
ABSTRACT
While anti-CEA antibodies have no direct effect on CEA-positive tumors, they can be used to direct potent anti-tumor effects as an antibody-IL-2 fusion protein (immunocytokine, ICK), and at the same time reduce the toxicity of IL-2 as a single agent. Using a fusion protein of humanized anti-CEA with human IL-2 (M5A-IL-2) in a transgenic murine model expressing human CEA, we show high tumor uptake of the ICK to CEA-positive tumors with additional lymph node targeting. ICK treated CEA-positive tumors exhibit significant tumor eradication. Analysis of tumor-infiltrating lymphocytes shows a high frequency of both CD8+ and CD4+ T cells along with CD11b positive myeloid cells in ICK treated mice. The frequency of tumor-infiltrating FoxP3+ CD4+ T cells (Tregs) is significantly reduced vs anti-CEA antibody-treated controls, indicating that ICK did not preferentially stimulate migration or proliferation of Tregs to the tumor. Combination therapy with anti-PD-1 antibody did not improve tumor reduction over ICK therapy alone. Since stereotactic tumor irradiation (SRT), commonly used in cancer therapy has immunomodulatory effects, we tested combination SRT+ICK therapy in two tumor model systems. Use of fractionated vs single high dose SRT in combination with ICK resulted in greater tumor inhibition and immunity to tumor rechallenge. In particular, tumor microenvironment and myeloid cell composition appear to play a significant role in the response rate to ICK+SRT combination therapy.
Introduction
A common drawback of many tumor-targeting antibodies is their lack of direct anti-tumor activity. This problem can be overcome by the generation of recombinant fusion proteins such as immunocytokines (ICKs). Since systemic immunotherapy with cytokines alone is often toxic,Citation1 the use of ICKs has the potential to solve two problems at once.Citation2 Given a large number of cytokines that affect the immune system, the choice of cytokine for fusion is an important consideration. Recombinant IL-2 is one of the first cytokine directed therapies in man; however, its significant toxicities limit its general use.Citation3 In terms of its application in ICK therapy, the preclinical and initial clinical results with L19-IL-2Citation4 and the anti-CD20-IL-2 fusion protein known as DI-Leu16 against Non-Hodgkins Lymphoma are impressive.Citation5 While immunotherapy of lymphomas has enjoyed widespread success with a variety of immunotherapeutic approaches, including anti-CTLA-4, anti-PD-1, and anti-PDL1,Citation6 immunotherapy of solid tumors is more challenging. Given the high expression of CEA, also known as CEACAM5,Citation7 in many solid tumors, including colonCitation8,Citation9 and breast,Citation10,Citation11 we asked if the clinically tested humanized anti-CEA antibody hT84.66-M5A (M5A)Citation12 would be effective against solid tumors in an ICK format. It should be noted that there are over 20 members of the CEA gene family, many of which have highly homologous protein sequences,Citation13 necessitating a careful choice of CEA specific antibodies, among which M5A and its parent murine antibody T84.66 have been shown to have exquisite CEA specificity.Citation14,Citation15
As the first step in this direction, we previously showed that the parent murine antibody to M5A (T84.66) fused to murine IL-2 was able to potently suppress CEA-positive tumor growth in CEA transgenic mice bearing CEA-positive tumors.Citation16 Although CEA is abundantly expressed on the apical lumen of the colons of the CEA transgenic mice, there was no evidence of anti-colon effects. Mechanistically, since CEA is expressed on the apical lumen of the colon, systemically administered ICK is unable to target this expression pattern, while CEA-positive tumor cells are accessible to the circulation. In accordance with this finding, the specific targeting of anti-CEA antibodies to CEA-positive tumors and not to normal colon in CEA transgenic animals is shown quantitatively by PET imaging with radiolabeled anti-CEA antibodies.Citation17 Furthermore, administration of radiolabeled chimeric anti-CEACitation17 or M5ACitation18 targets CEA-positive tumors and not normal colon in man.Citation19 In terms of therapeutic approaches, we have also shown that both the chimeric and humanized versions of this antibody have efficacy in treating CEA-positive tumors with Y-90 labeled antibody as single agentsCitation19 or in combination with chemotherapy.Citation20 Since the main drawback of radioimmunotherapy with Y-90 labeled antibodies is bone marrow immunosuppression,Citation19 ICKs are ideally positioned to take advantage of high tumor targeting, along with harnessing the immune system, rather than inhibiting it. Recently, Klein et al.Citation21 developed an anti-CEA-IL-2 ICK that showed excellent tumor regression in a CEA transgenic model. However, in their study, the binding of IL-2 to its cognate receptor CD25 was removed by genetic engineering. Thus, the role of a humanized anti-CEA ICK with an intact human IL-2 is an open question.
In the current study, we show that a fully bioactive anti-CEA-IL-2 ICK can be produced in clinically relevant amounts. Radiolabeled ICK targeted both CEA-positive tumors and lymph nodes, but not the colon in the CEA transgenic model, with reduced tumor uptake compared to radiolabeled M5A alone. When ICK was tested as a single agent against CEA-positive breast and colon tumors, significant tumor reduction was observed vs anti-CEA antibody controls in the CEA transgenic model. Analysis of lymphocyte fractions in blood, spleen, and tumor revealed significant increases in the frequency of CD4+, CD8+ T cells, and CD11b positive cells in CEA-positive tumors treated with ICK vs anti-CEA antibody-treated controls. Both CD8+ and CD4+ T cells exhibited increased IFNγ production, especially in the tumor infiltrates. PD-1 and FoxP3 levels were similar between ICK and antibody-treated controls for CD4+ T cells, but PD-1 only increased in CD8+ T cells in ICK treated tumor-bearing mice. When ICK therapy was combined with anti-PD-1 therapy only incremental tumor reduction was observed. When tumors were pretreated with stereotactic radiation (SRT) followed by ICK, infiltrating lymphocyte numbers were reduced and no further tumor reduction was observed, but when the mice were re-challenged with new CEA-positive tumors, these tumors failed to grow suggesting long-lasting tumor immunity had occurred. Taken together, these results suggest that the anti-CEA-IL-2 ICK is suitable for clinical trials, especially in the combination with SRT.
Results
Generation of a bioactive and immunoactive anti-CEA-IL-2 ICK
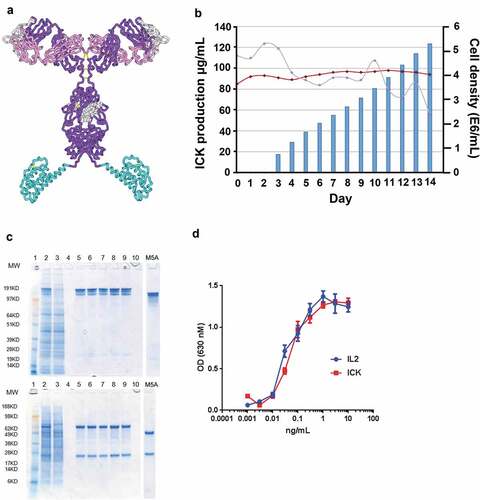
The ICK was generated by fusion of the gene for human IL-2 to the 3’-end of the gene of the heavy chain of M5A. Splice overlap extension was used to mutate 4 of the last 7 amino acid residues of the M5A heavy chain to remove a potential T cell epitope and proteolytic cleavage site at the heavy chain/IL-2 junction. A model of the ICK is shown in . Plasmids containing the fusion protein and the light chain of M5A were transfected into CHO-S cells as described in Methods. The ICK was expressed at the level of 0.12 g/L in 14 days with a purified yield of 0.435 g from a 20 L bioreactor run (). Purified ICK gave a single band for both heavy and light chains under reducing conditions on SDS gel electrophoresis () and high IL-2 activity in both an ELISA (data not shown) and in a bioassay (). Radiolabeled ICK had >95% immunoreactivity when tested in a size exclusion supershift assay with CEA and was stable in serum for >72 h (data not shown).
Figure 1. Generation of a bioactive and immunoactive anti-CEA-IL2 ICK. (a) A structure model of ICK. (b) ICK production in cell culture (bar graph), cell viability (red), cell density (blue). (c) SDS gel electrophoresis of purified ICK, non-reduced (top) and reduced (bottom). Lanes: 1. MW markers; 2. M5A-H-IL2 harvest; 3. Protein A flow through; 5. Protein A eluate; 6. Prior to low pH; 7. After low pH (30’); 8. After neutralization/filtration; 9. M5A-H-IL2 previous batch; 4 and 10. Empty. (d) IL-2 and ICK activity measured using a HEK-Blue IL-2 reporter cell line that expresses an inducible secreted embryonic alkaline phosphatase (SEAP), absorbance at 630 nm was measured after 2-h incubation
Murine models for CEA+ tumors in CEA transgenic mice
We previously showed that the murine colon carcinoma cell line MC38 transfected with CEA produced s.c. tumors that grew at comparable rates as the parental cell line in CEA transgenic mice.Citation22 To generate a second model, we transfected the murine breast carcinoma cell line E0771Citation23 with a CEA expressing plasmid () and showed that orthotopic tumors in the mammary fat pad also grew at comparable rates to the parental cell line in CEA transgenic mice (). Since both cell lines and the transgenic mice are in the C57/B6 background, they allow us to perform immunotherapy studies in immunocompetent animals in which human CEA is expressed in comparable tissues to man.Citation24 Importantly, CEA is a relevant tumor antigen for both colonCitation25 and breastCitation24 cancers.
Figure 2. Establishment and validation of CEA-positive mouse breast carcinoma model. (a) CEA expression in E0771 mouse breast carcinoma cells transfected with CEA plasmid shown using flow cytometry. (b) Tumor growth comparison of E0771 parental cells and CEA transfected. 1 × 105 cells injected in a mammary fat pad (n = 5–6 per group). (c) PET imaging of E0771/CEA bearing CEA transgenic mice using 64Cu-DOTA-M5A performed at indicated time points, two representative mice shown. (d) PET imaging of E0771/CEA bearing CEA transgenic mice using 64Cu-DOTA-ICK performed at indicated time points, two representative mice shown
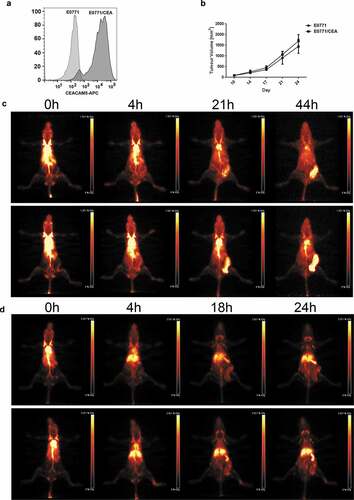
Since we wanted to treat CEA+ tumors with ICK, it was important to show that ICK bound to E0771/CEA cells, but not to parental E0177 cells, and to murine T cells (Supplemental Fig S2). We have previously used radiolabeled humanized anti-CEA antibody M5A to target CEA-positive tumors in preclinicalCitation12,Citation17 and patient studies.Citation20 Thus, it was also important to show that the ICK derived from M5A retained its ability to target CEA+ tumors in the murine model system. 64Cu-DOTA-M5A exhibited excellent targeting of E0771/CEA tumors in the CEA transgenic model (). While 64Cu-DOTA-ICK exhibited less targeting to E0771/CEA tumors, significant lymph node targeting was observed (), compared to M5A alone (). We conclude that while the binding of ICK to CD25+ T cells alters the biodistribution compared to M5A alone, there remains high tumor uptake with evidence that lymph node resident T cells are also targeted (Supplemental Fig S3). It should be noted that human IL-2 is able to bind both human and murine CD25 on their respective T cell populations.Citation26
Treatment of established E0771/CEA tumors with ICK leads to tumor regression and immune activation
When established orthotopic E0771/CEA tumors (200 mm3) were treated daily for five days with ICK (25 µg/mouse, i.p.) suppression of tumor growth was observed by day 15 and continued out to day 26 by which time all of the M5A treated control mice had to be euthanized due to the large size of their tumors (). Immune activation in the ICK vs M5A control treated tumor-bearing mice was evidenced by an increase of CD8+ T cells frequency in harvested tumors at day 26 (Fig S4A) as well as an increase of frequency of CD11b positive myeloid cells in the spleens (Fig S3B). Moreover, a reverse of CD4+ to CD8+ ratio in the spleen, draining lymph nodes and tumor tissues was also observed, with more CD8+ than CD4+ T cells in ICK treated mice (). Interestingly, even though the frequency of CD11b+ cells did not change between two groups, there was a significant reduction of CD11c and F4/80 double positive myeloid cells in tumors treated with ICK (Fig S4C). However, in our model we did not observe large changes in the presence of NK cells, showing only a decrease of this population in the blood and a slight increase in tumor-draining lymph nodes mice treated with ICK (Fig S4D).
Figure 3. ICK treatment significantly inhibits the growth of E0771/CEA mouse breast carcinoma. (a) Tumor-bearing CEA-Tg mice were injected I.P. with five daily injections of 25 µg of M5A or ICK antibodies. Representative experiment of three experiments shown (n = 4 per group). (b) CD8+ to CD4+ T cells ratios done by flow analysis in indicated tissues (pooled, n = 8–10 per group). (c) Increase of IFNγ producing CD4+ T cells frequency in ICK treated mice shown in indicated tissues, studied by intracellular staining and flow analysis (pooled, n = 8–10 per group). (d) Increase of IFNγ producing CD8+ T cells frequency in ICK treated mice shown in indicated tissues, studied by intracellular staining and flow analysis (pooled, n = 8–10 per group). (e) Flow analysis of IFNγ and PD-1 expression on tumor-infiltrating CD4+ T cells (n = 4–5 per group). (f) Flow analysis of IFNγ and PD-1 expression on tumor-infiltrating CD8+ T cells (n = 4–5 per group). (g) Significant decrease of FoxP3+ Treg population frequency among CD4+ T cells in tumor tissue by ICK treatment studied by intracellular staining and flow analysis (pooled, n = 8–10 per group). (h) Flow analysis of Foxp3 and CTLA4 expression on tumor-infiltrating CD4+ T cells (n = 4–5 per group). ****p < .0001; ***p < .001; **p < .01; *p < .05
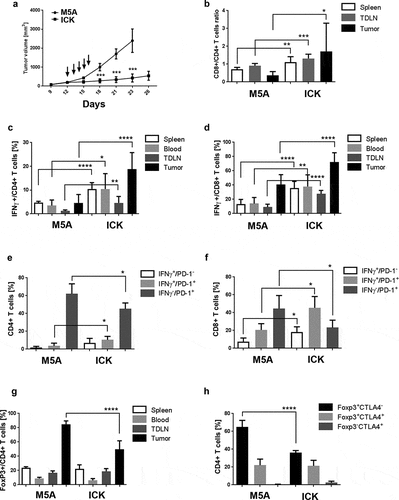
Increases in the production of IFNγ, an important marker of activated Th1/Tc1 cell, were observed for both CD4+ and CD8+ T cells in blood, spleen, tumor and tumor-draining lymph nodes (). These observations were further confirmed by analyzing the fractions of IFNγ+ CD4+ and CD8+ T cells in all live cells in the tumors, indicating an increase of both populations in ICK treated tumors (Fig S4E).
The total expression of PD-1 on CD4+ T cells from spleen, blood, and tumor-draining lymph nodes but not the tumor was increased with ICK treatment (Fig S4F), but was unchanged in CD8+ T cells (Fig S4G). Since PD-1 expression can be also attributed to general T-cell activationCitation27 we decided to analyze the simultaneous expression of both PD-1 and IFNγ. We found a shift in populations of both tumor-infiltrating CD4+ and CD8+ T cells expressing IFNγ and PD-1 that was indicated by an increase of IFNγ single positive cells and IFNγ/PD-1 double positive cells, and decreased PD-1 single positive cells T cells in mice treated with ICK (). Furthermore, tumor-draining lymph node CD8+ T cells from mice treated with ICK showed only an increase of IFNγ single positive cells (Fig S4H).
The frequency of FoxP3+ CD4+ T cells, Treg cells, was decreased in ICK vs M5A treated tumors (), indicating that ICK actually reversed Treg infiltration into tumors. Interestingly, the population that was reduced were Foxp3+ and CTLA4− (). In terms of IL-10 production by FoxP3+ tumor-infiltrating CD4+ T cells, less than 3% were double positive in both groups (data not shown).
Effect of stereotactic radiation therapy (SRT) on E0771/CEA tumor growth and immune activation
Since radiation therapy is a common treatment mode for breast cancer, with relatively unexplored effects on the immune system, we tested the effects of SRT ± ICK on E0771/CEA tumor growth. Tumors (200 mm3) were irradiated with 20 Gy using an image-guided irradiator followed by 4 doses of 25 µg of ICK administered i.p. (). The results show all untreated controls reached a maximum tumor size by day 19, the majority of the SRT only treated mice reached a maximum tumor size by day 25 [6/9], and the majority of the combined therapy show retarded tumor growth through day 25 (). After day 25, 7/9 of the SRT+ICK-treated tumors began to regrow, while 2/9 of the combined therapy exhibited no further tumor growth through day 32. Although we did not see a statistical increase of CD8+ T-cell frequency (Supplemental Fig S5A), the ratio of CD8+ to CD4+ T cells was higher in SRT+ICK vs SRT+M5A treatment groups (Supplemental Fig S5B). Analysis of IFNγ production of CD4+ and CD8+ T cells in the spleen, lung and tumor-draining lymph nodes revealed increases for combined SRT+ICK vs SRT+M5A alone therapy (). Moreover, similar increases were found in tumor tissues ( and Fig S5C, shown as fractions of both T cell populations and live cells, respectively). Analysis of the expression of checkpoint inhibitors PD-1, Tim3 and CTLA4, along with Foxp3 for Tregs within tumor-infiltrating CD4+ T cells, revealed highest levels for PD-1, and reduced levels for Foxp3 in the combined therapy group (). While we observed a reduction of Foxp3+ and CTLA4− CD4+ T cells, there was an increase of Foxp3+ and CTLA4+ CD4+ T cells in SRT+ICK vs SRT+M5A treated tumors (Supplemental Fig S5D).
Figure 4. Effect of stereotactic radiation therapy (SRT) on E0771/CEA tumor growth and immune activation. (a) Study design scheme indicating tumor injections, SRT (20 Gy) and immune therapy schedule. (b) SRT-ICK combination therapy shows highly significant tumor growth inhibition (Representative experiment of 2 shown, n = 3–4 per group). (c) Intracellular staining followed by flow analysis of CD4+ T cells shows an increase in IFNγ production in SRT/ICK group, shown in indicated tissues (pooled, n = 9 per group). (d) Increase of IFNγ production by CD8+ T cells in SRT/ICK group, shown in indicated tissues (pooled, n = 9 per group). (e) IFNγ production in tumor-infiltrating CD4+ and CD8+ T cells, studied by intracellular staining and flow analysis (pooled, n = 9 per group). (f) Changes in expression of inhibitory molecules on tumor-infiltrating CD4+ T cells, studied by extracellular and intracellular staining followed by flow analysis (pooled, n = 9 per group). (g) Increase of IFNγ producing and Tim3 tumor-infiltrating CD4+ and CD8+ T cells in SRT/ICK vs SRT/M5A studied by extracellular and intracellular staining followed by flow analysis (pooled, n = 9 per group). (h) In contrast to control tumors SRT/ICK and SRT/M5A treated tumors show decrease presence of PD-1+/Tim3+ double positive exhausted tumor-infiltrating CD8+/IFNγ+ T cells (n = 5 per group). ****p < .0001; ***p < .001; **p < .01; *p < .05
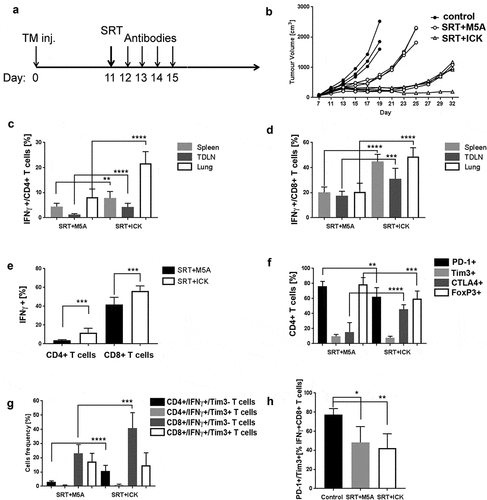
Although total expression of PD-1 and Tim3 on tumor-infiltrating CD8+ T cells was unchanged between both groups (Supplemental Fig S5E), we observed an increase of IFNγ producing and Tim3− CD4+ and CD8+ T cells in SRT+ICK vs SRT+M5A treatment groups (). Since it was previously described that the presence of two or more inhibitory molecules on the surface of activated T cells is associated with exhaustion,Citation28 we investigated the simultaneous expression of PD-1 and Tim3 on IFNγ producing CD8+ T cells in the tumors. We found a reduced frequency of these cells in both SRT+M5A and SRT+ICK treatment groups ( and Supplemental Fig S5F). We conclude that combined SRT+ICK therapy is more effective than SRT alone and that combined therapy has significant effects against aggressive tumors. Furthermore, if the elevation of checkpoint inhibitory markers on T cells plays a limiting role, the combination of PD-1 and Tim3 is found in this model.
Limited abscopal effect of SRT/ICK therapy of breast carcinoma
Since it has been reported that SRT can occasionally lead to abscopal effects,Citation29 orthotopic tumors were implanted in mammary fat pads on both sides of the mice. The treatment course (Supplemental Fig S6A) involved a single 20 Gy dose to tumors on the right flank only at day 10 when the tumors had reached an initial size of 200 mm3. Controls included no SRT or M5A treatment. Tumor-bearing mice received daily doses of ICK as before. Control mice receiving M5A only showed rapid tumor growth on the right flank by day 21 (Supplemental Fig S6B), and all had to be euthanized. Mice receiving SRT plus M5A or ICK only exhibited significant and similar tumor growth inhibition compared to M5A controls (Supplemental Fig S6B). Combination therapy of SRT and ICK led to complete tumor growth inhibition (Supplemental Fig S6B). Examination of tumor growth on the left flank showed poor evidence for an abscopal effect in this model system (Supplemental Fig S6C). All groups showed tumor escape with an indication that combined SRT + ICK was more effective than SRT or ICK alone. We conclude that the increased tumor burden (tumors on both flanks) overwhelmed the anti-tumor ability of the immune system even with the boost provided by SRT or ICK or the combination of SRT and ICK.
Tumors were collected at the end of the treatments (days 21, 27, or 30) and T cell infiltration into tumors compared (Supplemental Fig S6D and E). Although there was a trend toward higher CD4+ and CD8+ T cell frequency in the combined therapy group, no major differences were observed. Similarly, analysis of IFNγ levels in tumor-infiltrating T cell subgroups (Supplemental Fig S6F) showed a trend toward the combined therapy, but the variation prevented a statistically significant result. Interestingly, we found an increase of IFNγ producing CD4+ and CD8+ T cells in tumor-draining lymph nodes in ICK alone or in SRT-ICK combination therapy (Supplemental Fig S6G).
Fractionated SRT combined with ICK therapy significantly improves survival and anti-tumor immunity in a breast carcinoma model
While a single high dose of radiation can inhibit tumor growth, it can also limit the effectiveness of immune therapy by extensive damage to tumor vasculature, tumor intrinsic mechanisms that reduce the release of tumor antigens and killing of tumor-infiltrating immune cells.Citation30 It has been shown that using multiple lower doses of radiation is a better approach when immune therapy is combined with radiation.Citation31 To test this idea in our model system, mice received a single dose of SRT (10 Gy) or 4 daily fractions of 2.5 Gy, followed by four doses of ICK (). Both single dose and fractionation SRT combined with ICK initially inhibited tumor growth, but eventually, all single dose SRT treatment groups exhibited tumor regrowth. In contrast, half of the mice treated with the fractionated dose achieved long-lasting tumor inhibition and survival (). Moreover, when the best responding mice from the fractionated SRT+ICK group were rechallenged with a second tumor injection on the opposite flank, all the mice rejected tumors within 10 days post injection, indicating immune memory against tumor (). In the separate experiment, we investigated the efficacy of both radiation modalities alone and found no inhibition effects on tumor growth by fractionated radiation but there was a delay in tumor growth by the single dose of radiation (Supplemental Fig S7A). Next, we analyzed tumor-infiltrating immune cells by flow when tumors reached the maximum allowed size. Not surprising, we found no significant differences in T cell frequency (data not shown), but we did observe significant increases of IFNγ production in CD4+ T cells as well as reduced expression of PD-1 on CD8+ T cells in the SRT fractionation group (). Similarly, no changes in tumor infiltration by T cells were found in tumors irradiated only, with a modest increase of IFNγ production in both T cells populations in the 10 Gy treated tumors (Supplemental Fig S7B and C). In contrast to tumors, tumor-draining lymph nodes exhibited significant differences in expression of IFNγ and PD-1 on both T-cells populations in both single and fractionated SRT/ICK treatment groups vs controls (). Moreover, as shown in our previous experiments, we found less exhausted CD8+ T cells in the tumors in both SRT treated groups vs controls (). Interestingly, similar observations were found in CD8+ T cells infiltrating tumors treated with SRT only (Supplemental Fig S7E).
Figure 5. Fractionated SRT combined with ICK therapy significantly improves survival and anti-tumor immunity in E0771/CEA model. (a) Study design scheme indicating tumor injections, SRT and immune therapy schedule. (b) Fractionated SRT combined with ICK leads to tumor eradication in half of the treated mice (Representative experiment of two shown, n = 4–7 per group). (c) Fractionated SRT combined with ICK results in extended survival of treated mice (summary of two experiments, n = 7–13 per group). (d) Tumor rechallenge on the opposite flank in best responding mice treated with fractionated SRT and ICK (n = 4). (e) Flow analysis of tumor infiltrating CD4+ and CD8+ T cells for intracellular IFNγ staining and extracellular PD-1 expression (pooled, n = 7 per group). (f) Flow analysis of tumor-draining lymph nodes CD4+ and CD8+ T cells for intracellular IFNγ staining and extracellular PD-1 expression (pooled, n = 7 per group). (g) SRT+ICK therapy reduces exhaustion of tumor-infiltrating CD8+IFNγ+ T cells, shown as double positive PD-1 and Tim3 staining pre gated on CD8+IFNγ+ T cells (pooled, n = 7 per group). (h) Flow analysis of CD4+/FoxP3+ Treg population in tumor tissue (pooled, n = 7 per group). ***p < .001; *p < .05
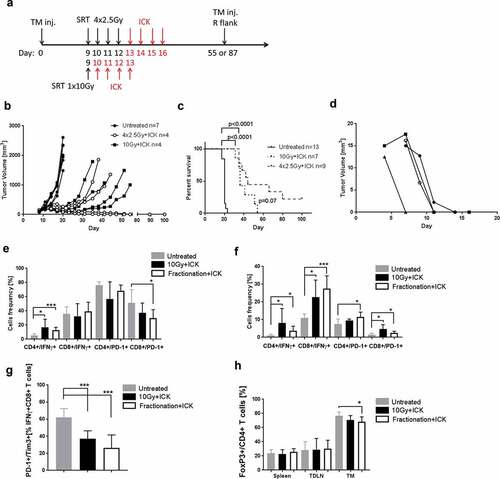
Regulatory T-cells frequency was decreased only in mice treated with a combination of fractionated SRT+ICK () and did not change in mice undergoing SRT only (Supplemental Fig S7D).
Effect of combined anti-PD-1 and ICK therapy in the E0771 model
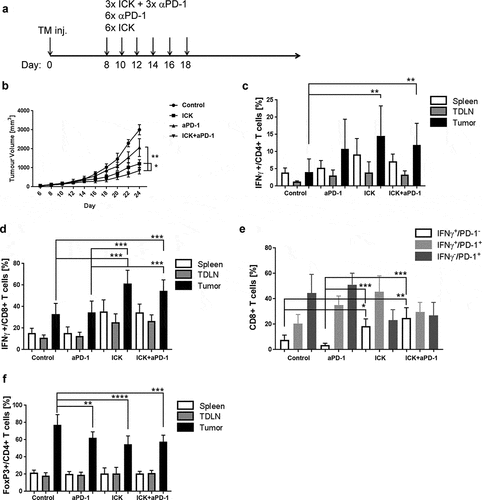
Since anti-PD-1 therapy is effective in the treatment of several solid tumors,Citation32 and PD-1 was the most conspicuous checkpoint inhibitor elevated in our model system, the two single and combined therapies were compared (). ICK alone was more effective than anti-PD-1 alone therapy, but the combined therapy was no better than the ICK monotherapy (). Analysis of IFNγ expression in CD4+ and CD8+ T-cells subsets from spleen, tumor and tumor-draining lymph nodes indicated a higher expression in ICK alone treated mice and in the combined therapy when compared to control or anti-PD-1 alone treated groups (). Similar to experiments shown in , in both groups treated with ICK we observed an increased frequency of tumor-infiltrating CD8+ T cells expressing IFNγ but not in the PD-1 alone treated mice (). In contrast, there was a reduced frequency of tumor-infiltrating Tregs in all treatment groups (). Nevertheless, there was little or no improvement in adding anti-PD-1 to ICK therapy.
Figure 6. Effect of combined anti-PD-1 and ICK therapy in the E0771 model. (a) Study design scheme indicating tumor injection and immune therapy schedule. (b) ICK alone or in combination with anti-PD-1 antibody shows the superior tumor growth inhibition in comparison to anti-PD-1 therapy alone (1 of 2 experiments shown, n = 4 per group). (c) Increase of IFNγ production in tumor-infiltrating CD4+ T cells from ICK alone or in combination with anti-PD-1-treated mice, analyzed by flow in indicated tissues (pooled, n = 8–9 per group). (d) Increase of IFNγ production in tumor-infiltrating CD8+ T cells from ICK alone or in combination with anti-PD-1-treated mice, analyzed by flow in indicated tissues (pooled, n = 8–9 per group). (e) Flow analysis of IFNγ and PD-1 expression on tumor-infiltrating CD8+ T cells (n = 5 per group). (f) Flow analysis of CD4+/FoxP3+ Treg population in indicated tissues (pooled, n = 8–9 per group). ****p < .0001; ***p < .001; **p < .01. TDLN – tumor-draining lymph nodes
SRT and SRT/ICK therapy in the MC38/CEA model
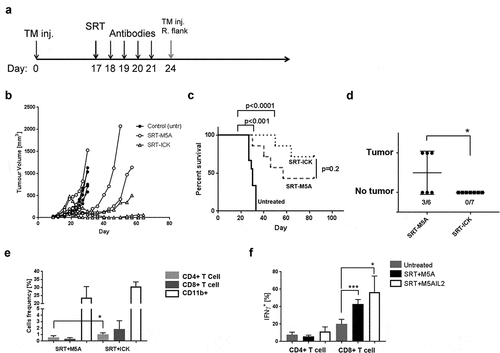
In order to evaluate SRT ± ICK therapy in a second CEA+ tumor model system, we examined the murine colon carcinoma MC38/CEA model that was previously studied by us using RITCitation33 and in all murine ICKCitation16 therapies. To test the possibility of immune memory, the mice were injected with MC38/CEA cells on the opposite flank on day 24 during therapy (). The results for SRT+M5A versus SRT+ICK combination were variable, with more (3/7 vs 5/7, respectively) mice showing tumor growth inhibition and increased survival for the SRT+ICK combined therapy (). The tumor rechallenge study resulted in the growth of secondary tumors in half (3/6) of SRT alone treated mice but in none (0/7) of the SRT+ICK treated group (). Since the analysis of immune cells infiltrating tumor tissues was limited to the mice that did not reject the tumors, the analysis is not fully representative to the biological outcome of therapy. Flow analysis of primary tumors indicated an increased frequency of CD4+ T cells in SRT+ICK treated mice (). In both treated groups, CD8+ T cells produced more IFNγ than untreated controls; however, there was no difference between SRT+M5A versus SRT+ICK (). Thus, the colorectal tumor model showed a higher radiosensitivity of the tumors compared to the breast cancer model with a major therapeutic effect of the SRT+ICK combined therapy that led to the establishment of anti-tumor immune memory in all of the SRT+ICK treated mice.
Figure 7. SRT and SRT/ICK therapy in the MC38/CEA model. (a) Study design scheme indicating tumor injections, SRT (20 Gy) and immune therapy schedule. (b) SRT-ICK combination inhibits primary MC38/CEA tumors in comparison to SRT alone (Representative experiment of 2 shown, n = 3–4 per group). (c) SRT combined with ICK results in extended survival of treated mice (summary of two experiments, n = 7–9 per group). (d) SRT-ICK completely stops implantation of secondary tumor injected on opposite, right flank (summary of two experiments, n = 6–7 per group). All but one SRT, SRT+ICK treated mice (the one that had to be terminated by day 24) were rechallenged. (e) Flow analysis of indicated immune cells infiltrating primary tumors, fractions of live cells shown (pooled, n = 3–4 per group). (f) Flow analysis of IFNγ producing tumor-infiltrating CD4+ and CD8+ T cells (pooled, n = 3–4 per group). ***p < .001; *p < .05
Comparison of myeloid infiltrates in E0771/CEA vs MC38/CEA tumor models
Besides the expression of T cell checkpoint inhibitors, it has been reported that immune inhibition of tumor growth can also be mediated by myeloid-derived suppressor cells as represented by inhibitory macrophages, granulocytes or monocytes.Citation34 Analysis of the tumor-infiltrating myeloid cells in the two model systems studied here reveals significant differences in their sub-populations (Supplemental Fig S8A and B). Quantitation of the analysis shows less total CD11b myeloid infiltration in E0771/CEA vs MC38/CEA tumors (Supplemental Fig S8C). Furthermore, MC38/CEA tumors were mainly infiltrated by F4/80+ macrophages (Supplemental Fig S8D) in contrast to E0771/CEA where Ly6CHi myeloid cells comprised the majority of CD11b+ cells (Supplemental Fig S8E). Interestingly, Ly6C-Ly6G double positive cells that could include neutrophils and granulocytic MDSCs represented an insignificant fraction of myeloid cells in both tumor models (Supplemental Fig S8F). Thus, myeloid infiltrates may play a role in particular in combination radio- and immunotherapies, indicating the increased infiltration of macrophages being a favorable feature in more radiosensitive MC38/CEA tumors.
Discussion
Activation and maintained survival of T cells are required for the generation of efficient immune responses against infectious agents as well as neoplasms. Among the major factors involved in this process are the production of cytokines such as IL-2 that stimulate the survival and expansion of T cells. The idea of using IL-2 in immune therapy of neoplasms is not new, but because of the risk of systemic toxicity and its rapid clearance from the circulation, the targeted delivery of this cytokine to the tumors was explored. One successful approach was the use of recombinant fusion proteins called immunocytokines (ICKs) that combined the targeting features of antibodies and activation of immune cells by cytokines. ICKs that combine tumor-targeting antibodies and IL-2 have exhibited significant therapeutic effects in multiple preclinical and clinical trial studies.Citation35 Clinical trials using the ICK L19-IL-2 for metastatic melanoma and renal cell carcinoma showed low toxicity and promising anti-tumor responses.Citation4,Citation36 DI-Leu16-IL-2, an anti-CD20 ICK, gave excellent results in a preclinical studyCitation5 and in a phase I/II trial of relapsed and refractory B-cell lymphoma (NCT01874288). In the case of CEA-positive tumors, an anti-CEA-IL-2 ICK was developed by Roche with a variant of IL-2 that does not bind to CD25, the α-subunit of the high-affinity IL-2R.Citation21 The rationale for this IL-2 variant was to reduce the expansion of Tregs that utilize CD25 and may otherwise compete with CD8 expansion. This ICK therapy is now in a phase I/II clinical trial in combination with anti-PD-L1 (NCT02350673). In contrast to their study where NK expansion occurred, no expansion of NK cells in the spleen and tumor was observed for our ICK treated mice.
In this study, we describe the generation, specificity and antitumor activity of a humanized anti-CEA-IL2 fusion antibody as monotherapy or combined with stereotactic radiation or with an anti-PD-1 antibody. We performed the study in immunocompetent mice expressing the full-length human CEA gene including its natural promoter and in an orthotopic breast cancer model to more closely mimic the clinical situation in breast cancer therapy. Our approach used an ICK with human IL-2 that retained binding to IL-2Rα to maximize its in vivo potency and to study the potential expansion of Tregs. Although IL-2 has high affinity to IL-2Rα,Citation37 in our studies we observed a decrease in the frequency of Treg cells in the tumors for groups treated with ICK alone or in combination with SRT. Our results are similar to those reported in which the ICK F8-IL-2 used to treat metastatic lung cancer in a mouse model resulted in significant tumor infiltration of both CD3+ T and NK cells but not of Treg cells.Citation38
To improve the anti-tumor efficacy of ICK therapy we combined it with SRT. Indeed, a growing number of preclinical studies, as well as clinical trials, show strong benefits in the treatment of solid tumors by the combination of radiation and immunotherapy.Citation39
Rekers et al. showed that colon cancer tumors in immune-competent mice treated with radiation in combination with the L19-IL-2 ICK resulted in long-lasting anti-tumor responses, including an abscopal effect.Citation40 Also in that study cured mice showed immune memory against tumor rechallenge. However, in our breast cancer model, immune memory responses were observed only in mice treated with fractionated SRT and ICK, suggesting that single high dose SRT has an immunosuppressive effect. It is known that radiation effects on tumors depend on the dose per fraction, the number of fractions and the total dose.Citation41 In agreement with this finding, Dewan et al. showed that fractionated vs single dose radiation combined with anti-CTLA4 immunotherapy produced an abscopal effect in an immunocompetent breast cancer model.Citation31 In this regard, we believe that the effectiveness of the immune response to therapy can be affected by the tumor location. In particular, the orthotopic mammary fat pad model is challenging, as shown by Meng et al. where irradiated adipose tissue produced numerous inflammatory factors that generated a wound healing-like environment that facilitated angiogenesis, favoring tumor growth.Citation42 In agreement with this finding, we observed higher response rates in the subcutaneous MC38/CEA tumor model treated by single high dose radiation compared to the orthotopic E0771/CEA breast tumor model. Mammary fat pad vs subcutaneous tumor implantation may have also played a role in the difference in myeloid cell tumor infiltration, that in orthotopic breast tumor model were predominantly Ly6CHi monocytic cells, whereas in the subcutaneous colon tumor model, macrophages were the major myeloid population. Some myeloid cells not only inhibit cytotoxic T cells but also support tumor angiogenesis, not only during tumor growthCitation43 but also after tumor irradiation.Citation44 Thus, the tumor microenvironment modulates T cell immunity and tumor responses to radiation. Since most solid tumors originate in or near adipose tissue, the tumor-supporting role of adipose tissue must be considered in designing effective tumor therapy, especially when immunotherapy is used.
Due to their positive results, clinical immunotherapy approaches have mainly focused on antibody-based, checkpoint inhibitors. Mechanistically, they rely on overcoming the natural tendency of the immune system to suppress anti-self immunity, especially where tumor antigenicity is weak. Since the production of humanized antibodies is routine, these reagents can be produced in the large quantities necessary for systemic therapy. However, these therapies are untargeted in that they may activate even self-reactive T cells to normal tissue. On the other hand, since IL-2-based ICKs are tumor targeted, they act more directly on T cells within the tumor. Since they are also delivered systemically, self-reactive T cells may be activated, but to a lesser extent than in anti-checkpoint therapy. The possible combination of anti-CEA-IL-2 ICK and anti-PD-1 therapy has shown some promising synergistic effects in the subcutaneous tumor model.Citation21 In addition, Schwager et al. found L19-IL-2 ICK treatment of subcutaneous CT26 colon tumors was inferior to the combination of ICK with anti-CTLA4 treatment.Citation45 However, in our study, the combination of ICK with anti-PD-1 antibody therapy had no improvement in the inhibition of orthotopic breast tumor growth, suggesting that ICK plus anti-checkpoint inhibitors may not be a universal approach. In this respect, co-expression of multiple inhibitory receptors like PD-1 and Tim-3 may play a more prominent role in the inhibition of tumor-infiltrating CD8+ T cells as described by Sakuishi et al.Citation28 Since IFNγ production is used as both a measure of T cell activation and induction of checkpoint inhibitors, measurement of IFNγ may offer a clue to therapy outcome. In this respect, we found that SRT and ICK reduced the numbers of exhausted IFNγ positive CD8+ T cells.
We conclude that ICK anti-CEA-IL-2 therapy, especially when used in combination with low dose, fractionated SRT, may be an effective strategy to treat CEA-positive tumors, in that induced immune responses may not only reduce tumor growth but also lead to prolonged tumor immunity.
Materials and methods
Generation and expression of the anti-CEA-IL-2 ICK
The protein sequences of M5A heavy and light chain have been previously published.Citation12 The protein sequence of the mature form of human IL-2 was from UniProt entry P60568 (residues 21–153). Murine codon-optimized synthetic cDNA genes were purchased from GeneArt. To create the M5A-H-IL-2 construct, the IL-2 gene was fused directly to the 3’-end of a mutated gene encoding the gamma-1 heavy chain. The mutation removes a potential T cell epitope and proteolytic cleavage site at the heavy chain/IL-2 junction (LSLSPGK→ATATPGA) as previously described.Citation5 Synthetic genes were PCR amplified using Platinum Taq DNA Polymerase and subcloned in expression vector pcDNA3.3-TOPO-TA (Life Technologies) and verified by Sanger sequencing. Large-scale transient transfection was performed using a Maxcyte STX transfection system (Gaithersburg, MD). Freedom CHO-S cells (Life Technologies) were grown in two 5 L shake flasks until 40 billion cells were obtained. After harvesting, cells were resuspended in electroporation buffer and mixed with separate plasmids encoding the antibody heavy chain-IL-2 and antibody light chain in a 3:1 molar ratio (final DNA concentration = 300 µg/mL). Duplicate 100 mL cell suspensions were electroporated using a MaxCyte STX instrument operating in flow mode. After a 20-minute recovery period, the transfected cells were pooled, transferred to a 20 L WAVE bag, and serum-free media added to a density of 5 million cells/mL. After one day at 37°C, sodium butyrate was added to a final concentration of 1 mM and the temperature was lowered to 32°C. Cells were fed daily with a nutrient-rich supplement that contained glucose, yeast hydrolyzate, EfficientFeed A and glutamine, formulated as per MaxCyte. After 14 days the supernatant was harvested and the product was purified using Protein A and cation exchange chromatography. Percent aggregation was determined using analytical size-exclusion chromatography. M5A was produced in CHO-S cells by the same protocol.
Purification
The cell culture harvests were clarified by batch treatment (5% w/v) with the anion exchanger, AG1x8 (Bio-Rad Laboratories, Hercules, CA). Initial ICK capture was on Prosep rA (Millipore-Sigma, St. Louis, MO, 117 mL CV). The Protein A eluted peak was held at pH 3.5 ± 0.1 for low pH viral inactivation and then loaded onto a Fractogel SO3 cation exchange column (Millipore; 61 ml CV) equilibrated with 20 mM MES, 50 mM NaCl, pH 5.0. The M5A-IL-2 was eluted with a linear gradient to 20 mM MES, 1 M NaCl, pH 6.0 to remove aggregates. The M5A-IL-2 was filtered through a Sartobind Q (Sartorius, Goettingen, Germany) membrane exchanger for removal of DNA contaminants and a Virosart CPV150 (Sartorius) for virus removal. The M5A-IL-2 was buffer exchanged by tangential flow filtration into a final formulation of 4% Sucrose, 100 mM Arginine, 5mM citric acid, pH 6.0. The M5A-IL-2 concentration was 1.27 mg/ml for a total of 435.3 mg. M5A and M5A-IL-2 were submitted for DNA, viral and endotoxin testing by BioReliance, Inc. and met their standard specifications.
Biochemical characterization
Aliquots of the purified M5A-IL2 were analyzed by SDS-PAGE, both reduced and non-reducedCitation46 and Novex isoelectric focusing gel (Thermo Fisher Scientific, Waltham, MA) Size-exclusion chromatography was carried out on a Superdex 200 HR10/30 column (GE Healthcare, 0.5 ml/min) run isocratic with PBS, 0.05% Sodium Azide. Protein concentration was determined by OD 280nm (Pharmacia Ultrospec III UV/Visible Spectrophotometer).
Immunological characterization
IL-2 activity was measured using a HEK-Blue IL-2 reporter cell line (InvivoGen) that expresses an inducible secreted embryonic alkaline phosphatase (SEAP) after IL-2 receptor activation. Cells (1x104 cells per well) were stimulated with respective concentrations of recombinant human IL-2 (Biolegend, San Diego, CA) or M5A-IL2. The supernatant was collected after 18 h and developed with QUANTI-Blue colorimetric enzyme assay reagent at 37°C. Absorbance at 630 nm was measured after 2 h with the CLARIOstar plate reader (BMG Labtech, Cary, NC).
Cell lines
Murine breast carcinoma cell line E0771 and colon cancer cell line MC38 were stably transfected with CEA expressing plasmid as previously described.Citation16,Citation47 Both cell lines were maintained in DMEM supplemented with 10% fetal bovine serum and 100 U/ml penicillin/streptomycin. Cell cultures were tested annually for the presence of mycoplasm using a mycoplasma detection kit (Universal Mycoplasma Detection Kit, ATCC).
Animal studies
All animal experiments were performed using CEA transgenic mice as previously described.Citation22 For the breast carcinoma model, E0771/CEA tumor cells were injected into the mammary fat pad at the concentration of 1 × 105 cells in matrigel (total volume 50 µl), on one or both flanks depending on the experiment performed. For the colon cancer model, MC38/CEA cells were injected subcutaneously at the concentration of 1 × 106 cells (total volume 50 µl). During the experiment, some of the mice received a second subcutaneous injection at the same concentration on the opposite flank. Twenty-five micrograms of M5A or ICK were delivered for each dose. Mouse care and experimental procedures were performed under pathogen-free conditions in accordance with established institutional guidance and approved protocols from the Institutional Animal Care and Use Committee of Beckman Research Institute at City of Hope National Medical Center.
PET imaging studies
Anti-CEA antibody M5A or ICK were conjugated with NHS-DOTA as previously described.Citation17 Immunoreactivity of both antibodies to CEA was confirmed and animal imaging studies were performed in CEA-Tg mice bearing E0771/CEA tumors as previously described.Citation17 PET scans were acquired using an Inveon microPET/CT scanner (Siemens Medical Solutions). Mice were anesthetized by inhalation of 2–4% isoflurane in oxygen, placed on the PET scanner and injected with a single intravenous dose of 100 µCi (10 µg) of 64Cu-DOTA-M5A or 64Cu-DOTA-ICK in 1% human serum albumin–buffered saline through a tail vein catheter. At the terminal time point, the mice were euthanized and biodistribution studies performed.
SRT studies
Tumor-bearing mice were treated with the Precision X-RAD SMART Plus/225cx (Precision X-Ray, North Branford, CT). The X-RAD SMART Plus/225cx is an x-ray irradiator able to image and treat small animals through modulation of its focal spot, and beam quality. It has a maximum tube potential of 225kv. Photons are filtered through a Beryllium window with an additional 2.0 mm Al filter for imaging and 0.32 mm Cu filter for treatment.Citation48 A Monte Carlo dose engine based on EGSnrc/DOSXYZnrcCitation49 is used for Dose calculations for small animal radiotherapy. Tumor volumes were determined by cone beam CT scan (40 kVp, 8 mA at a 200 µm resolution) and then treated with 10 × 10 mm square field with opposing lateral beams at 225 kVp, 13 mA x-rays. Beams were angled to match internal borders to minimize abdominal gut exposure. Groups were treated with single or 4 fractions at either 10 or 20 Gy. During treatment, animals were under anesthesia with 2% isoflurane. Dose was normalized to 102% of the prescription dose to the center of the tumor mass to ensure 100% of the tumor volume was receiving the prescription dose.
Leukocyte analysis
Tissues and blood were collected at the termination of the studies and leukocyte populations were analyzed by flow cytometry. Blood samples were used after red cell lysis. Tumor draining lymph nodes and spleens were pushed through a 40µm cell strainer and red cells were lysed. Lungs and tumors were dissociated by enzymatic digestion using gentleMacs Octo Dissociator and dissociation kit following the manufacture’s protocol (Miltenyi Biotec). Cell suspensions were stained with different combinations of fluorochrome-coupled antibodies to CD3, CD4, CD8, B220, CD19, CD11b, Ly6C, Ly6G, CD11c, F4/80, NK1.1, NKp46, PD-1, CTLA-4, Tim-3 (BioLegend). For IFNγ production cells from all tissues were re-stimulated using PMA (10 ng/ml; Sigma-Aldrich) and ionomycin (1 µg/ml; Sigma-Aldrich) in the presence of Brefeldin A (5 µg/ml; BioLegend, CA) in 10% FBS IMDM media for 4 h in 37°C. Next, cells were stained for surface markers and viability markers (Zombie UV, BioLegend) and fixed and permeabilized using Foxp3 Transcription Factor Fixation/Permeabilization kit (ThermoFisher) following the manufacture’s protocol. Finally, cells were stained for intracellular IFNγ (BioLegend) and analyzed by flow cytometry. For FoxP3 (ThermoFisher) expression cells were stained for surface markers, fixed and permeabilized as described above. The gating strategies for immune cells of interest are summarized in Fig S1.
Statistical analysis
To calculate two-tailed p values and estimate the statistical significance of differences between treatment groups we used an unpaired t-test and two-way ANOVA test. P values are indicated in figures as follows: ****P < .0001, ***P < .001, **P < .01, *P < .05. Data were analyzed using GraphPad Prism software.
Abbreviations
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Download ()Acknowledgments
The authors would like to thank Dr. Andrew Raubitschek for the production of the GMP grade cytokine and early experiments with the combination of radioimmunotherapy and ICK.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website..
Additional information
Funding
References
- Tarhini AA, Cherian J, Moschos SJ, Tawbi HA, Shuai Y, Gooding WE, Sander C, Kirkwood JM. Safety and efficacy of combination immunotherapy with interferon alfa-2b and tremelimumab in patients with stage IV melanoma. J Clin Oncol. 2012;30(3):322–14. doi:10.1200/JCO.2011.37.5394.
- List T, Neri D. Immunocytokines: a review of molecules in clinical development for cancer therapy. Clin Pharmacol. 2013;5:29–45. doi:10.2147/CPAA.S49231.
- Rosenberg SA. IL-2: the first effective immunotherapy for human cancer. J Immunol. 2014;192(12):5451–5458. doi:10.4049/jimmunol.1490019.
- Johannsen M, Spitaleri G, Curigliano G, Roigas J, Weikert S, Kempkensteffen C, Roemer A, Kloeters C, Rogalla P, Pecher G, et al. The tumour-targeting human L19-IL2 immunocytokine: preclinical safety studies, phase I clinical trial in patients with solid tumours and expansion into patients with advanced renal cell carcinoma. Eur J Cancer. 2010;46(16):2926–2935. doi:10.1016/j.ejca.2010.07.033.
- Gillies SD, Lan Y, Williams S, Carr F, Forman S, Raubitschek A, Lo K-M. An anti-CD20-IL-2 immunocytokine is highly efficacious in a SCID mouse model of established human B lymphoma. Blood. 2005;105(10):3972–3978. doi:10.1182/blood-2004-09-3533.
- Galanina N, Kline J, Bishop MR. Emerging role of checkpoint blockade therapy in lymphoma. Ther Adv Hematol. 2017;8(2):81–90. doi:10.1177/2040620716673787.
- Beauchemin N, Draber P, Dveksler G, Gold P, Gray-Owen S, Grunert F, Hammarström S, Holmes KV, Karlsson A, Kuroki M, Lin SH. Redefined nomenclature for members of the carcinoembryonic antigen family. Exp Cell Res. 1999;252(2):243–249.
- Duffy MJ. Carcinoembryonic antigen as a marker for colorectal cancer: is it clinically useful? Clin Chem. 2001;47(4):624–630. doi:10.1093/clinchem/47.4.624.
- Goldstein MJ, Mitchell EP. Carcinoembryonic antigen in the staging and follow-up of patients with colorectal cancer. Cancer Invest. 2005;23(4):338–351. doi:10.1081/CNV-58878.
- Shao Y, Sun X, He Y, Liu C, Liu H, Batra SK. Elevated levels of serum tumor markers CEA and CA15-3 are prognostic parameters for different molecular subtypes of breast cancer. PLoS One. 2015;10(7):e0133830. doi:10.1371/journal.pone.0133830.
- Lee JS, Park S, Park JM, Cho JH, Kim SI, Park BW. Elevated levels of preoperative CA 15-3 and CEA serum levels have independently poor prognostic significance in breast cancer. Ann Oncol. 2013;24(5):1225–1231. doi:10.1093/annonc/mds604.
- Yazaki PJ, Sherman MA, Shively JE, Ikle D, Williams LE, Wong JY, Colcher D, Wu AM, Raubitschek AA. Humanization of the anti-CEA T84.66 antibody based on crystal structure data. Protein Eng Des Sel. 2004;17(5):481–489. doi:10.1093/protein/gzh056.
- Thompson JA, Grunert F, Zimmermann W. Carcinoembryonic antigen gene family: molecular biology and clinical perspectives. J Clin Lab Anal. 1991;5(5):344–366. doi:10.1002/(ISSN)1098-2825.
- Bjerner J, Lebedin Y, Bellanger L, Kuroki M, Shively JE, Varaas T, Nustad K, Hammarström S, Børmer OP. Protein epitopes in carcinoembryonic antigen. Report of the ISOBM TD8 workshop. Tumour Biol. 2002;23(4):249–262. doi:10.1159/000067255.
- Wagener C, Clark BR, Rickard KJ, Shively JE. Monoclonal antibodies for carcinoembryonic antigen and related antigens as a model system: determination of affinities and specificities of monoclonal antibodies by using biotin-labeled antibodies and avidin as precipitating agent in a solution phase immunoassay. J Immunol. 1983;130:2302–2307.
- Xu X, Clarke P, Szalai G, Shively JE, Williams LE, Shyr Y, Shi E, Primus FJ. Targeting and therapy of carcinoembryonic antigen-expressing tumors in transgenic mice with an antibody-interleukin 2 fusion protein. Cancer Res. 2000;60:4475–4484.
- Li L, Bading J, Yazaki PJ, Ahuja AH, Crow D, Colcher D, Williams LE, Wong JYC, Raubitschek A, Shively JE, et al. A versatile bifunctional chelate for radiolabeling humanized anti-CEA antibody with In-111 and Cu-64 at either thiol or amino groups: PET imaging of CEA-positive tumors with whole antibodies. Bioconjug Chem. 2008;19(1):89–96. doi:10.1021/bc700161p.
- Nittka S, Krueger MA, Shively JE, Boll H, Brockmann MA, Doyon F, Pichler BJ, Neumaier M. Radioimmunoimaging of liver metastases with PET using a 64Cu-labeled CEA antibody in transgenic mice. PLoS One. 2014;9(9):e106921. doi:10.1371/journal.pone.0106921.
- Wong JY, Chu DZ, Williams LE, Liu A, Zhan J, Yamauchi DM, Wilczynski S, Wu AM, Yazaki PJ, Shively JE, et al. A phase I trial of (90)Y-DOTA-anti-CEA chimeric T84.66 (cT84.66) radioimmunotherapy in patients with metastatic CEA-producing malignancies. Cancer Biother Radiopharm. 2006;21(2):88–100. doi:10.1089/cbr.2006.21.88.
- Cahan B, Leong L, Wagman L, Yamauchi D, Shibata S, Wilzcynski S, Williams LE, Yazaki P, Colcher D, Frankel P, et al. Phase I/II trial of anticarcinoembryonic antigen radioimmunotherapy, gemcitabine, and hepatic arterial infusion of fluorodeoxyuridine postresection of liver metastasis for colorectal carcinoma. Cancer Biother Radiopharm. 2017;32(7):258–265. doi:10.1089/cbr.2017.2223.
- Klein C, Waldhauer I, Nicolini VG, Freimoser-Grundschober A, Nayak T, Vugts DJ, Dunn C, Bolijn M, Benz J, Stihle M, et al. Cergutuzumab amunaleukin (CEA-IL2v), a CEA-targeted IL-2 variant-based immunocytokine for combination cancer immunotherapy: overcoming limitations of aldesleukin and conventional IL-2-based immunocytokines. Oncoimmunology. 2017;6(3):e1277306. doi:10.1080/2162402X.2016.1277306.
- Clarke P, Mann J, Simpson JF, Rickard-Dickson K, Primus FJ. Mice transgenic for human carcinoembryonic antigen as a model for immunotherapy. Cancer Res. 1998;58:1469–1477.
- Ewens A, Mihich E, Ehrke MJ. Distant metastasis from subcutaneously grown E0771 medullary breast adenocarcinoma. Anticancer Res. 2005;25:3905–3915.
- Esteban JM, Felder B, Ahn C, Simpson JF, Battifora H, Shively JE. Prognostic relevance of carcinoembryonic antigen and estrogen receptor status in breast cancer patients. Cancer. 1994;74(5):1575–1583. doi:10.1002/(ISSN)1097-0142.
- Esteban JM, Paxton R, Mehta P, Battifora H, Shively JE. Sensitivity and specificity of Gold types 1 to 5 anti-carcinoembryonic antigen monoclonal antibodies: immunohistologic characterization in colorectal cancer and normal tissues. Hum Pathol. 1993;24(3):322–328. doi:10.1016/0046-8177(93)90044-H.
- Letourneau S, Krieg C, Pantaleo G, Boyman O. IL-2- and CD25-dependent immunoregulatory mechanisms in the homeostasis of T-cell subsets. J Allergy Clin Immunol. 2009;123(4):758–762. doi:10.1016/j.jaci.2009.02.011.
- Simon S, Labarriere N. PD-1 expression on tumor-specific T cells: friend or foe for immunotherapy? Oncoimmunology. 2017;7(1):e1364828. doi:10.1080/2162402X.2017.1364828.
- Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med. 2010;207(10):2187–2194. doi:10.1084/jem.20100643.
- Ng J, Dai T. Radiation therapy and the abscopal effect: a concept comes of age. Ann Transl Med. 2016;4(6):118. doi:10.21037/atm.
- Willers H, Azzoli CG, Santivasi WL, Xia F. Basic mechanisms of therapeutic resistance to radiation and chemotherapy in lung cancer. Cancer J. 2013;19(3):200–207. doi:10.1097/PPO.0b013e318292e4e3.
- Dewan MZ, Galloway AE, Kawashima N, Dewyngaert JK, Babb JS, Formenti SC, Demaria S. Fractionated but not single-dose radiotherapy induces an immune-mediated abscopal effect when combined with anti-CTLA-4 antibody. Clin Cancer Res. 2009;15(17):5379–5388. doi:10.1158/1078-0432.CCR-09-0265.
- Hu B, Jacobs R, Ghosh N. Checkpoint inhibitors hodgkin lymphoma and non-hodgkin lymphoma. Curr Hematol Malig Rep. 2018;13:543–554. doi:10.1007/s11899-018-0484-4.
- Kenanova V, Olafsen T, Williams LE, Ruel NH, Longmate J, Yazaki PJ, Shively JE, Colcher D, Raubitschek AA, Wu AM, et al. Radioiodinated versus radiometal-labeled anti-carcinoembryonic antigen single-chain Fv-Fc antibody fragments: optimal pharmacokinetics for therapy. Cancer Res. 2007;67(2):718–726. doi:10.1158/0008-5472.CAN-06-0454.
- Gabrilovich DI. Myeloid-derived suppressor cells. Cancer Immunol Res. 2017;5(1):3–8. doi:10.1158/2326-6066.CIR-16-0297.
- Mortara L, Balza E, Bruno A, Poggi A, Orecchia P, Carnemolla B. Anti-cancer therapies employing IL-2 cytokine tumor targeting: contribution of innate, adaptive and immunosuppressive cells in the anti-tumor efficacy. Front Immunol. 2018;9:2905. doi:10.3389/fimmu.2018.02905.
- Weide B, Eigentler TK, Pflugfelder A, Zelba H, Martens A, Pawelec G, Giovannoni L, Ruffini PA, Elia G, Neri D, et al. Intralesional treatment of stage III metastatic melanoma patients with L19-IL2 results in sustained clinical and systemic immunologic responses. Cancer Immunol Res. 2014;2(7):668–678. doi:10.1158/2326-6066.CIR-13-0206.
- Waldmann TA. The biology of interleukin-2 and interleukin-15: implications for cancer therapy and vaccine design. Nat Rev Immunol. 2006;6(8):595–601. doi:10.1038/nri1901.
- Wieckowski S, Hemmerle T, Prince SS, Schlienger BD, Hillinger S, Neri D, Zippelius A. Therapeutic efficacy of the F8-IL2 immunocytokine in a metastatic mouse model of lung adenocarcinoma. Lung Cancer. 2015;88(1):9–15. doi:10.1016/j.lungcan.2015.01.019.
- Kang J, Demaria S, Formenti S. Current clinical trials testing the combination of immunotherapy with radiotherapy. J Immunother Cancer. 2016;4:51. doi:10.1186/s40425-016-0156-7.
- Rekers NH, Olivo Pimentel V, Yaromina A, Lieuwes NG, Biemans R, Zegers CML, Germeraad WTV, Van Limbergen EJ, Neri D, Dubois LJ, et al. The immunocytokine L19-IL2: an interplay between radiotherapy and long-lasting systemic anti-tumour immune responses. Oncoimmunology. 2018;7(4):e1414119. doi:10.1080/2162402X.2017.1414119.
- Demaria S, Formenti SC. Radiation as an immunological adjuvant: current evidence on dose and fractionation. Front Oncol. 2012;2:153. doi:10.3389/fonc.2012.00153.
- Meng G, Tang X, Yang Z, Benesch MGK, Marshall A, Murray D, Hemmings DG, Wuest F, McMullen TPW, Brindley DN, et al. Implications for breast cancer treatment from increased autotaxin production in adipose tissue after radiotherapy. Faseb J. 2017;31(9):4064–4077. doi:10.1096/fj.201700159R.
- Kujawski M, Kortylewski M, Lee H, Herrmann A, Kay H, Yu H. Stat3 mediates myeloid cell-dependent tumor angiogenesis in mice. J Clin Invest. 2008;118(10):3367–3377. doi:10.1172/JCI35213.
- Kioi M, Vogel H, Schultz G, Hoffman RM, Harsh GR, Brown JM. Inhibition of vasculogenesis, but not angiogenesis, prevents the recurrence of glioblastoma after irradiation in mice. J Clin Invest. 2010;120(3):694–705. doi:10.1172/JCI40283.
- Schwager K, Hemmerle T, Aebischer D, Neri D. The immunocytokine L19-IL2 eradicates cancer when used in combination with CTLA-4 blockade or with L19-TNF. J Invest Dermatol. 2013;133(3):751–758. doi:10.1038/jid.2012.376.
- Laemmli U. Cleavage of structural protein during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi:10.1038/227680a0.
- Priceman SJ, Shen S, Wang L, Deng J, Yue C, Kujawski M, Yu H. S1PR1 is crucial for accumulation of regulatory T cells in tumors via STAT3. Cell Rep. 2014;6(6):992–999. doi:10.1016/j.celrep.2014.02.016.
- van Hoof SJ, Granton PV, Verhaegen F. Development and validation of a treatment planning system for small animal radiotherapy: smART-Plan. Radiother Oncol. 2013;109(3):361–366. doi:10.1016/j.radonc.2013.10.003.
- Faddegon BA, Kawrakow I, Kubyshin Y, Perl J, Sempau J, Urban L. The accuracy of EGSnrc, Geant4 and PENELOPE Monte Carlo systems for the simulation of electron scatter in external beam radiotherapy. Phys Med Biol. 2009;54(20):6151–6163. doi:10.1088/0031-9155/54/20/008.