
ABSTRACT
Hundreds of trials are being conducted to evaluate combination of newer targeted drugs as well as immunotherapy. Our aim was to compare efficacy and safety of combination versus single non-cytotoxic anticancer agents. We searched PubMed (01/01/2001 to 03/06/2018) (and, for immunotherapy, ASCO and ESMO abstracts (2016 through March 2018)) for randomized clinical trials that compared a single non-cytotoxic agent (targeted, hormonal, or immunotherapy) versus a combination with another non-cytotoxic partner. Efficacy and safety endpoints were evaluated in a meta-analysis using a linear mixed-effects model (guidelines per PRISMA Report).We included 95 randomized comparisons (single vs. combination non-cytotoxic therapies) (59.4%, phase II; 41.6%, phase III trials) (29,175 patients (solid tumors)). Combinations most frequently included a hormonal agent and a targeted small molecule (23%). Compared to single non-cytotoxic agents, adding another non-cytotoxic drug increased response rate (odds ratio [OR]=1.61, 95%CI 1.40-1.84)and prolonged progression-free survival (hazard ratio [HR]=0.75, 95%CI 0.69-0.81)and overall survival (HR=0.87, 95%CI 0.81-0.94) (all p<0.001), which was most pronounced for the association between immunotherapy combinations and longer survival. Combinations also significantlyincreased the risk of high-grade toxicities (OR=2.42, 95%CI 1.98-2.97) (most notably for immunotherapy and small molecule inhibitors) and mortality at least possibly therapy related (OR: 1.33, 95%CI 1.15-1.53) (both p<0.001) (absolute mortality = 0.90% (single agent) versus 1.31% (combinations)) compared to single agents. In conclusion, combinations of non-cytotoxic drugs versus monotherapy in randomized cancer clinical trials attenuated safety, but increased efficacy, with the balance tilting in favor of combination therapy, based on the prolongation in survival.
Introduction
Historically, cytotoxic agents have been the mainstay of treatment for advanced cancers. Many combinations of cytotoxic agents were tested in solid tumors, with additive/synergistic mechanisms of action. In general, a paradigm has been established for cytotoxic combinations, in which an increased response rate is often observed along with increased toxicities.Citation1-Citation4 Combining two different chemotherapeutic agents with distinct mechanisms of action is also used in an effort to minimize the evolution of the cancer and to attenuate development of drug resistance.Citation5 As a result, combinations of cytotoxic agents became the standard of care for many advanced solid tumors.
Treatment modalities in oncology are constantly evolving, with a recent increase in approvals of classes of agents other than chemotherapy. These newer agents include targeted drugs, hormonal agents and immunotherapies.Citation6 A better understanding of cancer biology and pathogenesis lead to molecular-targeted therapies. These agents are designed to block specific-altered proteins that stimulate cell growth in molecularly defined subsets of individuals. They may also target proteins preferentially expressed on tumor versus normal cells. Targeted treatments may evoke major tumor responses, usually with a better therapeutic index compared to conventional cytotoxic agents, especially when using a biomarker-driven strategy.Citation7,Citation8 Another cancer treatment approach is based on immunotherapy, taking advantage of the host immunological system to eradicate cancer cells. Long-term control or even complete eradication of cancer in a subset of patients is the great appeal associated with immunotherapy treatment.Citation9 In addition, many solid tumors, including breast and prostate cancer, are known to be sensitive to hormonal manipulations.
In addition to increasing tumor response rates, combination therapy is a strategy to circumvent resistance to treatment. Drug combinations can result in synergism, not only in efficacy parameters but also in delaying disease progression by impacting multiple intracellular escape pathways, crucial for tumor cell growth and survival. The development of new classes of anticancer agents ushered in a plethora of new drug combinations trials in oncology. However, the kinetics of tumor responses and safety profile of non-cytotoxic agents are distinct from those of chemotherapy. Previous studies suggested a possible rationale for dosing combinations with targeted and cytotoxic agents.Citation10-Citation12 Indeed, based on the crucial role that combination treatments are likely to play in improving outcome, more knowledge about the efficacy and safety of combining non-cytotoxic agents for the treatment of advanced solid tumors is needed.
We performed a systematic review of randomized trials designed to treat advanced/metastatic cancer. Our objective was to compare efficacy and safety of combination versus single non-cytotoxic anticancer agents for the management of advanced solid malignancies, establishing general parameters in common clinical trials endpoints for combinations.
Results
Search results
Our search yielded 2,551 trials according to our search criteria. We first excluded 2,400 after title review and another 63 were further excluded after abstract review (Figure S1). We included 88 published randomized trials and 4 additional trials presented as abstracts that evaluated immunotherapy combinations. This led to a total of 95 randomized comparisons, as three trials included more than one randomized comparison that fulfilled our criteria (Table S2). These trials enrolled a total of 28,704 patients (13,381 in arms testing single agents and 15,323 in arms testing combinations). Of the 95 randomized comparisons, 57 (60%) were phase II trials (). The three most frequent tumor types tested for combinations were breast (29%), non-small cell lung cancer (23%) and renal cancer (14%). The type of combination most frequently evaluated included a hormonal agent as the backbone drug combined to a targeted small molecule in 23% of randomized comparisons. The drug most frequently used as the backbone for the combinations was erlotinib (20%), followed by letrozole (9%) and bevacizumab (7%). Similarly, erlotinib was also the most frequent experimental drug and was present in 5% of combinations (Table S4).
Table 1. Characteristics of randomized clinical trials analyzed.a
Effects of combination on response rates
Of the 95 randomized comparisons, 88 reported results enabling RR analysis. Overall, RR was higher on combinations compared to single-agent drugs (OR = 1.61, 95%CI 1.40–1.84; p < .001) (). The overall response rate was 17.4% versus 24.8% (p < .001) in single agents and combination arms, respectively. The positive effect of combinations was observed regardless of other characteristics, including tumor type, line of therapy or the biomarker-based selection for the treatment. We observed that OR of RR for combination therapy increased over time according to the start of enrollment in each trials (p = .014).
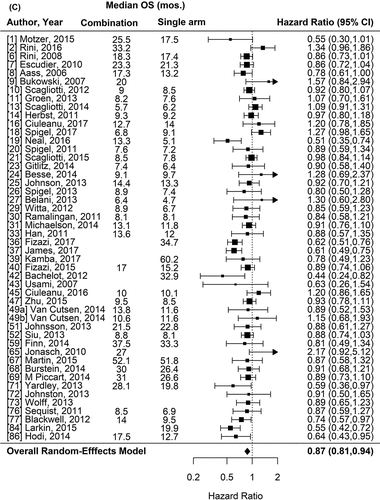
Figure 1. Forest plot representing the odds ratio for response rate (A) and hazard ratios for PFS (B) and OS (C) for experimental arms with combination of therapies compared to experimental arms with single-agent non-cytotoxic therapies. Studies are labeled by first author’s last name and year of publication and numbers in brackets are labeled according to supplementary references. Panel A shows odds ratio (95% confidence interval) for response rate for each randomized trial comparing combinations to single agents. The plot shows an overall increase in response rate for combinations: OR (95% CI) = 1.61 (1.40−1.84) (p < .001). Panel B shows hazard ratio (95% confidence interval) for PFS for each randomized trial comparing combinations to single agents. The plot shows an overall increase in PFS for combinations: HR (95% CI) = 0.75 (0.69–0.81) (p < .001). Panel C shows hazard ratio (95% confidence interval) for OS for each randomized trial comparing combinations to single agents. The plot shows an overall increase in OS for combinations: HR (95% CI) = 0.87 (0.81–0.94) (p < .001)
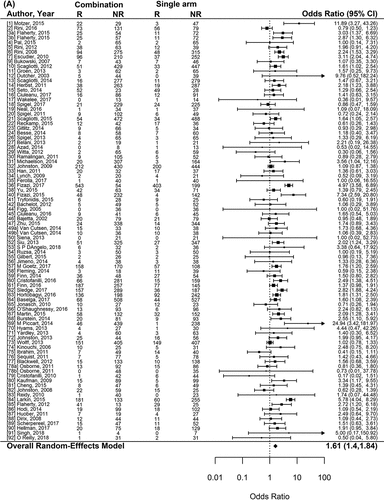
Figure 1. (continued)
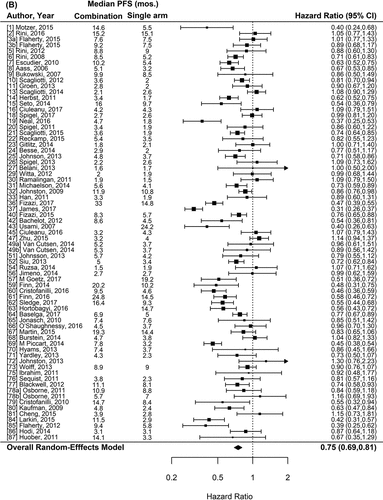
Figure 1. (continued)
The classes of the backbone drug had different effects upon the efficacy of combinations. Although statistically significant positive effects were observed for all classes, immunotherapies, and drugs not classified were more positively influenced vis-a-vis response rate by the addition of a second agent () while targeted small molecules were less impacted.
Table 2. Meta-analysis for the effects of combination therapies versus single agents on outcome in randomized trials (multivariate)a.
Effects of combinations on PFS
For this analysis, we included 71 randomized comparisons (24 were excluded, 6 because PFS was not reported in any form and 18 because only medians were published). Overall, PFS was significantly better with combinations compared to single-agent therapies (hazard ratio [HR] = 0.75, 95%CI 0.69–0.81; p < .001) (). Of the 18 trials that only reported median PFS, in 12 the outcome was numerically longer in combination arms, in 3 it was similar and in another 3 it was shorter for combinations. The class of the backbone agent and the tumor type had significant interaction with the effect observed and were included in the multivariate model. We observed a trend for a better effect of combinations on PFS for hormonal therapies as the backbone drug (HR 0.69, p < .001) and a significant positive pronounced effect in prostate (HR = 0.50, 95%CI 0.29–0.86; p = .01) and non-small cell lung cancers (HR = 0.83, 95%CI 0.73–0.94; p = .005) ().
Effects of combination on survival
For the survival analysis, we included 48 randomized comparisons (47 were excluded, 5 because OS estimates were not reached, 25 because OS was not reported in any form and 17 because only medians were published). Overall, OS was significantly better with combinations compared to single-agent therapies (HR = 0.87, 95%CI 0.81–0.94; p < .001) (). Of the 17 trials that only reported median OS, in 4 the outcome was numerically longer in combination arms, in 9 it was similar and in another 4 it was shorter for combinations. The class of the backbone agent, tumor type and the median of prior regimens used for the patients had significant interaction with the effect observed and were included on the multivariate model. We observed a significant positive pronounced effect of combinations when immunotherapies and monoclonal antibodies were selected as backbone drugs and a trend toward positive effect when “not classified” drugs were selected. Tumor types less sensitive to the positive effects of combinations included colorectal (trend), non-small cell lung cancer, and renal cell cancer, while prostate cancer presented a more pronounced positive effect of combinations. A positive effect was also observed for trials whose patients had a median of only one or at least three prior regimens (), with a trend toward positive effect with two prior therapies.
Effect of combinations on high-grade toxicities and treatment-related mortality
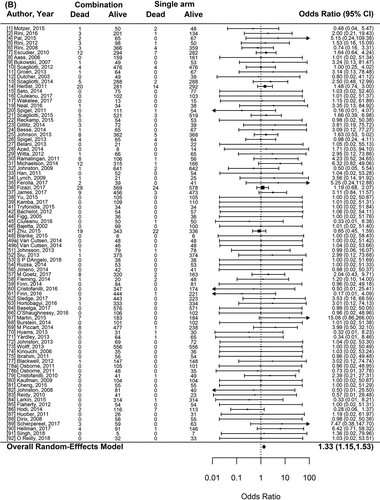
Seventy-six randomized comparisons were included for the high-grade toxicity analysis. Overall, combinations increased the risk of high-grade toxicities compared to single agents (OR = 2.42, 95%CI 1.98–2.97; p < .001) (). Each model had a decreasing linear dependence on the appropriate toxicity rate in the single-agent arm. Specifically that higher toxicity rates in the single-agent arm tended to have lower odds ratios for the experimental arm (as one would expect). After accounting for that covariate in an ANCOVA-like analysis in each case, the only characteristic that had an interaction with the effect and was included on the adjusted model was the class of the experimental agent (). The effects on increased toxicity rate were most pronounced with targeted small molecule and immunotherapy drugs added as an additional agent, with a smaller, but still statistically significant, increase for targeted monoclonal antibodies. The increases in hormonal and “not classified” experimental drugs were not statistically significant. Eighty-seven randomized comparisons were used for the treatment-related mortality analysis. Combinations significantly increased the risk of treatment-related mortality (OR: 1.33, 95%CI 1.15–1.53; p < .001) (). All classes of experimental drugs had statistically significant increases in treatment-related mortality in the experimental arms, with the exception of when drugs “not classified” were added as experimental agents, which had a trend in the opposite direction (OR 0.53, 95%CI 0.26–1.07; p = .08). The overall incidence of treatment-related deaths was 0.90% versus 1.31% (p < .001) in single agents and combinations arms, respectively. As safety objectives are different between phase II and phase III studies, we tested this variable in our model, but for both analysis, there was no statistical significance (high-grade toxicities, p-value 0.95; treatment-related mortality, p-value 0.08, )
Table 3. Meta-analysis for the effects of combination therapies versus single agentsa upon high-grade toxicities and treatment-related mortality (multivariateb)
Figure 2. Forest plot representing the odds ratio for high-grade toxicities (A), and for treatment-related mortality (B) for experimental arms with combination of therapies compared to experimental arms with single-agent non-cytotoxic therapies. Studies are labeled by first author’s last name and year of publication and numbers in brackets are labeled according to supplementary references. Panel A shows odds ratio (95% confidence interval) for high-grade toxicities for each randomized trial comparing combinations to single agents. The plot shows an overall increase in high-grade toxicities for combinations: OR (95% CI) = 2.42 (1.98−2.97) (p < .001). Panel B shows odds ratio (95% confidence interval) for treatment-related mortality for each randomized trial comparing combinations to single agents. The plot shows an overall increase in treatment-related mortality for combinations: OR (95% CI) = 1.33 (1.15–1.53) (p < .001)
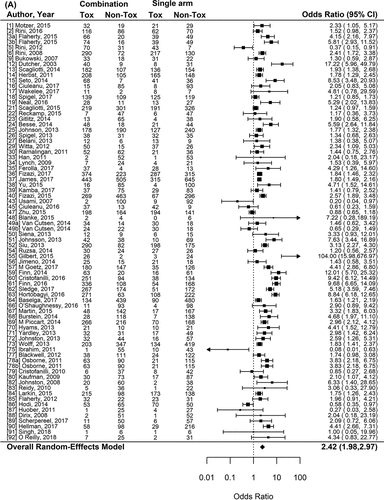
Figure 2. (continued)
Discussion
The present meta-analyses of randomized trials of monotherapy versus combination therapy in the non-cytotoxic setting yielded several important findings. First, combinations of non-cytotoxic drugs overall increased efficacy compared to single-agent therapies, as demonstrated by higher RR (OR = 1.61, 95% CI 1.40–1.84; p < .001) and a PFS and OS benefit (HR = 0.75, 95% CI 0.69–0.81 and HR = 0.87, 95%CI 0.81–0.94, respectively; both p < .001) (). However, along with this benefit, toxicity and treatment-related mortality were also increased, although the absolute incidence of deaths at least possibly related to drugs given in combinations is low (~1.3%) and did not obviate the OS benefit of the combinations. Indeed, one of the main findings from this study was the fact that survival, the ultimate goal of cancer treatment, was improved with the combination treatment strategy.Citation13,Citation14
Power to demonstrate a statistically significant gain in OS can be limited among individual studies, with a need for large sample size and long follow-up time. Disease-specific characteristics can also interfere with information gained from a single clinical trial that attempts to demonstrate a survival benefit. As an example, in breast cancer, which was the most frequent tumor type tested in combinations in our analysis, median OS can be long, and patients frequently try multiple therapies after progression on a clinical trial.Citation15 As a consequence, the chance of detecting a statistically significant difference in OS after a clinical trial may be attenuated or confounded by subsequent interventions. Specifically, combinations of cyclin-dependent kinase 4 and 6 (CDK4/6) (palbociclib, ribociclib, and abemaciclib) with endocrine therapy was well evaluated in our meta-analysis (a total of six randomized comparisons) (for metastatic hormone receptor-positive HER-2 negative breast cancer). Addition of these drugs to endocrine therapy in the first-line setting led to an absolute median PFS gain of about 10 to 15 months, but OS benefit was not statistically significant or was not reached in the individual trials.Citation16-Citation18 A previous analysis demonstrated that the power of these pivotal first-line trials to demonstrate a statistically significant improvement in OS is less than 70% if the prolongation in median OS is less than 12 months, whatever the OS data maturity, probably because these patients receive many follow-up treatments.Citation19 Therefore, a meta-analysis of time-to-event outcomes, such as that presented herein, using hazard ratio as a measure to evaluate the impact of a treatment, can be of value in aggregating available evidence from different clinical trials.Citation20
Concerning the combinations included in our analysis, a hormonal agent combined with a targeted agent, and targeted plus targeted agent combinations were the most common treatment arrangements. An important rationale of anti-cancer drug combinations is avoidance of treatment resistance.Citation21,Citation22 Indeed, it is well known that emerging resistance mediated by activation of alternative pathways is commonly seen during treatment with hormonal and targeted therapies.Citation23,Citation24 Also, the distinct mechanism of action of each drug used in the combination could theoretically mitigate the impact of tumor heterogeneity. These considerations may be operative in the prolongation of PFS and OS seen with such combinations.
The data presented here is relevant for tailoring drug combinations. Although efficacy and safety endpoints are extremely important, combinations should also be based on biological rationale. Previous studies (mainly focused on monotherapies) demonstrated that a biomarker-based rationale was associated with increased efficacy of anticancer drugs.Citation8,Citation25 In our current data, only 9% of randomized comparisons included a biomarker-based rationale for the addition of the second agent. We adopted the definition of a biomarker-based rationale as previously reported in these studies. A differential benefit in efficacy was not detected for this group, perhaps because of the paucity of biomarker-based combination studies. Indeed, a previous report including prospective clinical trials over a 5-year period, demonstrated that there was a significant reduction in using biological or molecular criteria for patient selection from phase I to phase III studies (phase I: 41.1% vs. phase II: 29.3% vs. phase III: 3.1%).Citation26 Hence, there is a need for future trials that explore the value of biomarkers that select optimized combinations in individual patients.
Our meta-analysis identified only a few immunotherapy combinations, and the majority included two immunotherapies. During immunotherapy drug development, monotherapy was the initial approach. Pairing immunotherapy with other immunotherapeutic or targeted agents, as well as with radiation or cytotoxic chemotherapy, is a more recent strategy, and many clinical trials exploring this dynamic field are currently ongoing and/or are still in phase I or phase II stages.Citation27 The rationale for immunotherapy combinations include bypassing immune evasion as well as targeting non-redundant pathways, such as CTLA-4 and PD-L1.Citation28,Citation29 As far as immunotherapy combinations, our meta-analysis demonstrated that OS was significantly improved (HR 0.49 [0.33–0.74] p = .001), whereas PFS for combinations was not superior compared to monotherapy (HR 0.64 [0.36–1.12] p = .11). Recognition that unusually long duration of responses can occur, responses after initial progression (pseudoprogression) are possible, and also that responses may remain durable after treatment withdrawal in patients receiving immunotherapy, are important in understanding this discrepancy.Citation30,Citation31 More recently, hyperprogression was also described as a pattern of response to immune checkpoint inhibitors and might compromise PFS as well.Citation32,Citation33 Further, traditional imaging response criteria may not be suitable to detect PFS benefit of immunotherapeutic agents.Citation34 A previous pooled meta-analysis evaluated correlation between PFS and OS outcomes for patients who received PD-1 inhibitors and showed that, using a random-effects meta-analysis, the protective effects of treatment were greater for OS than for PFS, and there was no significant correlation between OS and PFS in terms of medians and gains in medians.Citation35 Our meta-analysis supports this observation, suggesting that OS should be included as a primary endpoint in future phase 3 trials of immunotherapy agents. It is important to recognize that inter- and intra-tumor heterogeneity of the immune-microenvironment may affect the results of immunotherapy combinations in each tumor type. Unfortunately, due to the low number of trials, specifically addressing immunotherapy combinations, we could not proceed with sub-analysis by tumor type in this topicFor toxicity, combination therapy clearly increased treatment-related side effects and mortality, which was associated with the type of drug added to the combination, and was most significant for targeted small molecule inhibitors combined with immunotherapy drugs. Nevertheless, it is important to emphasize that the incidence of deaths at least possibly related to single-agent versus combination therapy was extremely low (0.95% and 1.40%, respectively). Interestingly, in our analysis, drug types that more intensively increased toxicity when added to the single-agent backbone were targeted small molecules and immunotherapies. Of relevance in this regard, phase I trials with monoclonal antibodies frequently reveal an absence of dose-limiting toxicities and dosing not limited by safety parameters.Citation36 On the other hand, targeted small molecules are associated with more frequent dose-limiting toxicities,Citation37 and immunotherapies in combinations also amplify toxicities that may limit dosing.Citation38
The current study has several limitations. Amongst individual trials, the totality of efficacy data, including RR, PFS, and OS, were not uniformly reported, which may limit direct comparisons of the effects on each efficacy endpoint. In some trials, time-to-event endpoints were not the objective of analysis, but it is unclear if possible omission of PFS or OS (in part due to different follow-up times) would impact our results. Additionally, we excluded randomized controlled studies that contained cytotoxic chemotherapy in any arm. Therefore, studies with chemotherapy and immunotherapy were excluded. A plethora of trials are currently being conducted with these types of combinations, exploiting potential immunomodulatory effects from chemotherapy.Citation39 Such cytotoxic combination trials warrant further study in order to assess overall impact on efficacy and safety endpoints. Our analysis may also be affected by publication bias, which usually favors positive trials. To minimize this bias we searched a long period of time and included journals regardless of their impact, but further updates in this analysis may be warranted. Finally, our study does not address the mechanisms of improved response and whether or not sequencing strategies would be just as good as combinations. Of interest in this regard, previous modeling has shown that, for many combinations, the effects obtained could be explained by each of the drugs working on an independent subset of patients, rather than a synergistic mode of action.Citation40 Very few trials have been designed in oncology to test the hypotheses of sequencing therapies,Citation41 and this is a question that remains open for future trials.
In conclusion, our findings provide the first evidence of the overall impact of non-cytotoxic combinations on efficacy and safety outcomes in solid tumors. Using data from 96 randomized comparisons of single versus combination treatments focused on immunotherapy, hormonal agents and gene-targeted agents (29,175 patients), we demonstrated that adding another non-cytotoxic drug considerably increased response rate, and prolonged progression-free and overall survival compared to a single non-cytotoxic agent. Although non-cytotoxic agents in combinations also increased toxicities, the weight of the evidence points to the advantages of combinations based on an overall survival gain. Our findings highlight the importance of non-cytotoxic combinations in the treatment strategy of solid tumors. A second implication of the results is that the effects of combinations on efficacy and safety vary according to the class of drugs, with this important finding demonstrated with immunotherapies. Future trial development should focus on improving the effectiveness of cancer therapies exploring combinations of non-cytotoxic agents, taking in account the different class effects described herein.
Material and methods
Search strategy
We searched PubMed for published, randomized prospective clinical trials that compared a single non-cytotoxic agent (targeted therapy, hormonal agent or immunotherapy) with the same agent combined with another non-cytotoxic agent. We used the following terms: ((cancer OR neoplasm OR carcinoma) AND (metastatic OR advanced)) and randomized Clinical Trial, Phase III, Clinical Trial, Phase II. Our search covered the period from 01/01/2001 to 03/06/2018. Trials including a cytotoxic agent or radiotherapy in any arm were excluded, as well as trials for a supportive or adjuvant setting, or in pediatric population. Only advanced/metastatic solid tumor trials were included. We categorized drug classes as targeted small molecules, targeted monoclonal antibodies, immunotherapy, hormonal therapy and as not classified (defined in Table S1). We considered a “backbone drug” as the agent that was present in both arms of the randomized trial. The drug that was added to the backbone and present only on one arm was considered as an “experimental drug.” Whenever appropriate, we followed the PRISMA (Preferred Reporting Items for Systematic Reviews and Meta-Analysis) statement.Citation13,Citation42
For immunotherapy combinations, randomized trials presented as abstracts were also searched through ASCO and ESMO meetings between 2016 and March 2018.Citation14,Citation15,Citation43,Citation44 Only randomized trials comparing combination vs. single-agent immunotherapies were selected for inclusion. The reason for this expanded approach for immunotherapy is that results of numerous of trials testing newer immunotherapies have just been released and only few of them were published in manuscript form. We included drug combinations regardless of their regulatory approval, to avoid a selection bias toward successful strategies (Table S2).
Data extraction
Data extraction was conducted independently by two investigators (DLJ and DMG) and any discrepancies were resolved by consensus in frequent meetings in the presence of the principal investigator (RK). We considered response rate (RR), progression-free survival (PFS) or time to tumor progression (TTP) when PFS was not available, and overall survival (OS) as acceptable efficacy endpoints for analysis. For the analysis of time to event endpoints (OS and PFS), only trials reporting hazard rations were included. Responses were recorded according to the response criteria adopted in the trial (Table S3). For the safety analysis, we extracted the total percent of high-grade treatment-related adverse events (AEs) described in each trial and treatment-related mortality. Classification of the grade of toxicity was per investigator's description. Alternatively, when high-grade AEs were not described we extracted the percent of treatment-related serious AEs, considering that the nomination was used for both experimental and control arm. Trials that only described the percent of high toxicity per type of toxicity (and total incidence) were excluded. All deaths reported by investigators as “possibly”, “probably”, or “definitely” related to treatment were considered toxicity-related deaths.
Statistical analysis
Statistical analysis was performed by our biostatistician (DAB). The statistical meta-analysis was done using a linear mixed-effects model with potential fixed-effects of backbone drug class, experimental drug class, diagnosis, whether there was a biomarker rationale for the combination, and median prior regimens for patients on the trial. In addition, for the odds ratio (OR) tests (response rate, high-grade toxicity rate, and treatment-related mortality rate), a polynomial dependence on the appropriate rate in the backbone arm was considered as a possible covariate. The models were fit using the metafor package (version 2.0–0) in R (version 3.5.1), as described in Viechtbauer et al.Citation16,Citation45 The meta-analysis models were fit using restricted maximum likelihood and the Knapp and Hartung method.Citation17,Citation46 The final model was chosen using forward selection with entry p-value 0.1. The risk of bias was not applicable to this meta-analysis.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Authors’ contributions
DLJ, DMG and RK conceived and designed the work; DLJ and DMG participate in data acquisition; DAB analyzed the data; DLJ, MN, and RK proceeded with interpretation of data; DLF, DMG, MN, DAB, and RK have drafted the work. All authors approved the submitted version.
Conflict of interests
Dr. Jardim receives speaker fees from Roche, Janssen, Astellas, MSD, Bristol-Myers Squibb and Libbs, as well as consultant fees from Janssen, Bristol-Myers Squibb and Libbs. Dr. Gagliato receives speaker fees from Roche, Astra Zeneca, United and Libbs, as well as consultant fees from Libbs. Dr. Kurzrock has research funding from Incyte, Genentech, Merck Serono, Pfizer, Sequenom, Foundation Medicine, Guardant Health, Grifols, and Konica Minolta, as well as consultant fees from LOXO, X-Biotech, Actuate Therapeutics, Genentech and NeoMed. She receives speaker fees from Roche and has an ownership interest in IDbyDNA and Curematch, Inc.
Data Availability
All data reported in this manuscript are found in the literature as referenced in the text.
Ethical approval and consent to participate
Not applicable to this study
Supplemental Material
Download ()Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
References
- Carmo-Pereira J, Costa FO, Henriques E. Single-drug vs combination cytotoxic chemotherapy in advanced breast cancer: a randomized study. Eur J Cancer. 1980;16:1621–12. doi:10.1016/0014-2964(80)90037-7.
- Biganzoli L, Minisini A, Aapro M, Di Leo A. Chemotherapy for metastatic breast cancer. Curr Opin Obstet Gynecol. 2004;16:37–41. doi:10.1097/00001703-200402000-00008.
- Vale, C. L., Tierney, J., Bull, S. J., & Symonds, P. R. Chemotherapy for advanced, recurrent or metastatic endometrial carcinoma. Cochrane Database Syst Rev. 2012. 15;(8) CD003915.
- Zer A, Prince RM, Amir E, Abdul Razak AR. Multi-agent chemotherapy in advanced soft tissue sarcoma (STS) - A systematic review and meta-analysis. Cancer Treat Rev. 2018;63:71–78. doi:10.1016/j.ctrv.2017.12.003.
- Hu Q, Sun W, Wang C, Gu Z. Recent advances of cocktail chemotherapy by combination drug delivery systems. Adv Drug Deliv Rev. 2016;98:19–34. doi:10.1016/j.addr.2015.10.022.
- de la Torre BG, Albericio F. The pharmaceutical industry in 2017. An analysis of FDA drug approvals from the perspective of molecules. Molecules. 2018;23:533. doi:10.3390/molecules23030533.
- Druker BJ, David A. Karnofsky award lecture. Imatinib as a paradigm of targeted therapies. J Clin Oncol. 2003;21:239s–245s. doi:10.1200/JCO.2003.10.589.
- Jardim DL, Schwaederle M, Wei C, Lee JJ, Hong DS, Eggermont AM, Schilsky RL, Mendelsohn J, Lazar V, KurzrockR. Impact of a biomarker-based strategy on oncology drug development: a meta-analysis of clinical trials leading to FDA approval. J Natl Cancer Inst. 2015;107:djv253.
- Fang L, Lonsdorf AS, Hwang ST. Immunotherapy for advanced melanoma. J Invest Dermatol. 2008;128:2596–2605. doi:10.1038/jid.2008.101.
- Liu S, Nikanjam M, Kurzrock R. Dosing de novo combinations of two targeted drugs: towards a customized precision medicine approach to advanced cancers. Oncotarget. 2016;7:11310–11320. doi:10.18632/oncotarget.7023.
- Nikanjam M, Liu S, Kurzrock R. Dosing targeted and cytotoxic two-drug combinations: lessons learned from analysis of 24,326 patients reported 2010 through 2013. Int J Cancer. 2016;139:2135–2141. doi:10.1002/ijc.v139.9.
- Nikanjam M, Liu S, Yang J, Kurzrock R. Dosing three-drug combinations that include targeted anti-cancer agents: analysis of 37,763 patients. Oncologist. 2017;22:576–584. doi:10.1634/theoncologist.2016-0357.
- Fiteni F, Bonnetain F. Surrogate end points for overall survival in breast cancer trials: A review. Breast. 2016;29:44–48. doi:10.1016/j.breast.2016.06.005.
- Pazdur R. Endpoints for assessing drug activity in clinical trials. Oncologist. 2008;13(Suppl 2):19–21. doi:10.1634/theoncologist.13-S2-19.
- Lebwohl D, Kay A, Berg W, Baladi JF, Zheng J. Progression-free survival: gaining on overall survival as a gold standard and accelerating drug development. Cancer J. 2009;15:386–394. doi:10.1097/PPO.0b013e3181b9c5ec.
- Finn RS, Martin M, Rugo HS, Jones S, Im S-A, Gelmon K, Harbeck N, Lipatov ON, Walshe JM, Moulder S, et al. Palbociclib and letrozole in advanced breast cancer. N Engl J Med. 2016;375:1925–1936. doi:10.1056/NEJMoa1607303.
- Hortobagyi GN, Stemmer SM, Burris HA, Yap YS, Sonke GS, Paluch-Shimon S, Campone M, Petrakova K, Blackwell KL, Winer EP, et al. Updated results from MONALEESA-2, a phase III trial of first-line ribociclib plus letrozole versus placebo plus letrozole in hormone receptor-positive, HER2-negative advanced breast cancer. Ann Oncol. 2018;29:1541–1547. doi:10.1093/annonc/mdy155.
- Goetz MP, Toi M, Campone M, Sohn J, Paluch-Shimon S, Huober J, Park IH, Trédan O, Chen S-C, Manso L, et al. MONARCH 3: abemaciclib as initial therapy for advanced breast cancer. J Clin Oncol. 2017;35:3638–3646. doi:10.1200/JCO.2017.75.6155.
- Tanguy ML, Cabel L, Berger F, Pierga J-Y, Savignoni A, Bidard F-C. Cdk4/6 inhibitors and overall survival: power of first-line trials in metastatic breast cancer. NPJ Breast Cancer. 2018;4:14. doi:10.1038/s41523-018-0068-4.
- Wei Y, Royston P, Tierney JF, Parmar MKB. Meta-analysis of time-to-event outcomes from randomized trials using restricted mean survival time: application to individual participant data. Stat Med. 2015;34:2881–2898. doi:10.1002/sim.v34.21.
- Bayat Mokhtari R, Homayouni TS, Baluch N, Morgatskaya E, Kumar S, Das B, Yeger H. Combination therapy in combating cancer. Oncotarget. 2017;8:38022–38043. doi:10.18632/oncotarget.16723.
- Zhao B, Pritchard JR, Lauffenburger DA, Hemann MT. Addressing genetic tumor heterogeneity through computationally predictive combination therapy. Cancer Discov. 2014;4:166–174. doi:10.1158/2159-8290.CD-13-0465.
- Silberstein JL, Taylor MN, Antonarakis ES. Novel insights into molecular indicators of response and resistance to modern androgen-axis therapies in prostate cancer. Curr Urol Rep. 2016;17:29. doi:10.1007/s11934-016-0584-4.
- Attarian S, Rahman N, Halmos B. Emerging uses of biomarkers in lung cancer management: molecular mechanisms of resistance. Ann Transl Med. 2017;5:377. doi:10.21037/atm.
- Schwaederle M, Zhao M, Lee JJ, Eggermont AM, Schilsky RL, Mendelsohn J, Lazar V, Kurzrock R. Impact of precision medicine in diverse cancers: a meta-analysis of phase II clinical trials. J Clin Oncol. 2015;33:3817–3825. doi:10.1200/JCO.2015.61.5997.
- Ottaiano A, Mollo E, Di Lorenzo G, Pisano C, Di Maio M, Barletta E, Pensabene M, Segati R, Bullian P, Nasti G, et al. Prospective clinical trials of biotherapies in solid tumors: a 5-year survey. Cancer Immunol Immunother. 2005;54:44–50. doi:10.1007/s00262-004-0567-z.
- Nikanjam M, Patel H, Kurzrock R. Dosing immunotherapy combinations: analysis of 3,526 patients for toxicity and response patterns. Oncoimmunology. 2017;6:e1338997. doi:10.1080/2162402X.2017.1338997.
- Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, Segal NH, Ariyan CE, Gordon R-A, Reed K, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369:122–133. doi:10.1056/NEJMoa1302369.
- Long GV, Atkinson V, Cebon JS, Jameson MB, Fitzharris BM, McNeil CM, Hill AG, Ribas A, Atkins MB, Thompson JA, et al. Standard-dose pembrolizumab in combination with reduced-dose ipilimumab for patients with advanced melanoma (KEYNOTE-029): an open-label, phase 1b trial. Lancet Oncol. 2017;18:1202–1210. doi:10.1016/S1470-2045(17)30428-X.
- Queirolo P, Spagnolo F. Atypical responses in patients with advanced melanoma, lung cancer, renal-cell carcinoma and other solid tumors treated with anti-PD-1 drugs: A systematic review. Cancer Treat Rev. 2017;59:71–78. doi:10.1016/j.ctrv.2017.07.002.
- Lipson EJ, Sharfman WH, Drake CG, Wollner I, Taube JM, Anders RA, Xu H, Yao S, Pons A, Chen L, et al. Durable cancer regression off-treatment and effective reinduction therapy with an anti-PD-1 antibody. Clin Cancer Res. 2013;19:462–468. doi:10.1158/1078-0432.CCR-12-2625.
- Kato S, Goodman A, Walavalkar V, Barkauskas DA, Sharabi A, Kurzrock R. Hyperprogressors after immunotherapy: analysis of genomic alterations associated with accelerated growth rate. Clin Cancer Res. 2017;23:4242–4250. doi:10.1158/1078-0432.CCR-16-3133.
- Champiat S, Dercle L, Ammari S, Massard C, Hollebecque A, Postel-Vinay S, Chaput N, Eggermont A, Marabelle A, Soria J-C, et al. Hyperprogressive disease is a new pattern of progression in cancer patients treated by anti-PD-1/PD-L1. Clin Cancer Res. 2017;23:1920–1928. doi:10.1158/1078-0432.CCR-16-1741.
- Hoering A, Durie B, Wang H, Crowley J. End points and statistical considerations in immuno-oncology trials: impact on multiple myeloma. Future Oncol. 2017;13:1181–1193. doi:10.2217/fon-2016-0504.
- Gyawali B, Hey S, Kesselheim AS. A comparison of response patterns for progression-free survival and overall survival following treatment for cancer with pd-1 inhibitors: A meta-analysis of correlation and differences in effect sizes. JAMA Network Open. 2018;1:e180416.
- Tosi D, Laghzali Y, Vinches M, Alexandre M, Homicsko K, Fasolo A, Del Conte G, Durigova A, Hayaoui N, Gourgou S, et al. Clinical development strategies and outcomes in first-in-human trials of monoclonal antibodies. J Clin Oncol. 2015;33:2158–2165. doi:10.1200/JCO.2014.58.1082.
- Jardim DL, Hess KR, Lorusso P, Kurzrock R, Hong DS. Predictive value of phase I trials for safety in later trials and final approved dose: analysis of 61 approved cancer drugs. Clin Cancer Res. 2014;20:281–288. doi:10.1158/1078-0432.CCR-13-2103.
- Jardim DL, de Melo Gagliato D, Giles FJ, Kurzrock R. Analysis of drug development paradigms for immune checkpoint inhibitors. Clin Cancer Res. 2018;24:1785–1794. doi:10.1158/1078-0432.CCR-17-1970.
- Emens LA, Middleton G. The interplay of immunotherapy and chemotherapy: harnessing potential synergies. Cancer Immunol Res. 2015;3:436–443. doi:10.1158/2326-6066.CIR-15-0064.
- Palmer AC, Sorger PK. Combination cancer therapy can confer benefit via patient-to-patient variability without drug additivity or synergy. Cell. 2017;171:1678–1691 e13. doi:10.1016/j.cell.2017.11.009.
- Motzer RJ, Barrios CH, Kim TM, Falcon S, Cosgriff T, Harker WG, Srimuninnimit V, Pittman K, Sabbatini R, Rha SY, et al. Phase II randomized trial comparing sequential first-line everolimus and second-line sunitinib versus first-line sunitinib and second-line everolimus in patients with metastatic renal cell carcinoma. J Clin Oncol. 2014;32:2765–2772. doi:10.1200/JCO.2013.54.6911.
- Liberati A, Altman DG, Tetzlaff J, Mulrow C, Gøtzsche PC, Loannidis J, Clarke M, Devereaux PJ, KleijnenJ, Moher D. The prisma statement for reporting systematic reviews and meta-analyses of studies that evaluate health care interventions: explanationand elaboration. Ann Intern Med. 2009;151:W65–94.
- ASCO Meeting Library. 2018. https://meetinglibrary.asco.org.
- Conferences ESMO. 2018. https://cslide.ctimeetingtech.com/library/esmo/home.
- Viechtbauer W. Conducting meta-analyses in r with the metafor package. J Stat Softw. 2010;36(3):1-48. doi:10.18637/jss.v036.i03.
- Knapp G, Hartung J. Improved tests for a random effects meta-regression with a single covariate. Stat Med. 2003;22:2693–2710.