were obtained from the Pathology Department, Union Hospital of Fujian Medical University. All IHC staining followed the protocol described previously.Citation37 Briefly, endogenous peroxidase activity, unspecific binding activity, and endogenous avidin/biotin binding activity were blocked by incubating the tissue sections with 0.3% hydrogen peroxide solution, Block AEC solution (AbD Serotec), and Avidin Blocking Buffer (Vector Laboratories), respectively. The sections were then stained with a commercial mAb (clone 6A1, Abcam) at final concentration of 5 μg/mL against B7-H3 at 4°C for overnight. Subsequently, the HRP polymer conjugated anti-biotin secondary Ab in Catalyzed Signal Amplification System (DAKO, K1500) was used for staining.
Tumor models and treatment
For the subcutaneous mouse model, NCG mice were injected in the flank subcutaneously with 0.5 × 106 Pulmonary giant cell carcinoma (PG) cells, and the mice were randomly divided into groups consisted of n = 5 per group. When the tumors reached a mean diameter of approximately 4 mm at days 4–5 after injection, each group mice were intravenously treated with PBS, or 5 × 106 CAR-T cells (Control CAR-T, H3 CAR-T, H3/DS CAR-T, H3/PD28 CAR-T, or H3/PDmut7R CAR-T) per mouse on day 5, 10 and 15, respectively. In the experiments of combination therapy, mice (n = 5 per group) were treated with three doses of B7-H3 specific CAR-T cells alone as described above, or in combination with two doses of anti-PD-1 antibody (clone M3, produced by our lab) by i.v. administration on day 6 and 11, respectively. Tumor sizes were measured twice a week with calipers. Blood were drawn from the mice 1 week after treatments to quantify persistent circulating CD4+ and CD8+ T cells and cytokine-producing in the serum.
For the subcutaneous ovarian carcinoma mouse model, 1 × 106 SKOV3 tumor cells were subcutaneously injected to the flank of female NCG mice and were randomly grouped. When the tumors reached a mean diameter of approximately 3–4 mm, the mice were intravenously administered with 5 × 106 CAR-T cells on day 12, 20 and 29, respectively.
For the re-challenge model of subcutaneous tumors, NCG mice were s.c. inoculated with 0.5 × 106 PG-B7-H3 cells and were randomly separated into seven independent groups. When the tumors reached a diameter of approximately 3–4 mm, the mice were i.v. treated with of 2 × 106 CAR-T cells on day 7. At day 30, tumors grew in the mice treated with control T cells were surgically excised, and this group of mice were used as control cohort in the following tumor re-challenge experiments. At day 44, all groups of mice were re-challenged by subcutaneously injecting with 2.5 × 106 tumor cells. Tumor sizes and body weight were measured. At day 71, mice were sacrificed; the tissues and blood samples were obtained; single-cell suspensions were analyzed by flow cytometry.
For the metastatic mouse model, NCG mice were intravenously injected with 1 × 105 PG cells or PG cells expressing Firefly-luciferase. The mice were then intravenously treated with 5 × 106 CAR-T cells on day 3, 7 and 15, respectively. Tumor growth in each treatment group of mice (n = 4 per group) was assessed by bioluminescent imaging weekly. For the survival analysis, separated experiments were performed with same treatments to the groups of mice consisted of n = 8 per group.
Graphs and statistical analysis
The graphs and data analysis were generated using GraphPad Prism Software v.7.0. Each experiment presented in this report was repeated at least three times. All data are presented as the means ± SD. Significant difference was analyzed by two-way ANOVA, two-tailed unpaired t-test, unpaired and non-parametric Mann-Whitney test with two-tailed P value calculation, or log-rank (Mantel-Cox) test, accordingly. P-values are represented as * (P < .05), **(P < .01), ***(P < .001) and ****(P < .0001); NS, not significant.
Results
The generation and characterization of mouse monoclonal antibody against human B7-H3
To specifically target B7-H3 cancer antigen, we generated a panel of mouse anti-human B7-H3 hybridomas. The mAb derived from hybridoma clone, 7E12, was shown to bind to CHO cells transfected with human 4Ig-B7-H3 protein (CHO-hB7-H3), but not to mock-transfected CHO control cells (CHO-Mock) (Figure S1A). The binding affinity and specificity of scFv derived from clone 7E12 was validated using recombinant scFv-Fc fusion protein. The scFv bound specifically to CHO-hB7-H3, not to CHO cells expressing human B7-H1 (CHO-hB7-H1), human B7-H4 (CHO-hB7-H4), mouse B7-H3 (CHO-mB7-H3), nor to CHO-Mock cells (Figure S1A). The binding of scFv to CHO cells expressing human 4Ig-B7-H3 protein was dose-dependent (Figure S1B). The scFv exhibited slightly lower but comparable binding affinity to B7-H3 protein compared with the mAb-7E12 (scFv, KD = 0.168nM vs. mAb, KD = 0.0244nM, Table S1; Figure S1B). These data demonstate the specificity of the mAb of clone 7E12 against human B7-H3 and confirm that the scFv retains high affinity and specificity to human B7-H3. The mAb-7E12 and its scFv were thus chosen for further experiments.
B7-H3 cell surface protein is widely expressed on various solid human tumors
Using flow cytometry analysis, high levels of B7-H3 were detected on various tumor cell lines derived from solid tumors, including melanoma, colon cancer, lung cancer, hepatocellular carcinoma, ovarian cancer, renal cancer, pancreatic cancer, and prostate cancer by using mAb-7E12 (, Table S2). Interestingly the majority of tumor lines derived from hematological malignancies were found to be negative or to have a low level of B7-H3 expression (Table S2).
Figure 1. B7-H3 expression on human tumors. (a) Cell-surface expression of B7-H3 on cell lines and in solid human tumors from patient tissue. Flow cytometry analyses using 7E12-mAb were performed to detect cell-surface B7-H3 on numerous human tumor cell lines, including melanoma (624Mel), lung cancer (PG, A549), liver cancer (Huh7, HepG2), breast cancer (MDA-MB-231), ovarian cancer (SKOV3), cervical cancer (HeLa), squamous carcinoma (SCC-47), and colon cancer (HT-29, SW620). HLB100, a human epithelial cell line which is tumorigenic in nude mice. Gray area: isotype; Dotted line: B7-H3. (b) The microarray tumor and normal tissue slides (US Biomax or Zhuoli Biotech) were analyzed by IHC using anti-B7-H3 mAb (clone 6A1, Abcam). Representative immunohistochemical staining of B7-H3 expression in the normal tissues verse tumor tissues from a variety of solid human tumors including colon cancer, gastric carcinoma, ovarian cancer, breast cancer, lung cancer, endometriasl cancer, melanoma and prostate cancer. Images were taken under x400 magnification
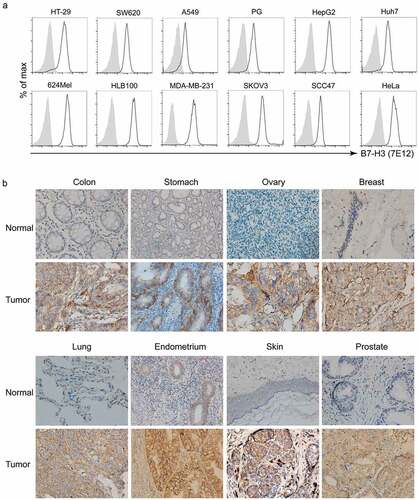
Using immunohistochemical analysis, B7-H3 expression was also detected on microarray tissue specimens from various human tumors including colon cancer, gastric cancer, ovarian cancer, breast cancer, lung cancer, endometrial cancer, melanoma, and prostate cancer, but was either absent or very low level on normal tissues (). The IHC staining of tumor microarray tissues also showed a high percentage of B7-H3 expression from multiple solid tumors, including esophageal cancer (20/20 = 100%), gastric cancer (6/20 = 30%), hepatocellular carcinoma (11/20 = 55%), colorectal cancer (29/40 = 72.5%) and breast cancer (14/20 = 70%) (Table S3). Normal liver tissue was focally positive for B7-H3 staining, however, positive expression was predominantly intracellular and rarely on the cell surface (Figure S2A). Single human liver cells were isolated from human liver tissue samples after surgical intervention and were stained with biotin labeled anti-human B7-H3 scFv-Fc (7E12). No positive staining was noted by FACS analysis (Figure S2A), indicating that B7-H3 protein is predominantly limited to the cytoplasm in normal liver tissue. IHC staining on surgical tumor specimans also showed that normal epithelial cells of the colon and stomach, adjacent to tumor tissues, expressed cytoplasmic B7-H3, but with significantly weaker staining than tumor tissues (Figure S2B).
CAR-T cells based on scFv of mAb-7E12 are effective against tumor growth
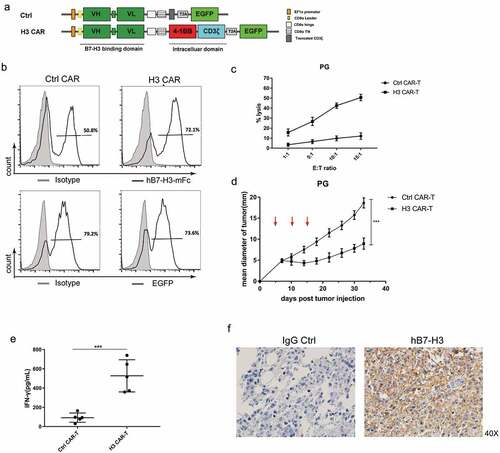
B7-H3 specific CAR was engineered by linking scFv to intracellular 4-1BB’s co-stimulating domain and CD3ζ’s activation domain; CAR, containing a truncated form of CD3ζ lacking activation signal domain was engineered as a control (). Transduction of human pan T cells with CAR expressing lentivirus resulted in an average of approximately 70% CAR expression (). When co-culturing effector cells to target cells at different ratios (E:T), B7-H3 specific CAR-T cells showed sufficient in vitro cytotoxic activity to targeted pulmonary giant cell carcinoma (PG) cells expressing B7-H3 (). To test the antitumor activity of B7-H3 specific CAR-T cells in vivo, a subcutaneous tumor model was generated by implanting PG tumor cells (0.5 x 106/mouse) in non-obese diabetic SCID gamma (NCG) mice. When tumors reached a mean diameter of approximately 5 mm around days 4–5, three infusions of B7-H3 specific or control CAR-T cells (5 x 106/mouse) were i.v. administered, on days 5, 10, and 15. The B7-H3 specific CAR-T cells, but not the control CAR-T cells, significantly inhibited PG tumor growth (). Consistently, infusion with B7-H3 specific CAR-T cells produced a significantly higher level of human IFN-gamma in mouse serum compared to control CAR-T cells (). Meanwhile, IHC staining of tumor tissues revealed an absence of B7-H3 negative cancer cells, indicating that there was no B7-H3 antigen escape in this model (). Taken together, these results demonstrate a high level of efficacy and the specificity of B7-H3 CAR-T cells.
Figure 2. The function of B7-H3 specific CAR-T cells. (a) Schematic structure of the B7-H3 targeted CARs. H3 CAR consisting of anti-B7-H3 scFv and following the CD8 hinge and transmembrane domain, 4-1BB intracellular domain and the CD3ζ intracellular domain. A control CAR was designed by replacing CD3ζ domain in the H3 CAR with a truncated form of CD3ζ. VH: variable heavy chain; VL: variable light chain; L: linker; T2A: 2A oligopeptides of Thosea asigna virus. (b) Expression of B7-H3-specific CAR or EGFP on human T cells was detected by flow cytometry using hB7-H3-mFc protein (black line); Flag-mFc protein served as an isotype control (gray). (c) Cytotoxicity of CAR-T cells was evaluated by incubating CAR-T cells with FarRed-labeled PG cells at different effector/target (E/T) ratios ranging from 1:1 to 15:1 for 12 h. The cells were stained with DAPI and the viability of the PG cells was analyzed by Flow cytometer. Cytotoxicity was then calculated as described in Materials and methods. (d) PG tumors were subcutaneously established in the right flanks of NCG mice. Control or H3 CAR-T cells were i.v. injected to treat the mice at day 5, 10, and 15 (red arrows). The mean tumor diameters in each group (n = 5) are shown. Error bars denote SD. P values by a two-way ANOVA. ***(P < .001). (e) IFN-gamma levels in the serum of the mice were measured by ELISA at day 15 after last treatment. All data are presented as mean ± standard deviation, and error bars denote SD (c-e). P values by a two-way ANOVA (d) or a two-tailed unpaired t-test (e). ***(P < .001). (f) Representative IHC staining using anti-B7-H3 mAb (clone 6A1, Abcam) for B7-H3 protein on the tumor tissues remained in the mice treated with H3 CAR-T cells
Co-expression of PD-1 chimeric receptors improves the anti-tumor activities of B7-H3 specific CAR-T cells
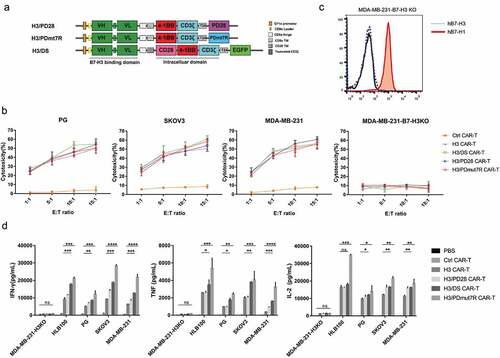
B7-H1/PD-1 inhibitory signaling may attenuate the function of activated B7-H3 specific CAR-T cells in the TME of solid tumors. Our data show that anti-PD-1 antibody helped to significantly slow tumor growth in mice treated with B7-H3 specific CAR-T cells (Figure S3A), indicating that blocking the B7-H1/PD-1 pathway could augment the therapeutic effects of B7-H3 specific CAR-T cells. Thus, as an alternative strategy, co-expression of engineered chimeric receptor such as PD1-CD28, which switched PD-1 mediated signaling from inhibitory to stimulatory by engaging B7-H1 expressed on the TME, may improve the antitumor activity of CAR-T cells for solid tumors.Citation35 In this study, two types of PD-1 chimeric receptors were engineered for investigation; one is that the cell surface domain of PD-1 was linked to the intracellular domain of CD28 (PD28), and the another was the cell surface domain of PD-1 linked to the intracellular domain of IL-7 receptor (PDmut7R) (). Human pan T cells were transduced with lentiviral vectors co-expressing B7-H3 specific CAR with either PD28 (called H3/PD28 CAR-T cells) or PDmut7R (called H3/PDmut7R CAR-T cells), resulting in 60-80% transduction efficiency, determined by flow cytometry (Figure S3B). The B7-H3 specific CAR-T cells containing both intracellular 4-1BB and CD28 domains (called H3/DS CAR-T cells) were used as the control (, S3B). The H3, H3/DS, H3/PD28 and H3/PDmut7R CAR-T cells showed equivalent levels of cytolytic activity against B7-H1+/B7-H3+ tumor cells (PG, SKOV3, and MDA-MB-231, Figure S3C) in a 12h in vitro cytotoxicity assay () and equivalent tumor inhibition effect in an in vivo Winn assay (Figure S4); in contrast, none of these CAR-T cells showed cytolytic activity and tumor inhibition function against B7-H1+/B7-H3- tumor cells (MDA-MB-231- H3KO, , c), and the control CAR-T cells did not respond to any tumor cell type regardless of B7-H3 expression (). Moreover, the H3/PDmut7R CAR-T cells secreted the highest levels of Th1 cytokines (IFN-gamma, TNF and IL-2) in response to B7-H3+ tumor cells (HLB100, PG, SKOV3, and MDA-MB-231) than that of H3/DS, H3/PD28 and H3 CAR-T cells, while the H3/PD28 CAR-T cells exhibiting more productions of IFN-gamma, TNF (when in response to MDA-MB-231), and IL-2 (when in response to SKOV3 and MDA-MB-231) compared to H3 CAR-T cells (). These results suggested an equivalent B7-H3 specific cytolytic activity for H3, H3/DS, H3/PD28 and H3/PDmut7R CAR-T cells; however, the H3/PD28 and H3/PDmut7R CAR-T cells exhibited more cytokine productions than the H3 CAR-T cells, suggesting that co-expression of PD-1 decoy receptors, particually PDmut7R, in B7-H3 specific CAR-T cells may have more potent anti-tumor activities.
Figure 3. Antitumor activity of B7-H3 targeted CAR-T cells coexpressing PD-1 chimeric receptors in vitro. (a) Schematic representation of H3/DS CAR, H3/PD28 CAR and H3/PDmut7R CAR constructs. PD28: extracellular PD-1 fused with CD28 domains; PDmut7R: extracellular PD-1 fused with IL-7R domains. (b) CAR-T cells were co-incubated with the FarRed labeled tumor cells at different effector: target (E:T) ratios ranging from 1:1 to 15:1 for 12 h. Cells were stained with DAPI and the viability of tumor cells was analyzed via Flow cytometer. Cytotoxicity was then calculated as described in Materials and Methods. The data are presented as mean ± standard deviation of triplicates. (c). B7-H1 expression on MDA-MB-231-H3KO cell line were validated by flow cytometry analysis. (d) The ability of CAR-T cells to release cytokines upon antigen recognition was evaluated in vitro. CAR-T cells (1 × 106) were co-incubated with 5 × 105 irradiated B7-H3 positive or negative tumor cells. Supernatants were collected after 24 h and analyzed for cytokines using a cytometric bead array. The data are presented as mean ± standard deviation of triplicates. Error bars denote SD. P values by a two-tailed unpaired t-test. * (P < .05), **(P < .01), ***(P < .001) and **** (P < .0001); ns, not significant
Co-expression of PD-1 chimeric receptors augments the therapeutic efficacy of B7-H3 specific CAR-T cells on established xenograft solid tumors
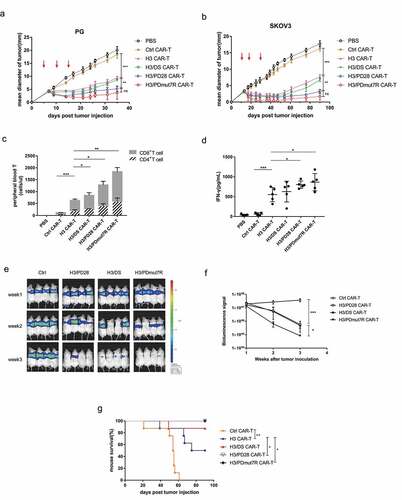
To evaluate the antitumor activity of B7-H3 specific CAR-T cells in established tumors, we tested the therapeutic efficacy of CAR-T cells using a subcutaneous tumor model. NCG mice were subcutaneously implanted with B7-H1+/B7-H3+ PG tumor cells () or SKOV3 tumor cells (). When tumors reached approximately 3–4 mm, triple doses of 5 × 106 CAR-T cells were administered via i.v. on days 5, 10 and 15 (in PG tumor model), and on days 12, 20, and 29 (in SKOV3 tumor model), respectively, and tumor sizes were monitored. The results showed that each type of H3, H3/DS, H3/PD28, and H3/PDmut7R CAR-T cells significantly inhibited tumor growth, however, compared with the H3, H3/DS CAR-T cells, the efficacies of H3/PD28 and H3/PDmut7R CAR-T cells were superior (, ). The antitumor activity of CAR-T cells was further supported by the presence of expanded effector T cells and increased cytokine levels. Compared with control CAR-T cells, each of the H3, H3/DS, H3/PD28, and H3/PDmut7R CAR-T cells significantly increased the level of CD8+ T cells and IFN-gamma in the peripheral blood of mice as assessed on day 7 after CAR-T cell infusions, and among which the H3/PDmut7R CAR-T cells were most effective and H3/PD28 CAR-T cells were secondary (,). To evaluate the capability of infiltration of the H3/PDmut7R CAR-T cells, tissues from the site of tumor regression were examined using histological and IHC analyses. The presence of CD3+ T cells confirmed tumor-directed infiltration and accumulation of these CAR-T cells (Figure S5). Taken together, these results indicate a superior antitumor activity for B7-H3 directed CAR-T cells that co-express a PD-1 chimeric receptor, especially H3/PDmut7R CAR-T cells, in an established B7-H1+/B7-H3+ tumor model.
Figure 4. The antitumor activity of B7-H3 targeted CAR-T cells co-expressing PD-1 chimeric receptors in vivo. (a) PG tumors were s.c. established in the right flank of NCG mice and randomly grouped (n = 5 per group). When tumors reached a diameter of approximately 3–4 mm, mice were treated with three intravenous injections of 5 × 106 CAR-T cells on day 5, 10 and 15 (red arrows). (b) SKOV3 tumors were s.c. established in the right flank of NCG mice and randomly grouped (n = 5 per group). When the tumors reached a mean diameter of approximately 3–4 mm, mice were treated with three intravenous injections of 5 × 106 CAR-T cells on day 12, 20 and 29 (red arrows). (c) The quantity of persistently circulating CD4+ and CD8+ T cells from mice bearing PG tumors 1 week after being treated with the indicated T cells. (d) Serum cytokine productions from mice bearing PG tumors treated with the indicated T cells were analyzed by a cytometric bead array. (e) BLI illustrating tumor growth in PG-luc metastatic mouse model. NCG mice were intravenously injected with 105 Firefly-luciferase expressing PG cells and randomly grouped (n = 4 per group). Then mice were treated with three intravenous injections of 5 × 106 CAR-T Cells. Tumor growth was assessed by BLI weekly. (f) BLI kinetics of tumor growth were calculated using Living Image software. (g) The Kaplan Meier survival analysis of PG i.v. challenged mice treated with the indicated T cells (n = 8 per group). Error bars denote SD. P values by a two-way ANOVA (a, b, f); Unpaired and non-parametric Mann-Whitney test with two-tailed P value calculation (c); P values by a two-tailed unpaired t-test (d); P values by a log-rank (Mantel-Cox) test (g). * (P < .05), **(P < .01), ***(P < .001) and **** (P < .0001); ns, not significant
Co-expression of PD-1 chimeric receptors augments the therapeutic effect of B7-H3 specific CAR-T cells on metastatic xenograft tumors
Having observed the superior antitumor activities of H3/PD28 and H3/PDmut7R CAR-T cells in the subcutaneous tumor models, we further investigated the effects of CAR-T cells on a metastatic model of lung carcinoma. NCG mice were i.v. injected with B7-H1+/B7-H3+ PG-Luc tumor cells. Mice were treated with three doses of 5 × 106 CAR-T cells on day 3, 7 and 15, respectively, after tumor injection. Tumor burden was evaluated by bioluminescence imaging (BLI) and survival was monitored. We found that treatment with each of the H3/DS, H3/PD28 and H3/PDmut7R CAR-T cells resulted in a significant reduction in tumor burden compared with control CAR-T cells, whereas the H3/PDmut7R CAR-T cells showed the highest effectiveness (, ). Both H3/PD28 and H3/PDmut7R CAR-T cells significantly extended mouse survival (). Therefore, both H3/PDmut7R and H3/PD28 CAR-T cells demonstrated superior antitumor activity again in a B7-H1+/B7-H3+ lung metastatic tumor model.
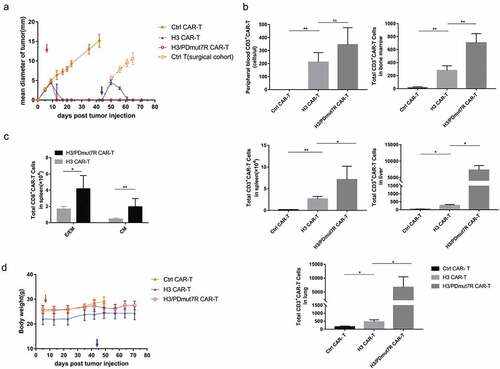
Co-expression of PDmut7R enhanced the protective efficacy of B7-H3 specific CAR-T cells in a re-challenged tumor model
IL-7/IL-7R signaling plays an essential role in regulating memory T cell genesis.Citation38,Citation39 Having observed that H3/PDmut7R CAR-T cells preferentially function during antitumor efficacy in vivo, we further investigated the potential immunological memory response of H3/PDmut7R CAR-T cells compared with other groups of CAR-T cells in a re-challenged tumor model (). Subcutaneous tumors of B7-H1+ PG cells overexpressing human 4Ig-B7-H3 (PG-hB7-H3) were established to reach a mean diameter of 3–4 mm by incolation of 5 × 105 tumor cells, followed by a single dose of 2 × 106 CAR-T cells was i.v. infused on day 7. H3/PDmut7R CAR-T cells completely eradicated tumors on day 12, while H3 CAR-T cells eradicated tumors at day 15; control CAR-T cells failed to restrain tumor growth. Importantly, when mice were re-challenged with tumor cells at a dose of 2.5 × 106 (5-fold the initial dose) on day 44, the H3/PDmut7R CAR-T cells exhibited a significant efficacy in preventing tumor growth compared to H3 CAR-T cells. In contrast, when control CAR-T cell treated tumors were excised and mice were re-challenged using the same quantity of tumor cells on day 44, the tumors grew rapidly. Moreover, when peripheral blood, bone marrow (BM), liver, lung, and spleen were assessed for the persistence and memory phenotype of CAR-T cells, it was found that, compared with the H3 CAR-T cell treatment group, the H3/PDmut7R CAR-T cell treatment group had a significantly greater absolute number of CD3+ CAR-T cells in bone marrow, liver, spleen and lung with the exception of peripheral blood (). While a majority of the CD8+ CAR-T cells in the spleen were TE/EM subsets, the H3/PDmut7R CAR-T cell group had a greater absolute number of CD8+CAR-TE/EM and CD8+ CAR-TCM cells than that of the H3 CAR-T cell group (). Taken together, these data indicate that the H3/PDmut7R CAR-T cells developed a greater number of memory T cells. The antitumor activity to re-challenge tumor was not due to an allogeneic effect because the control T cells could not suppress tumor growth. The absence of an allogeneic effect was further evidenced by the observation that there was no a decrease in mouse body weight (), and no histological tissue damage in the brain, heart, liver, lung and kidney of the mice treated with CAR-T cells (Figure S6).
Figure 5. H3/PDmut7R CAR-T cells are prone to promoting anti-tumor persistence in a re-challenge tumor model. (a) NCG mice were inoculated subcutaneously with 5 × 105 PG-B7-H3 cells. When the tumors reached a diameter of approximately 3–4 mm, mice were randomly grouped (n = 5 per group) and intravenously treated with different groups of CAR-T cells, 2 × 106 per mouse, on day 7 (red arrow). At day 30, tumors in the control CAR-T cell treated group were surgically excised. At day 44, all mice were re-challenged with 2.5 × 106 tumor cells (blue arrow). Tumor growth was monitored by measuring tumor size. (b) The number of CD3+ CAR-T cells in peripheral blood, spleen, liver, lung and BM were determined by flow cytometry at day 71, the end of experiments. (c) The phenotypes of the CAR-T cells obtained from the spleen were analyzed by flow cytometry at day 71. Effector/effector memory (E/EM; CD44+ CD62L-) and central memory (CM; CD44+ CD62L+) subsets of CD8+ CAR-T cells were counted. (d) Mouse body weight was measured once a week. Error bars denote SD. Data were analyzed by an unpaired and non-parametric Mann-Whitney test with two-tailed P value calculation (b, c). * (P < .05), **(P < .01), ***(P < .001) and **** (P < .0001); ns, not significant
Discussion
CAR-T cells rely on target molecules expressed on the cell surface to specifically recognize and kill tumor cells. In this study, we developed a new monoclonal antibody (mAb-7E12) that is highly specific to human 4Ig-B7-H3 protein which is overexpressed in a variety of solid tumors. We investigated the function of B7-H3 directed CAR-T cells with an scFv derived from mAb-7E12 and demonstrated sufficient antitumor activity for these CAR-T cells against B7-H3+ solid tumors in vitro and in vivo. Moreover, we found that co-expression of a PD-1 decoy receptor, which potentially overcomes the inhibitory signaling of B7-H1/PD1 in the TME of solid tumors, significantly improved the therapeutic efficacy of B7-H3 specific CAR-T cells in an established xenograft tumor model and in a re-challenge lung metastatic model. These findings validate a potential strategy for the treatment of multiple solid tumors using B7-H3 specific CAR-T cells directed by the scFv of the mAb-7E12.
Tumor exclusive antigens are ideal therapeutic targets but are rarely available. B7-H3 that is aberrantly upregulated on the cell surface of many solid tumors has served as a therapeutic target for tumor associated antigen. Several mAb based therapies targeting B7-H3 have been tested in patients with refractory neoplasms (MGA271, clone 84D),Citation40 SCCHN and NSCLC (combination of MGA271+ Anti-PD-1),Citation41 unresectable or metastatic B7-H3-expressing tumors (MGD009, bispecific antibody for anti-CD3 and anti-B7-H3 (clone 84D)),Citation42 and diffuse intrinsic pontine glioma (antibody drug conjugate, clone 8H9).Citation31 However, the therapeutic efficacies of these trials have yet to be determined. Currently, to the best of our knowledge, there is no ongoing clinical trial for testing B7-H3 specific CAR-T cell therapy. It would be important to evaluate the potential of CAR-T cell-based therapy in B7-H3 positive solid tumors in future clinical trials.
The specificity of cell-surface target antigens greatly affects the safety of CAR-T cell therapy. Monoclonal antibody therapies targeting B7-H3 have shown tolerant toxicities in different clinical trials in general.Citation31,Citation40,Citation43,Citation44 In addition, the difference in epitope and affinity of a specific antibody may also influence its safety profile. In this study, the mAb-7E12 as well as its scFv recognized membranous B7-H3 in a broad array of human tumor cell lines. Meanwhile, we showed overexpression of membranous B7-H3 in a variety of human malignancies, but minimal staining of B7-H3 protein in the cytoplasm of normal tissues that were largely limited in focal liver cells and a few luminal epithelial cells of the stomach and colon, which is consistent with previous reports.Citation12 Therefore, B7-H3 specific CAR-T cells are expected to have minimal toxicity in normal tissues. Nevertheless, potential toxicities upon recognition of the antigen expressed in normal tissues require further investigation. Since mAb-7E12 only recognizes human B7-H3, not mouse B7-H3, developing a B7-H3-Tg mouse model that expresses a human B7-H3 would be a valuable tool for further evaluating the safety profile of B7-H3 specific CAR-T cell therapy.
In an early trial, a fatal acute respiratory failure was reported in a patient with advanced colon cancer after receiving 1 × 1010 HER2 targeted CAR-T cells with a scFv derived from trastuzumab, likely due to low-level expression of HER2 in normal lung epithelial cells.Citation36 Since then, increased precautions have been undertaken to reduce on-target off-tumor toxicity caused by CAR-T cell therapy. Using low dose CAR-T cell infusion, decreasing the affinity of a CAR for the target, or optimizing costimulatory design have been attempted to decrease toxicity.Citation45–Citation48 For example, in a phase I/II clinical trial reported in 2015, Ahmed and colleagues used another form of HER2 targeted CAR-T cells to treat 12 sarcoma patients achieving no significant toxicity, in which lower affinity scFv derived from mAb FRP5 rather from trastuzumab was used to generate CAR-T cells, and lower doses of maximal 1 × 108 cells/m2 CAR-T cells were infused to treat patients. Furthermore, regional delivery of CAR-T cells may limit systemic toxicity by reducing the migration of CAR-T cells in normal tissues, and has shown favorable safety and improved therapeutic efficacy in several preclinical investigations.Citation37,Citation49–Citation52 It was reported that intrathecally or intracranially delivery of IL-13Rα2 specific CAR-T cells to patients with glioblastoma resulted in antitumor responses and safe outcomes.Citation53 Regional delivery of B7-H3 specific CAR-T cells to primary or localized metastatic tumor sites may be an optimal therapeutic strategy in the future. Further study to minimize the potential for off-tumor toxicity is expected to promote B7-H3 specific CAR-T cell therapy.
The potent antitumor activity of CAR-T cells may be impaired by inhibitory PD-1/B7-H1 signaling induced in the TME, a demonstrated immune escape mechanism within the TME.Citation32,Citation54–Citation57 Overcoming the immune suppressive TME is necessary to improve CAR-T cell therapy in advanced solid tumors. Several strategies have been attempted to interrupt PD-1/B7-H1 signaling in CAR-T cells, including combinations of anti-PD antibody with CAR-T cells, engineering anti-PD antibody that produces CAR-T cells or silencing PD-1 in CAR-T cells.Citation51–Citation53 In addition, coexpression of the PD-1/CD28 chimeric receptor has been shown to augment the function of PSCA specific CAR-T cells in a prostate cancer model.Citation35 Consistently, in this study, we found that PD-1/CD28 (PD28) improved the functions of B7-H3 specific CAR-T cells by enhancing the anti-tumor activity and the therapeutic effects against B7-H1+/B7-H3+ tumors. B7-H1 binding to a PD28 chimeric receptor might induce CD28 activation instead of an inhibitory signal in T cells, leading to enhanced antitumor activity in B7-H3 specific CAR-T cells. Furthermore, in this study, we generated another new chimeric PD-1 decoy receptor, PDmut7R, which outperformed PD28 in improving both antitumor activity and cytokine secretion for B7-H3 specific CAR-T cells in the setting of tumors that express both B7-H1 and B7-H3. More importantly, the H3/PDmut7R CAR-T cell offered a more potent therapeutic effect in established B7-H1+/B7-H3+ tumors and exhibited superiority in preventing the growth of the re-challenged tumors. The superior antitumor activities of the H3/PDmut7R CAR-T cells were correlated with an increased number of effector CD3+ CAR-T cells and CD8+ CAR-T cell memory subsets in mice. Thus, PDmut7R promoted an increase in the memory phenotype of CAR-T cells and developed a more sustained and persistent antitumor activity for CAR-T cells. As PDmut7R was composed of an extracellular PD-1 domain fused to a constitutive IL-7R activation domain, it most likely functions by competing with B7-H1 and the activation signaling of IL-7R.
In summary, we have demonstrated that B7-H3 directed CAR-T cells with an scFv derived from mAb-7E12 possess efficient antitumor activity against multiple solid tumors in vitro and in vivo, and further modification of these CAR-T cells by expressing chimeric PD-1 decoy receptors enhances their therapeutic efficacy against B7-H3+/B7-H1+ tumors in mouse models. B7-H3 targeted CAR-T cells provide a potential therapeutic option for advanced solid tumors.
Abbreviations
| B7-H3 | = | B7 homolog three |
| CAR-T cell | = | chimeric antigen receptor T cell, |
| mAb | = | monoclonal antibody |
| PD-1 | = | programmed cell death one |
| scFv | = | single-chain variable fragment |
Disclosure of potential conflicts of interest
In the past 12 months, LC was or has been a consultant/advisory board member for NextCure, Pfizer, Junshi, Vcanbio and GenomiCare; is a scientific founder of NextCure and Tayu Biotech Group and has sponsored research grants from NextCure and Tayu. LC was also an uncompensated adjunct faculty member at Sun Yat-sen University and Fujian Medical University. GH is partially compensated by Tcelltech Bioscience & Technology Inc. Other authors declare no competing financial interest.
Acknowledgments
We thank Beth Cadugan for editing the manuscript. This study is supported partially by grants from Guangdong Province Innovative Research Program Project and 863 Project grants to Sun Yat-sen University; Fujian Province Department of Science and Technology Research Program, and an endowed professorship from United Technologies Corporation to Yale University.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
References
- Chmielewski M, Hombach AA, Abken H. Antigen-specific T-cell activation independently of the MHC: chimeric antigen receptor-redirected T cells. Front Immunol. 2013;4:371. doi:10.3389/fimmu.2013.00371.
- Gross G, Waks T, Eshhar Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci U S A. 1989;86(24):10024–13. doi:10.1073/pnas.86.24.10024.
- Johnson LA, June CH. Driving gene-engineered T cell immunotherapy of cancer. Cell Res. 2017;27(1):38–58. doi:10.1038/cr.2016.154.
- Maus MV, Grupp SA, Porter DL, June CH. Antibody-modified T cells: cARs take the front seat for hematologic malignancies. Blood. 2014;123(17):2625–2635. doi:10.1182/blood-2013-11-492231.
- Park JH, Geyer MB, Brentjens RJ. CD19-targeted CAR T-cell therapeutics for hematologic malignancies: interpreting clinical outcomes to date. Blood. 2016;127(26):3312–3320. doi:10.1182/blood-2016-02-629063.
- Lanitis E, Dangaj D, Irving M, Coukos G. Mechanisms regulating T-cell infiltration and activity in solid tumors. Ann Oncol. 2017;28(suppl_12):xii18–xii32. doi:10.1093/annonc/mdx238.
- Munn DH, Bronte V. Immune suppressive mechanisms in the tumor microenvironment. Curr Opin Immunol. 2016;39:1–6. doi:10.1016/j.coi.2015.10.009.
- Liu B, Yan L, Zhou M. Target selection of CAR T cell therapy in accordance with the TME for solid tumors. Am J Cancer Res. 2019;9:228–241.
- Martinez M, Moon EK. CAR T cells for solid tumors: new strategies for finding, infiltrating, and surviving in the tumor microenvironment. Front Immunol. 2019;10:128. doi:10.3389/fimmu.2019.00128.
- Watanabe K, Kuramitsu S, Posey AD Jr, June CH. Expanding the therapeutic window for CAR T cell therapy in solid tumors: the knowns and unknowns of CAR T cell biology. Front Immunol. 2018;9:2486. doi:10.3389/fimmu.2018.02486.
- Chapoval AI, Ni J, Lau JS, Wilcox RA, Flies DB, Liu D, Dong H, Sica GL, Zhu G, Tamada K, et al. B7-H3: a costimulatory molecule for T cell activation and IFN-gamma production. Nat Immunol. 2001;2(3):269–274. doi:10.1038/85339.
- Du H, Hirabayashi K, Ahn S, Kren NP, Montgomery SA, Wang X, Tiruthani K, Mirlekar B, Michaud D, Greene K. Antitumor responses in the absence of toxicity in solid tumors by targeting B7-H3 via chimeric antigen receptor T cells. Cancer Cell. 2019;35(2):221–237.e8. doi:10.1016/j.ccell.2019.01.002.
- Mesri M, Birse C, Heidbrink J, McKinnon K, Brand E, Bermingham CL, Feild B, Fitzhugh W, He T, Ruben S, et al. Identification and characterization of angiogenesis targets through proteomic profiling of endothelial cells in human cancer tissues. PLoS One. 2013;8(11):e78885. doi:10.1371/journal.pone.0078885.
- Boorjian SA, Sheinin Y, Crispen PL, Farmer SA, Lohse CM, Kuntz SM, Leibovich BC, Kwon ED, Frank I. T-cell coregulatory molecule expression in urothelial cell carcinoma: clinicopathologic correlations and association with survival. Clin Cancer Res. 2008;14(15):4800–4808. doi:10.1158/1078-0432.CCR-08-0731.
- Crispen PL, Sheinin Y, Roth TJ, Lohse CM, Kuntz SM, Frigola X, Thompson RH, Boorjian SA, Dong H, Leibovich BC. Tumor cell and tumor vasculature expression of B7-H3 predict survival in clear cell renal cell carcinoma. Clin Cancer Res. 2008;14(16):5150–5157. doi:10.1158/1078-0432.CCR-08-0536.
- Ingebrigtsen VA, Boye K, Tekle C, Nesland JM, Flatmark K, Fodstad O. B7-H3 expression in colorectal cancer: nuclear localization strongly predicts poor outcome in colon cancer. Int J Cancer. 2012;131(11):2528–2536. doi:10.1002/ijc.27566.
- Qin X, Zhang H, Ye D, Dai B, Zhu Y, Shi G. B7-H3 is a new cancer-specific endothelial marker in clear cell renal cell carcinoma. Onco Targets Ther. 2013;6:1667–1673. doi:10.2147/OTT.S53565.
- Roth TJ, Sheinin Y, Lohse CM, Kuntz SM, Frigola X, Inman BA, Krambeck AE, McKenney ME, Karnes RJ, Blute ML.. B7-H3 ligand expression by prostate cancer: a novel marker of prognosis and potential target for therapy. Cancer Res. 2007;67(16):7893–7900. doi:10.1158/0008-5472.CAN-07-1068.
- Sun J, Guo Y, Li X, Zhang Y, Gu L, Wu P, Bai G, Xiao Y. B7-H3 expression in breast cancer and upregulation of VEGF through gene silence. Onco Targets Ther. 2014;7:1979–1986. doi:10.2147/OTT.
- Sun Y, Wang Y, Zhao J, Gu M, Giscombe R, Lefvert AK, Wang X. B7-H3 and B7-H4 expression in non-small-cell lung cancer. Lung Cancer. 2006;53(2):143–151. doi:10.1016/j.lungcan.2006.05.012.
- Wu C-P, Jiang J-T, Tan M, Zhu Y-B, Ji M, Xu K-F, Zhao J-M, Zhang G-B, Zhang X-G. Relationship between co-stimulatory molecule B7-H3 expression and gastric carcinoma histology and prognosis. World J Gastroenterol. 2006;12(3):457–459. doi:10.3748/wjg.v12.i3.457.
- Yamato I, Sho M, Nomi T, Akahori T, Shimada K, Hotta K, Kanehiro H, Konishi N, Yagita H, Nakajima Y, et al. Clinical importance of B7-H3 expression in human pancreatic cancer. Br J Cancer. 2009;101(10):1709–1716. doi:10.1038/sj.bjc.6605375.
- Zang X, Sullivan PS, Soslow RA, Waitz R, Reuter VE, Wilton A, Thaler HT, Arul M, Slovin SF, Wei J, et al. Tumor associated endothelial expression of B7-H3 predicts survival in ovarian carcinomas. Mod Pathol. 2010;23(8):1104–1112. doi:10.1038/modpathol.2010.95.
- Zang X, Thompson RH, Al-Ahmadie HA, Serio AM, Reuter VE, Eastham JA, Scardino PT, Sharma P, Allison JP. B7-H3 and B7x are highly expressed in human prostate cancer and associated with disease spread and poor outcome. Proc Natl Acad Sci U S A. 2007;104(49):19458–19463. doi:10.1073/pnas.0709802104.
- Seaman S, Zhu Z, Saha S, Zhang XM, Yang MY, Hilton MB, Morris K, Szot C, Morris H, Swing DA, et al. Eradication of tumors through simultaneous ablation of CD276/B7-H3-positive tumor cells and tumor vasculature. Cancer Cell. 2017;31(4):501–515.e8. doi:10.1016/j.ccell.2017.03.005.
- Benzon B, Zhao SG, Haffner MC, Takhar M, Erho N, Yousefi K, Hurley P, Bishop JL, Tosoian J, Ghabili K, et al. Correlation of B7-H3 with androgen receptor, immune pathways and poor outcome in prostate cancer: an expression-based analysis. Prostate Cancer Prostatic Dis. 2017;20(1):28–35. doi:10.1038/pcan.2016.49.
- Tekle C, Nygren MK, Chen YW, Dybsjord I, Nesland JM, Maelandsmo GM, Fodstad O. B7-H3 contributes to the metastatic capacity of melanoma cells by modulation of known metastasis-associated genes. Int J Cancer. 2012;130(10):2282–2290. doi:10.1002/ijc.v130.10.
- Ye Z, Zheng Z, Li X, Zhu Y, Zhong Z, Peng L, Wu Y. B7-H3 overexpression predicts poor survival of cancer patients: a meta-analysis. Cell Physiol Biochem. 2016;39(4):1568–1580. doi:10.1159/000447859.
- Zhao X, Li DC, Zhu XG, Gan WJ, Li Z, Xiong F, Zhang ZX, Zhang GB, Zhang XG, Zhao H. B7-H3 overexpression in pancreatic cancer promotes tumor progression. Int J Mol Med. 2013;31(2):283–291. doi:10.1126/scitranslmed.aad7118.
- Hofmeyer KA, Ray A, Zang X. The contrasting role of B7-H3. Proc Natl Acad Sci U S A. 2008;105(30):10277–10278. doi:10.1073/pnas.0805458105.
- Souweidane MM, Kramer K, Pandit-Taskar N, Zhou Z, Haque S, Zanzonico P, Carrasquillo JA, Lyashchenko SK, Thakur SB, Donzelli M, et al. Convection-enhanced delivery for diffuse intrinsic pontine glioma: a single-centre, dose-escalation, phase 1 trial. Lancet Oncol. 2018;19(8):1040–1050. doi:10.1016/S1470-2045(18)30322-X.
- Zou W, Wolchok JD, Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: mechanisms, response biomarkers, and combinations. Sci Transl Med. 2016;8(328):328rv4. doi:10.1126/scitranslmed.aad7118.
- Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, Roche PC, Lu J, Zhu G, Tamada K. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8(8):793–800. doi:10.1038/nm730.
- Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. 2013;14(10):1014–1022. doi:10.1038/ni.2703.
- Liu X, Ranganathan R, Jiang S, Fang C, Sun J, Kim S, Newick K, Lo A, June CH, Zhao Y, et al. A chimeric switch-receptor targeting PD1 augments the efficacy of second-generation CAR T cells in advanced solid tumors. Cancer Res. 2016;76(6):1578–1590. doi:10.1158/0008-5472.CAN-15-2524.
- Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18(4):843–851. doi:10.1038/mt.2010.24.
- Adusumilli PS, Cherkassky L, Villena-Vargas J, Colovos C, Servais E, Plotkin J, Jones DR, Sadelain M. Regional delivery of mesothelin-targeted CAR T cell therapy generates potent and long-lasting CD4-dependent tumor immunity. Sci Transl Med. 2014;6(261):261ra151. doi:10.1126/scitranslmed.3010162.
- Bradley LM, Haynes L, Swain SL. IL-7: maintaining T-cell memory and achieving homeostasis. Trends Immunol. 2005;26(3):172–176. doi:10.1016/j.it.2005.01.004.
- Kaech SM, Tan JT, Wherry EJ, Konieczny BT, Surh CD, Ahmed R. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat Immunol. 2003;4(12):1191–1198. doi:10.1038/ni1009.
- Powderly J, Cote G, Flaherty K, Szmulewitz RZ, Ribas A, Weber J, Loo D, Baughman J, Chen F, Moore P, et al. Interim results of an ongoing Phase I, dose escalation study of MGA271 (Fc-optimized humanized anti-B7-H3 monoclonal antibody) in patients with refractory B7-H3-expressing neoplasms or neoplasms whose vasculature expresses B7-H3. J Immunother Cancer. 2015;3(Suppl 2):O8. doi:10.1186/2051-1426-3-S2-O8.
- Charu A, Desantes K, Maris JM, McDowell K, Mackall C, Shankar S, Vasselli J, Chen F, Loo D, Moore PA, et al. A phase 1, open-label, dose escalation study of enoblituzumab in combination with pembrolizumab in patients with select solid tumors. The Society for Immunotherapy of Cancer (SITC) 33rd Annual Meeting; 2018; Washington, DC.
- Shankar S, Spira AI, Strauss J, Liu L, La Motte-Mohs R, Wu T, Johnson S, Bonvini E, Moore PA, Wigginton JM, et al. A phase 1, open label, dose escalation study of MGD009, a humanized B7-H3 x CD3 DART protein, in combination with MGA012, an anti-PD-1 antibody, in patients with relapsed or refractory B7-H3-expressing tumors. J Clin Oncol. 2018;36(15_suppl):TPS2601–TPS2601. doi:10.1200/JCO.2018.36.15_suppl.TPS2601.
- Kramer K, Kushner BH, Modak S, Pandit-Taskar N, Smith-Jones P, Zanzonico P, Humm JL, Xu H, Wolden SL, Souweidane MM, et al. Compartmental intrathecal radioimmunotherapy: results for treatment for metastatic CNS neuroblastoma. J Neurooncol. 2010;97(3):409–418. doi:10.1007/s11060-009-0038-7.
- Loo D, Alderson RF, Chen FZ, Huang L, Zhang W, Gorlatov S, Burke S, Ciccarone V, Li H, Yang Y, et al. Development of an Fc-enhanced anti-B7-H3 monoclonal antibody with potent antitumor activity. Clin Cancer Res. 2012;18(14):3834–3845. doi:10.1158/1078-0432.CCR-12-0715.
- Arcangeli S, Rotiroti MC, Bardelli M, Simonelli L, Magnani CF, Biondi A, Biagi E, Tettamanti S, Varani L. Balance of anti-CD123 chimeric antigen receptor binding affinity and density for the targeting of acute myeloid leukemia. Mol Ther. 2017;25(8):1933–1945. doi:10.1016/j.ymthe.2017.04.017.
- Drent E, Poels R, Ruiter R, van de Donk NWCJ, Zweegman S, Yuan H, de Bruijn J, Sadelain M, Lokhorst HM, Groen RWJ, et al. Combined CD28 and 4-1BB costimulation potentiates affinity-tuned chimeric antigen receptor-engineered T cells. Clin Cancer Res. 2019;25:4014–4025. doi:10.1158/1078-0432.CCR-18-2559.
- Pan J, Yang JF, Deng BP, Zhao XJ, Zhang X, Lin YH, Wu YN, Deng ZL, Zhang YL, Liu SH, et al. High efficacy and safety of low-dose CD19-directed CAR-T cell therapy in 51 refractory or relapsed B acute lymphoblastic leukemia patients. Leukemia. 2017;31(12):2587–2593. doi:10.1038/leu.2017.145.
- Turatti F, Figini M, Balladore E, Alberti P, Casalini P, Marks JD, Canevari S, Mezzanzanica D. Redirected activity of human antitumor chimeric immune receptors is governed by antigen and receptor expression levels and affinity of interaction. J Immunother. 2007;30(7):684–693. doi:10.1097/CJI.0b013e3180de5d90.
- Brown CE, Badie B, Barish ME, Weng L, Ostberg JR, Chang WC, Naranjo A, Starr R, Wagner J, Wright C. Bioactivity and safety of IL13Ralpha2-redirected chimeric antigen receptor CD8+ T cells in patients with recurrent glioblastoma. Clin Cancer Res. 2015;21(18):4062–4072. doi:10.1158/1078-0432.CCR-15-0428.
- Katz SC, Point GR, Cunetta M, Thorn M, Guha P, Espat NJ, Boutros C, Hanna N, Junghans RP. Regional CAR-T cell infusions for peritoneal carcinomatosis are superior to systemic delivery. Cancer Gene Ther. 2016;23(5):142–148. doi:10.1038/cgt.2016.14.
- Nellan A, Rota C, Majzner R, Lester-McCully CM, Griesinger AM, Mulcahy Levy JM, Foreman NK, Warren KE, Lee DW. Durable regression of Medulloblastoma after regional and intravenous delivery of anti-HER2 chimeric antigen receptor T cells. J Immunother Cancer. 2018;6(1):30. doi:10.1186/s40425-018-0340-z.
- Priceman SJ, Tilakawardane D, Jeang B, Aguilar B, Murad JP, Park AK, Chang WC, Ostberg JR, Neman J, Jandial R. Regional delivery of chimeric antigen receptor-engineered T cells effectively targets HER2(+) breast cancer metastasis to the brain. Clin Cancer Res. 2018;24(1):95–105. doi:10.1158/1078-0432.CCR-17-2041.
- Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, Ostberg JR, Blanchard MS, Kilpatrick J, Simpson J, et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N Engl J Med. 2016;375(26):2561–2569. doi:10.1056/NEJMoa1610497.
- Cherkassky L, Morello A, Villena-Vargas J, Feng Y, Dimitrov DS, Jones DR, Sadelain M, Adusumilli PS. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest. 2016;126(8):3130–3144. doi:10.1172/JCI83092.
- Chong EA, Melenhorst JJ, Lacey SF, Ambrose DE, Gonzalez V, Levine BL, June CH, Schuster SJ. PD-1 blockade modulates chimeric antigen receptor (CAR)-modified T cells: refueling the CAR. Blood. 2017;129(8):1039–1041. doi:10.1182/blood-2016-09-738245.
- Rafiq S, Yeku OO, Jackson HJ, Purdon TJ, van Leeuwen DG, Drakes DJ, Song M, Miele MM, Li Z, Wang P, et al. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nat Biotechnol. 2018;36(9):847–856. doi:10.1038/nbt.4195.
- Yin Y, Boesteanu AC, Binder ZA, Xu C, Reid RA, Rodriguez JL, Cook DR, Thokala R, Blouch K, McGettigan-Croce B. Checkpoint blockade reverses anergy in IL-13Ralpha2 humanized scFv-based CAR T cells to treat murine and canine gliomas. Mol Ther Oncolytics. 2018;11:20–38. doi:10.1016/j.omto.2018.08.002.