
ABSTRACT
Mutations of the gene coding for calreticulin (CALR) that cause the loss of the C-terminal KDEL motif abolish its retention in the endoplasmic reticulum and cause CALR to be secreted from cells. Specific CALR mutants bearing a novel C-terminus can precipitate the manifestation of myeloproliferative diseases via the autocrine activation of the thrombopoietin receptor. We recently employed the retention using selective hooks (RUSH) technology to monitor CALR trafficking and demonstrated the secretion of C-terminally truncated variants of CALR in vitro and in vivo. Of note, extracellular CALR inhibited the phagocytosis of dying cancer cells by dendritic cells (DC). Via this mechanism, mutant CALR induced immunosuppression, which decreased the efficacy of immunogenic anticancer chemotherapies and PD-1 blockade.
Exon 9 mutations in the CALR gene affect approximately 30% of patients with myeloproliferative neoplasms (MPNs) such as thrombocythemia and myelofibrosis.Citation1 These mutations typically give rise to CALR variants with a C terminus lacking the endoplasmic reticulum (ER) retention signal KDEL but possessing novel properties resulting from a frameshifted mutation that yields a different C-terminal motif.Citation2 Ligation of the thrombopoietin receptor (MPL) by this mutant CALR protein leads to the activation of JAK2 and a premalignant disease that can ultimately progress toward acute myeloid leukemia.Citation3
We investigated the fate of mutant CALR in (pre-)malignant cells employing the retention using selective hooks (RUSH) technology which allows monitoring the trafficking of fluorescent CALR mutants. In this approach, the protein of interest is fused with a streptavidin-binding peptide (SBP) and hence is sequestered by streptavidin in the endoplasmic reticulum until biotin is added to the system to competitively disrupt the SBP-streptavidin interaction and to liberate the protein of interest.Citation4 The most common MPN-related CALR mutations occur in exon 9 of the gene, most often as del52 and ins5 mutations, yielding C-terminally modified proteins that lack the KDEL ER retention signal and are consequently secreted from the cells via brefeldin A-inhibitable Golgi-mediated exocytosis.Citation5 Consistently, MPN patients affected by CALR mutations exhibited increased plasma levels of CALR as compared to age-matched controls. CALR in those patients was also found to be bound to circulating monocytes and lymphocytes.Citation6
This raised the question if the excess of soluble CALR might saturate CALR-receptor binding sites on phagocytic cells. Indeed, we found that soluble CALR inhibits phagocytosis of CALR- exposing cells.Citation6
Excess amounts of soluble recombinant CALR protein inhibited the phagocytosis of oxaliplatin-treated cancer cells (which expose CALR on the surface) by bone-marrow-derived dendritic cells (BMDCs). More importantly the in vivo uptake of oxaliplatin-treated cancer cells by splenic dendritic cells was inhibited by intravenously injected recombinant CALR protein. A similar inhibition of phagocytosis was observed in mice that contained high levels of circulating CALR due to a genetic manipulation leading to the expression of mtCALR (CALRdel52) in bone marrow cells. In sum, the elevated abundance of soluble CALR inhibits phagocytosis of CALR-exposing cells both in vitro and in vivo.Citation6
Certain chemotherapeutic agents such as mitoxantrone (MTX) or oxaliplatin (OXA) induce immunogenic cell death (ICD) that is accompanied by the exposure of CALR protein on the surface of stressed and dying cells, where it serves as an ‘eat-me’ signal for antigen presenting cells (APCs), in particular immature dendritic cells.Citation7,Citation8 In therapeutic settings, ICD triggers cytotoxic T-lymphocyte-mediated adaptive anticancer immunity against tumor associated antigens.Citation9,Citation10 We thus evaluated the effects of systemically elevated soluble CALR on anticancer immunity in the context of ICD. To this aim, we augmented systemic levels of soluble CALR by the intravenous injection of recombinant CALR or by the reconstitution of the hematopoietic system via bone marrow transfer from mtCALR (CALRdel52) mice along with the subcutaneous inoculation of MTX-treated tumor cells. The capacity of this ICD vaccine to induce a protective immune response that would prevent the outgrowth of live tumor cells upon rechallenge was abrogated by elevated levels of soluble CALR. of note, augmented levels of soluble CALR led to an expansion of immunosuppressive myeloid-derived suppressor cells (MDSCs) in the spleen and in peripheral blood.Citation6
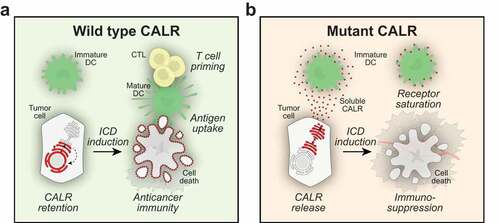
Figure 1. Immunosuppression by soluble calreticulin. Calreticulin (CALR) can be exposed on the cell surface of cells undergoing immunogenic cell death. Surface-exposed CALR then serves as an uptake signal for dendritic cells (DC), thereby facilitating the transfer of tumor-associated antigens to DC and ultimately the priming of cytotoxic T lymphocytes (CTL) (a). Mutations of the (CALR) gene occur in many forms of cancer and the loss of the KDEL retention signal can lead to the expression of mutant CALR that is secreted from the cells. Extracellular CALR binds to DC and inhibits phagocytosis, presumably by saturating a specific receptor for CALR. Through this mechanism, soluble CALR exerts immunosuppressive effects, hence subverting the effects of anticancer immunotherapy (b)
Next, RUSH-controlled local release of mutant CALR from genetically engineered cancer cells was used to examine immunosuppressive effects of CALR mutations during immunogenic anticancer therapy. Mice bearing subcutaneous tumors that were expressing mutant CALR were treated with the immunogenic chemotherapeutics MTX or OXA in conditions in which CALR was retained by cancer cells or released into the extracellular space upon intraperitoneal injection of biotin. In this in vivo model, the biotin-mediated release of CALR interfered with MTX- and OXA-mediated tumor growth reduction, supporting the hypothesis that soluble CALR released from malignant cells can cause local immunosuppressive effects that abrogate therapeutic effects of chemotherapeutic ICD inducers. Next, we engineered cells that carry a knockin mutation in the CALR gene resembling the genetic alterations associated with MPL. In contrast to wildtype cells, clones expressing secreted CALR mutants, failed to reduce their growth in response to treatment with MTX or OXA. Of note, spontaneous release of CALR from gene-edited cells in vivo also caused a systemic increase in the frequency of MDSC.
In the following step, we identified sporadic mutations of the CALR genes in human carcinomas. We then engineered mouse tumor cells carrying CALR mutants including E405* and X352, which are representative of solid-tumor-associated mutations. Both mutants exhibited the loss of the KDEL retention signal, yet in contrast to MPL-associated alterations do not carry a frameshifted C-terminus. Sarcoma and carcinoma cells bearing E405* and X352 CALR mutants exhibited spontaneous secretion of soluble CALR and, when implanted into immunocompetent hosts, failed to reduce their growth in response to treatment with MTX and OXA. Chemotherapy with MTX usually induces T cell-dependent anticancer immunity that is associated with an increase in the ratio of CD8+ over FOXP3+ regulatory T cells in the tumor bed. Neoplasias expressing del52, ins5 or E405* CALR mutants failed to exhibit such signs of anticancer immunity and exhibited reduced tumor antigen uptake by dendritic cells in vivo. Moreover, CALR protein purified from cell culture supernatants of tumor cells expressing E405* or X352 CALR mutants inhibited phagocytosis of dying cancer cells by immature dendritic cells.
Finally, we compared wild type tumors with tumors bearing MPL-associated mutations (del52 or ins5) or mutations found in solid tumors (wE405* or X352) with respect to their response to immunotherapy with PD-1 blockade. All mutations causing the secretion of soluble CALR protein significantly reduced the efficacy of immune checkpoint blockade-based immunotherapy, underlining the immunosuppressive nature of CALR secretion.
In summary, we established chemical biology and genetic tools that allowed to explore the trafficking of mutant forms of CALR. The inducible release of mutant CALR in vivo revealed its immunosuppressive functions, explaining how CALR-secreting tumors escape from anticancer immune responses.
Disclosure of potential conflicts of interest
OK and GK are scientific co-founders of Samsara Therapeutics.
Acknowledgments
GK is supported by the Ligue contre le Cancer (équipe labellisée); Agence National de la Recherche (ANR) – Projets blancs; ANR under the frame of E-Rare-2, the ERA-Net for Research on Rare Diseases; Association pour la recherche sur le cancer (ARC); Cancéropôle Ile-de-France; Chancelerie des universités de Paris (Legs Poix), Fondation pour la Recherche Médicale (FRM); a donation by Elior; European Research Area Network on Cardiovascular Diseases (ERA-CVD, MINOTAUR); Fondation Carrefour; Institut National du Cancer (INCa); Inserm (HTE); Inserm Transfert; Institut Universitaire de France; LeDucq Foundation; the LabEx Immuno-Oncology; the RHU Torino Lumière; the Seerave Foundation; the SIRIC Stratified Oncology Cell DNA Repair and Tumor Immune Elimination (SOCRATE); the SIRIC Cancer Research and Personalized Medicine (CARPEM).
References
- Klampfl T, Gisslinger H, Harutyunyan AS, Nivarthi H, Rumi E, Milosevic JD, Them NCC, Berg T, Gisslinger B, Pietra D, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. 2013;369(25):2379–3. doi:10.1056/NEJMoa1311347.
- Grinfeld J, Nangalia J, Baxter EJ, Wedge DC, Angelopoulos N, Cantrill R, Godfrey AL, Papaemmanuil E, Gundem G, MacLean C, et al. Classification and personalized prognosis in myeloproliferative neoplasms. N Engl J Med. 2018;379(15):1416–1430. doi:10.1056/NEJMoa1716614.
- Marty C, Pecquet C, Nivarthi H, El-Khoury M, Chachoua I, Tulliez M, Villeval J-L, Raslova H, Kralovics R, Constantinescu SN, et al. Calreticulin mutants in mice induce an MPL-dependent thrombocytosis with frequent progression to myelofibrosis. Blood. 2016;127(10):1317–1324. doi:10.1182/blood-2015-11-679571.
- Zhao L, Liu P, Boncompain G, Loos F, Lachkar S, Bezu L, Chen G, Zhou H, Perez F, Kepp O, et al. Identification of pharmacological inhibitors of conventional protein secretion. Sci Rep. 2018;8(1):14966. doi:10.1038/s41598-018-33378-y.
- Han L, Schubert C, Köhler J, Schemionek M, Isfort S, Brümmendorf TH, Koschmieder S, Chatain N. Calreticulin-mutant proteins induce megakaryocytic signaling to transform hematopoietic cells and undergo accelerated degradation and golgi-mediated secretion. J Hematol Oncol. 2016;9(1):45.
- Liu P, Zhao L, Loos F, Marty C, Xie W, Martins I, Lachkar S, Qu B, Waeckel-Énée E, Plo I, et al. Immunosuppression by mutated calreticulin released from malignant cells. Mol Cell. 2019. doi:10.1016/j.molcel.2019.11.004.
- Galluzzi L, Buqué A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol. 2017;17:97–111.
- Kepp O, Menger L, Vacchelli E, Locher C, Adjemian S, Yamazaki T, Martins I, Sukkurwala AQ, Michaud M, Senovilla L, et al. Crosstalk between ER stress and immunogenic cell death. Cytokine Growth Factor Rev. 2013;24(4):311–318. doi:10.1016/j.cytogfr.2013.05.001.
- Liu P, Zhao L, Pol J, Levesque S, Petrazzuolo A, Pfirschke C, Engblom C, Rickelt S, Yamazaki T, Iribarren K, et al. Crizotinib-induced immunogenic cell death in non-small cell lung cancer. Nat Commun. 2019;10(1):1486. doi:10.1038/s41467-019-09415-3.
- Kepp O, Senovilla L, Kroemer G. Immunogenic cell death inducers as anticancer agents. Oncotarget. 2014;5(14):5190–5191. doi:10.18632/oncotarget.v5i14.