
ABSTRACT
Despite considerable progress, the treatment of acute leukemia continues to be a challenge for a significant majority of patients. Using a well-characterized preclinical mouse model of acute promyelocytic leukemia (APL), we evaluated here the antileukemic efficacy of RT53, an anticancer peptide with potential immunological properties. Our results indicate that RT53 possesses a direct antileukemic effect, even at a late stage. We also demonstrate that a single injection of a vaccine consisting of leukemic blasts exposed to RT53, which induces the hallmarks of immunogenic cell death, was highly effective in preventing leukemia development in both prophylactic and therapeutic settings. The vaccine comprising RT53-treated APL cells generated long-term antileukemic protection and depletion experiments indicated that CD4 + T cells were of crucial importance for vaccine efficacy. Combined, our results provide the rationale for the exploration of RT53-based therapies for the treatment of acute leukemia.
Introduction
A major strategy in anticancer efforts is to target oncogenic signaling networks that are required for tumor cell growth and survival. However, intrinsic or acquired resistance often limits the efficacy of these targeted therapies.Citation1,Citation2 In parallel to the development of molecular-targeted agents, cancer immunotherapies are changing the therapeutic landscape for cancer patients by providing a clinically beneficial alternative to conventional treatments.Citation3,Citation4 Despite the improved molecular characterization of malignancies and development of target therapies, acute leukemia is not curable and a minority of patients survive more than 10 years after diagnosis. Among leukemia subtypes, prognosis is particularly poor in adult acute myeloid leukemia (AML).Citation5 Therefore, new agents and novel regimens are clearly needed to improve outcomes for AML patients. We previously reported that RT53, a cell-permeable peptide derived from the survival protein AAC-11, selectively killed cancer cells in vitro and prevented tumor growth in melanoma mouse models.Citation6,Citation7 RT53 mechanism of action includes the inhibition of AAC-11 anti-apoptotic properties, through disruption of protein–protein interactions between AAC-11 and protein partners such as Acinus, as well as a non-regulated membranolysis of the cancer cells plasma membrane.Citation6–Citation8 Interestingly, RT53 treatment of melanoma cells induces all the hallmarks of immunogenic cell death and B16F10 mouse cells treated by RT53 were able to mediate protective effects in a tumor vaccination model.Citation6,Citation8 In this study, we evaluated the therapeutic potential of RT53 as a treatment for acute leukemia. We found that RT53 possesses a direct antileukemic effect, both in vitro and in a preclinical acute promyelocytic leukemia (APL) mouse model. A vaccine consisting of RT53-exposed leukemic blast cells was highly effective at preventing leukemia development both prophylactically and therapeutically through the induction of CD4 + T cell-dependent long-term response. These results make RT53-based antileukemic therapies attractive for clinical development.
Material and methods
Peptides
Peptides were synthesized by Proteogenix (Strasbourg, France) and were >95% pure as verified by HPLC and mass spectrographic analysis. The peptide sequence of RT53 is the following: RQIKIWFQNRRMKWKKAKLNAEKLKDFKIRLQYFARGLQVYIRQLRLALQGKT
The penetratin sequence is underlined.
Cell lines and chemicals
NB4 (purchased from ATCC), UF-1 (provided by Dr. Y. Ikeda, Tokyo, Japan), HUT-78 (provided by Dr. A. Marie-Cardine, INSERM U976, Paris, France) and B16F10 (provided by Dr. M. Dutreix, CNRS UMR3347, INSERM 1021, Paris, France) were used for the experiments. Cells were grown in RPMI 1640 medium supplemented with 10% fetal calf serum, L-glutamine (2 mM), 1 mM Hepes and 200 ug/ml penicillin/streptomycin antibiotics (Gibco). All cells were maintained at 37°C in humidified 5% CO2 atmosphere. All chemicals were purchased from Sigma.
Lactate dehydrogenase, ATP and HMGB1 release assays
Release of lactate dehydrogenase (LDH) and ATP in the culture medium was assessed with the CytoTox 96 Non-Radioactive Cytotoxicity Assay and Enliten ATP Assay, respectively (Promega, Madison, WI, USA). HMGB1 release in the culture medium was assessed with the HMGB1 ELISA kit (IBL International, Hamburg, Germany).
Electron microscopy
Samples were fixed in 3% glutaraldehyde in phosphate buffer, pH 7.4 for 1 h, washed, post-fixed with 1% osmium tetroxide in 0.1 M phosphate buffer and then gradually dehydrated in 70%, 90% and 100% ethanol. After 10 min in a 1:2 mixture of epoxy propane and epoxy resin and 10 min in epon, samples were embedded in epoxy resin and polymerized at 60°C for 24 h. After polymerization, ultrathin sections of 90 nm were cut with an ultra-microtome (Reichert ultracut S), stained with uranyl acetate and Reynold’s lead and observed with a transmission electron microscope (JEOL 1011). Acquisition was performed with a Gatan Orius 1000 CCD camera.
Apoptosis assays
Leukemic blasts apoptosis assessment was performed using fluorescein isothiocyanate (FITC) Annexin V Apoptosis Detection Kit I (BD Biosciences, Waltham, MA) and propidium iodide (PI) as per the manufacturer’s instructions, followed by flow cytometry analysis with a CytoFLEX Flow Cytometer (Beckman and Coulter). Viable cells were defined as annexin V/PI.
Autophagy assessment
The formation of autophagic vesicles was assessed using the Cyto-ID Autophagy Detection Kit (EnzoLife Sciences, Antwerpen, Belgium) using a 488-nm excitable fluorescent probe that selectively stains autophagic compartments, according to manufacturer’s protocol. Serum starvation (1 h) was used as a positive control for autophagic flux. Flow cytometry analysis was performed with a CytoFLEX Flow Cytometer (Beckman and Coulter).
Determination of surface-exposed CRT
CRT exposure was assessed by surface immunostaining and flow cytometry. In brief, APL blast cells (106 cells per mL) in RPMI 1640 medium supplemented with 10% fetal calf serum, L-glutamine (2 mM), 1 mM Hepes and 200 ug/ml penicillin/streptomycin antibiotics plated in 24 well plates were treated for 6 h with 5 µM of RT53 peptide. Cells were washed with PBS (Phosphate-Buffered Saline), harvested and plated in 96-well round-bottomed microtiter plates and incubated in blocking solution for 45 min (Blocking Solution Image-iT® Fixation/Permeabilization Kit Cat. #R37602, Thermo Fischer Scientific). After 1X wash with PBS, cells were stained with anti-calreticulin primary antibody (Calreticulin (D3E6) XP® Rabbit mAb #12238, Cell Signaling). Goat anti-rabbit Alexa Fluor 488 was used as a secondary antibody after another PBS wash (Alexa Fluor® 488 goat anti-rabbit IgG secondary antibody Cat. #A11034, Thermo Fischer Scientific). Cells were then analyzed with a CytoFLEX Flow Cytometer (Beckman and Coulter) by gating on propidium iodide-negative cells for the exclusion of dead cells.
Ethics statement
This study has been carried out in accordance with the EC Directive 86/609/EEC for animal experiments and was approved by the Committee for Experimental Animal Studies of the University of Paris 7 Institute Board Ethics (Protocol Number: 2303.01). Animals were housed and bred at our animal facility (Institut de Recherche Saint-Louis, Saint Louis Hospital, Paris, France) in vented animal cabinets under controlled temperature (22°C) and 12 h light-dark cycle under pathogen-free conditions and were allowed food and water ad libitum.
Preclinical acute promyelocytic leukemia-transplantable mice model
APL blasts (provided by Drs. M. Bishop and S. Kogan, UCSF, USA) origin from the spleen of mice bearing the human PML-RARA cDNA construct driven by a myeloid lineage specific promoter (hMRP8) in the FVB/N inbred strain of mice. For amplification, cells (1 × 105 or 1 × 106) were suspended in PBS and transplanted by intravenous (i.v) tail injection (200 uL) into female syngeneic recipient mice (5–6 weeks old). The establishment of leukemia was assessed by a decrease in blood platelet counts approximately 3 weeks after graft. Spleen cells from a primary recipient were collected, washed, re-suspended in PBS and injected (104 cells/mouse; 200 uL) into the tail veins of male FVB/N mice (7–8 weeks old) for experiments. For direct treatment experiments, mice were treated daily or every other day (i.p) with normal saline or RT53 at 2.4 mg/kg in normal saline starting from day 10 or day 20 for a total of 7 injections.
RT53-treated APL blast vaccination assay
Two millions live cells from primary recipients’ spleens or the indicated cells were washed in PBS and resuspended in 200 µl of serum-free RPMI medium. The cells were then exposed to 30 µM RT53 for 3 h for cell death induction and the whole suspension of RT53-treated cells was injected subcutaneously (2 × 106 cells) into the left flanks of FVB/N syngeneic mice. For leukemia induction, the mice were injected i.v. with 1 × 104 blast cells from primary recipients’ spleens at the indicated time.
T cell depletion
Mice were depleted of either CD4+, CD8+ or both T cell populations by bi-weekly i.p. injection of 0.2 mg of ascites fluids containing an anti-CD4 or -CD8 antibody starting 2 weeks before experiments. Injections were then performed 2 times per week during the study period. Blood was collected by submandibular bleeding. PBMC were labeled with a mix of anti-CD3Ɛ-APC (MACS), anti-CD4-PE (MACS) and anti-CD8-APC-cy7 (BD Biosciences) (2.5 µl, 30 min, 4°C). Red blood cells were then lysed in ACK buffer for 7 min at RT. The efficacy of depletion was monitored using Canto II (BD Biosciences) cytometer and data analyzed with FlowJo software.
Results and discussion
RT53 possess direct antileukemic properties
To test the antileukemic properties of RT53, we first exposed human all-trans retinoic acid ATRA-sensitive (NB4) and ATRA-resistant (UF-1) acute promyelocytic leukemia (APL) cells as well as mouse APL spleen blast cells derived from hMRP8-PML-RARA transgenic miceCitation9 to increasing concentrations of RT53. As shown in ), RT53 decreased viability of all the tested cells through the rapid loss of plasma membrane integrity, as detected by the release of the intracellular enzyme lactate dehydrogenase (LDH). Importantly, no cell death was detected in spleen cells from healthy mice even at the highest concentration tested, indicating that RT53 exhibits selective cytotoxicity toward leukemic cells, but not normal cells. The observed LDH release upon RT53 treatment indicates that leukemic cells death is associated with loss of outer cell membrane integrity and cytoplasmic leakage, which are characteristic features of necrosis. In line with this hypothesis, ultrastructural analysis of APL spleen cells exposed to RT53 revealed an obvious necrotic morphology of the leukemic blasts, with loss of plasma membrane integrity and cytoplasmic swelling without morphological signs of nuclear apoptosis ()). No changes in cellular integrity or ruptured cells could be detected in the normal spleen cells, confirming that RT53 exhibits high specificity toward leukemic cells ()). To further define the RT53 mechanism of action and response kinetics, we assessed cell death induced by the peptide using annexin V and propidium iodide (PI) staining over time of treatment. This double labeling allows to distinguish early apoptotic (annexin V positive and PI negative) and late apoptotic/necrotic (both annexin V and PI positive) cells. As shown in ), RT53 exposure of APL spleen cells induced concomitant annexin V and PI positivity, even at early points in the treatment. These data, together with the electron microscopy analysis, indicate that RT53 does not induce the characteristic morphological and biochemical features of apoptosis, but rather mediates a necrotic form of cell death. In line with this hypothesis, inhibition of caspases activity with the pan-caspase inhibitor zVAD-fmk, while efficiently interfering with cell death induced by staurosporine, failed to prevent cell killing mediated by RT53 ()), confirming that RT53 cytotoxic effects do not involve apoptosis. We next investigated whether RT53 could trigger necroptosis, an alternative, caspase-independent form of cell death that is regulated by receptor-interacting protein kinases (RIPK) 1 and 3.Citation10 As shown in ), the RIPK1 inhibitor necrostatin-1 was unable to interfere with RT53-mediated APL cells death, although it had a marked cytoprotective effect on TNF-α–induced necroptosis ()). However, IM-54, a potent and selective inhibitor of unregulated necrosis induced by oxidative stress,Citation11 efficiently prevented cell death induced by RT53 ()), indicating that RT53 treatment triggers necrosis of the leukemic cells through the induction of oxidative stress. Of note, RT53 treatment did not promote autophagy, as peptide exposure failed to upregulate formation of autophagic compartments ()), and, consistently, the autophagy inhibitor 3-Methyladenine had no effect on RT53 cytotoxicity toward APL cells ()). Altogether, these results indicate that RT53 kills the APL blasts via a rapid, unregulated necrosis mechanism induced by oxidative stress, without relying on the major regulated cell death pathways.
Figure 1. RT53 treatment increases APL mice survival. (a) The indicated cells were left untreated or exposed to increasing concentrations of RT53 for 20 h. Cell death induced by peptide treatment was measured by lactate dehydrogenase (LDH) release. Data are means ± s.e.m. (n = 3). (b) Ultrastructural analysis of RT53-treated spleen cells. Spleen cells from healthy or APL mice were left either untreated or exposed to 10 µM of RT53 for 1 h. Cells were then analyzed by transmission electron microscopy following osmium tetroxide staining. (c) APL spleen cells were exposed to 5 µM RT53 for the indicated periods of time and phosphatidylserine exposure (annexin V labeling) and cell membrane permeabilization (propidium iodide (PI) labeling) were analyzed by flow cytometry. The percentages of viable cells (annexin V−/PI−), early apoptotic cells (annexin V+/PI−) and late apoptotic/necrotic cells (annexin V+/PI+) are represented (average of two independent experiments). (d) APL spleen cells were exposed to 5 µM of RT53 in the presence or absence of 50 μM zVAD-fmk for 3 h. Cell death was measured by flow cytometry as in (c). Staurosporine (5 µM) was used as a control. Average of two independent experiments. (e) APL spleen cells were exposed to 5 µM of RT53 in the presence or absence of 50 μM Necrostatin-1 (Nec-1) for 3 h and cell membrane permeabilization (PI labeling) was analyzed by flow cytometry. [TNF-α (30 ng/ml) + zVAD-fmk (40 μM) + 10 µg/mL cycloheximide (CHX)] treatment was used as a control. (f) APL spleen cells were exposed to 5 µM of RT53 in the presence or absence of 10 μM of IM-54 for 3 h. Cell death was measured as in (e). H2O2 (100 µM) treatment was used as a control. Average of two independent experiments. (g) APL spleen cells were exposed to 5 µM RT53 for 1 h in the presence or absence of 10 µM 3-Methyladenine (3-MA) and autophagic activity was determined by flow cytometry analysis using the Cyto-ID autophagy detection reagent. Serum starvation (1 h) was used as a control. Data are expressed as the mean fluorescence intensity of Cyto-ID (average of two independent experiments). (h) APL spleen cells were exposed to 5 µM of RT53 for 1 h in the presence or absence of 10 µM 3-Methyladenine (3-MA). Cell death was measured as in (e). Average of two independent experiments. (i) 104 APL blasts were inoculated intravenously (i.v.) into FVB/N mice at day 0. Mice were then either left untreated (n = 6), treated with ATRA (5 mg, subcutaneous implantation of 21-day release pellets, n = 6) at day 6 or injected intraperitoneally (i.p.) with RT53 (2.4 mg/kg in normal saline) at day 10 every day for a total of seven doses (n = 6). Survival curves were analyzed with the Mantel–Cox test. (j) APL mice obtained as in (i) were either left untreated (n = 6), injected i.p. with RT53 (2.4 mg/kg in normal saline) at day 10 every other day for a total of seven doses (D10 Q2D schedule, n = 6) or at day 20 every day for a total of seven doses (D20 schedule, n = 4). Survival curves were analyzed with the Mantel–Cox test
![Figure 1. RT53 treatment increases APL mice survival. (a) The indicated cells were left untreated or exposed to increasing concentrations of RT53 for 20 h. Cell death induced by peptide treatment was measured by lactate dehydrogenase (LDH) release. Data are means ± s.e.m. (n = 3). (b) Ultrastructural analysis of RT53-treated spleen cells. Spleen cells from healthy or APL mice were left either untreated or exposed to 10 µM of RT53 for 1 h. Cells were then analyzed by transmission electron microscopy following osmium tetroxide staining. (c) APL spleen cells were exposed to 5 µM RT53 for the indicated periods of time and phosphatidylserine exposure (annexin V labeling) and cell membrane permeabilization (propidium iodide (PI) labeling) were analyzed by flow cytometry. The percentages of viable cells (annexin V−/PI−), early apoptotic cells (annexin V+/PI−) and late apoptotic/necrotic cells (annexin V+/PI+) are represented (average of two independent experiments). (d) APL spleen cells were exposed to 5 µM of RT53 in the presence or absence of 50 μM zVAD-fmk for 3 h. Cell death was measured by flow cytometry as in (c). Staurosporine (5 µM) was used as a control. Average of two independent experiments. (e) APL spleen cells were exposed to 5 µM of RT53 in the presence or absence of 50 μM Necrostatin-1 (Nec-1) for 3 h and cell membrane permeabilization (PI labeling) was analyzed by flow cytometry. [TNF-α (30 ng/ml) + zVAD-fmk (40 μM) + 10 µg/mL cycloheximide (CHX)] treatment was used as a control. (f) APL spleen cells were exposed to 5 µM of RT53 in the presence or absence of 10 μM of IM-54 for 3 h. Cell death was measured as in (e). H2O2 (100 µM) treatment was used as a control. Average of two independent experiments. (g) APL spleen cells were exposed to 5 µM RT53 for 1 h in the presence or absence of 10 µM 3-Methyladenine (3-MA) and autophagic activity was determined by flow cytometry analysis using the Cyto-ID autophagy detection reagent. Serum starvation (1 h) was used as a control. Data are expressed as the mean fluorescence intensity of Cyto-ID (average of two independent experiments). (h) APL spleen cells were exposed to 5 µM of RT53 for 1 h in the presence or absence of 10 µM 3-Methyladenine (3-MA). Cell death was measured as in (e). Average of two independent experiments. (i) 104 APL blasts were inoculated intravenously (i.v.) into FVB/N mice at day 0. Mice were then either left untreated (n = 6), treated with ATRA (5 mg, subcutaneous implantation of 21-day release pellets, n = 6) at day 6 or injected intraperitoneally (i.p.) with RT53 (2.4 mg/kg in normal saline) at day 10 every day for a total of seven doses (n = 6). Survival curves were analyzed with the Mantel–Cox test. (j) APL mice obtained as in (i) were either left untreated (n = 6), injected i.p. with RT53 (2.4 mg/kg in normal saline) at day 10 every other day for a total of seven doses (D10 Q2D schedule, n = 6) or at day 20 every day for a total of seven doses (D20 schedule, n = 4). Survival curves were analyzed with the Mantel–Cox test](/cms/asset/318742b2-abae-47e3-838a-6ab70a8f8c3e/koni_a_1728871_f0001a_b.gif)
Figure 1. (continued)
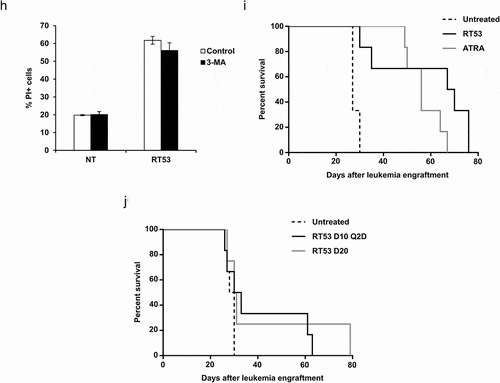
To explore the in vivo therapeutic potential of RT53 as a treatment of acute leukemia, we used a well-characterized preclinical APL model bearing the human PML-RARA oncogene which mimics human APL, both in its characteristics and its response to conventional therapeutic drugs such as ATRA and arsenic trioxide.Citation9,Citation12 In this model, 100% of the syngeneic mice (FVB/N) transplanted with 104 primary recipients’ spleen blasts developed an APL and succumbed within 30 days (). APL mice treated with RT53 (2.4 mg/kg) for 7 days starting on day 10 after leukemia engraftment had a significantly extended survival compared to control mice (P < .0001). The survival of APL mice treated with RT53 was significantly (P < .0001) superior to that of mice treated by ATRA (). Similar survival advantage was obtained when RT53 was administered every other day for a total of seven administrations (D10 Q2D schedule, ). RT53 treatment starting on day 20 after leukemia engraftment, when leukemia is fully establishedCitation13 as shown by standardized minimal residual disease (MRD) monitoring (high level of PML-RARα transcripts in PBL, bone marrow and spleen), also prolonged the survival of leukemic mice (D20 schedule, ). To our knowledge, no therapeutic approach has demonstrated such a pronounced effect on the survival of mice with comparable advanced disease stage in this preclinical model. No organ toxicity (macroscopic or microscopic) was noted with either treatment schedule (data not shown). Therefore, these results indicate that RT53 possesses robust antileukemic activity both in vitro and in vivo.
A vaccine comprising RT53-treated APL cells induces long-term survival
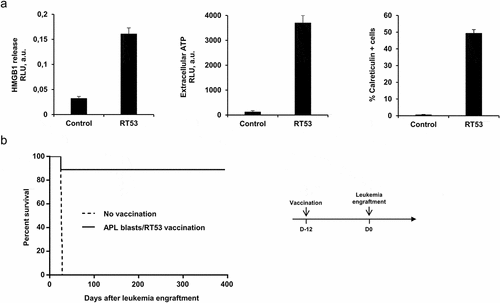
Anticancer chemotherapies are particularly effective when they induce immunogenic cell death (ICD), thus eliciting an antitumor immune response.Citation14 We, therefore, investigated whether RT53 treatment of APL spleen cells would be able to induce the key known biomarkers of ICD, which include the endoplasmic reticulum (ER) chaperone calreticulin (CRT) surface exposure and the release of the chromatin protein high mobility group box1 protein (HMGB1) as well as ATP.Citation15 As showed in , RT53 treatment triggered the release of both HMGB1 and ATP in the culture medium, detected by ELISA and ATP-bioluminescence assays, respectively, as well as surface exposure of CRT, detectable by FACS analysis, indicating that RT53 can induce all tested characteristics of ICD. Similar data were obtained with the human APL cells NB4 (not shown). To further investigate the capacity of RT53 to induce an antileukemic response, we took advantage of the APL preclinical model, which is based on immunocompetent FVB/N mice, to develop a prophylactic tumor vaccination model (). Interestingly, 7 out of 8 mice vaccinated subcutaneously with RT53-exposed APL spleen blast cells did not develop the disease after APL engraftment (), indicating the prophylactic effect of RT53-exposed APL cells. Eight months after APL engraftment, surviving animals were found disease-free by MRD monitoring. These data indicate that a simple vaccine constituted by RT53-treated APL cells triggered a very effective prophylaxis, protecting against the development of leukemia.
Figure 2. Inhibition of APL progression by prophylactic vaccination with RT53-treated APL blasts. (a) APL blasts in basal RPMI medium were left untreated or treated with either 5 μM of RT53 for 6 h (CRT exposure analysis) or 10 µM of RT53 for 1 h (HMGB1 and ATP release analysis). Extracellular HMGB1 (left) and ATP (middle) were then measured in the culture supernatant by ELISA and ATP-bioluminescence assays, respectively, and surface exposure of CRT (right) detected by FACS analysis. (b) APL blasts were exposed to 30 µM RT53 in basal RPMI medium for 3 h for cell death induction and the whole suspension was injected subcutaneously (2 × 106 cells) into the left flanks of FVB/N mice. Twelve days later, the vaccinated (n = 8) or control mice (n = 7) were injected i.v. with live 104 APL blasts. Survival curves were analyzed with the Mantel–Cox test. The schematic protocol used is illustrated (right)
The prophylactic effect generated by the RT53-treated APL cells vaccine is tumor type-specific and long-lasting
To demonstrate the specificity of RT53-exposed APL cells prophylactic effect, mice were vaccinated subcutaneously with various human (NB4, HUT78) or mouse (B16F10) cancerous cells treated with RT53. As shown in , none of the tumor cells generated protection against leukemia development, indicating that the protection induced by RT53-treated APL cells is tumor specific. Moreover, the absence of protection observed following vaccination with RT53-treated NB4 cells suggests that immune clearance of leukemic cells in RT53-exposed APL cells vaccinated animals does not rely on the recognition of the unique PML-RARα fusion protein, which is also expressed in NB4 cells. Finally, vaccination with RT53-treated spleen cells from healthy FVB/N mice generated no protection (), indicating that the prophylactic effect against APL was exclusively triggered by the RT53-treated leukemic APL cells.
Figure 3. Tumor specificity and long-lasting effect of prophylactic vaccination with RT53-treated APL blasts. (a) FVB/N mice were vaccinated with RT53-treated APL blasts (n = 10), RT53-treated spleen cells from healthy mice (n = 5), or the indicated RT53-treated cells (n = 5 per group) using the same protocol as in . Twelve days later, the vaccinated or control mice were injected i.v. with live 104 APL blasts. Survival curves were analyzed with the Mantel–Cox test. (b) Surviving mice from were challenged with 104 live APL spleen blasts 107 days (group 1, n = 5) or 226 days (group 2, n = 4) after initial APL engraftment. Survival curves were analyzed with the Mantel–Cox test. The schematic protocol used is illustrated (lower panel)
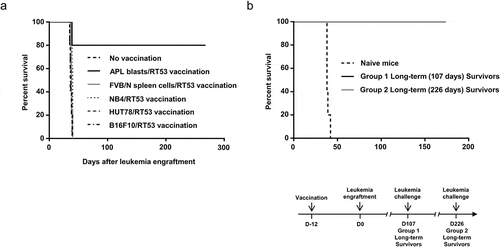
We next determined whether surviving mice developed long-lasting antileukemic protection. At 107 or 226 days after initial leukemia engraftment, survivors from ) or control mice were challenged with 104 live APL spleen blast cells, in the absence of any further therapy. Strikingly, all vaccinated animals that received vaccination were protected from APL cells challenge, whereas all control mice succumbed to leukemia within 40 days (). These results indicate that single vaccination with RT53-exposed APL cells induces eradication of a rapidly fatal tumor burden and evokes effective, long-lasting prophylaxis capable of preventing leukemia. Moreover, when we inoculated 107 spleen cells originating from long-term survivors vaccinated with RT53-treated APL blasts into secondary recipients, none of the injected mice developed APL (followed up >200 days; not shown), suggesting that this vaccine leads to eradication of APL-initiating cells. Injection of 104 spleen cells from unvaccinated APL mice was sufficient to establish APL and all recipients died (not shown).
CD4 + T cells are critical for the induction of prolonged survival induced by the RT53-treated APL cells vaccine
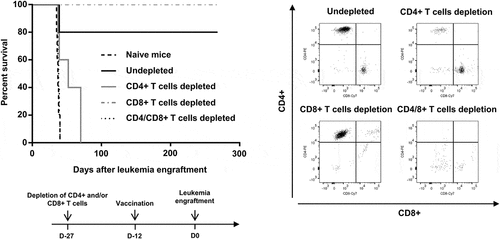
To determine the cells involved in the prophylactic effect of RT53-treated APL cells vaccination, mice were depleted of CD4+ T, CD8+ T or both T cell populations using cell type-specific antibodies. Depletion of CD4+ T cells notably reduced vaccine-induced protection, whereas depletion of CD8+ T cells had no effect on vaccine efficacy (), demonstrating the essential role of CD4+ T cells in the induction of effective antileukemic immunity. As CD4+ T cells are crucial in the establishment of immune memory,Citation16 our results might explain the protective effect observed in ). However, complete loss of protection was observed in mice that were depleted of both T-cell populations (), indicating that the antileukemic response generated by RT53-treated APL cells vaccination required the presence of both CD4+ and CD8+ T cells. Although the precise mechanisms involved in vaccination-induced protection remain to be defined, the observation that leukemia development is effectively contained in mice depleted for CD8+ T cells is suggestive of the induction of innate immune cells, such as macrophages and natural killer cells, which can be activated by CD4+ T cells.Citation17 A cytotoxic activity of CD4+ T cells toward the leukemic cells is also possible, as witnessed in different experimental tumor settings.Citation18–Citation20 Of note, depletion of either CD4+, CD8+ or both T cell populations in long-term survivors from ) did not result in APL (Supplementary Figure 1), indicating that CD4+ and/or CD8 + T cells were not necessary for the maintenance of the antileukemic effects of RT53-treated APL cells vaccination and suggesting strongly that this vaccination leads to a cure of the mice.
Figure 4. Requirement of CD4+ and CD8 + T cells for prolonged survival induced by vaccination with RT53-treated APL blasts. FVB/N mice were depleted of either CD4+, CD8+ or both T cell populations by bi-weekly i.p. injection of 0.2 mg of T cell type-specific monoclonal antibodies starting 2 weeks before experiments. Injections were then performed 2 times per week during the study period. The efficacy of depletion was monitored by flow cytometric analysis (right panel). Depleted (n = 5 per group) or naive mice (n = 10) were then vaccinated with RT53-treated APL blasts and injected i.v. with live 104 APL blasts (left, upper panel). Survival curves were analyzed with the Mantel–Cox test. The schematic protocol used is illustrated (left, lower panel)
The RT53-treated APL cells vaccine is effective in mice with well-established leukemia
Having shown that RT53-treated APL cells vaccination can elicit an efficient prophylactic antileukemic effect, we next tested the therapeutic benefit of the vaccine in mice with well-established leukemia. For that purpose, mice received the RT53-treated APL cells 3 (rising disease) or 10 (well-established disease)Citation13 days after APL cell engraftment and the onset of the disease was compared to that of non-vaccinated controls. Very interestingly, 100% of the vaccinated mice were protected from leukemia development and remained disease-free through 80 days of observation, irrespective of the immunization schedule (, lower panel). These data indicate that the therapeutic administration of the RT53-treated APL cells vaccine resulted in the eradication of leukemic cells in all the tested mice, even when vaccination was delayed until 10 days after tumor inoculation, indicating the effectiveness of this approach.
Figure 5. Therapeutic efficacy of RT53-treated APL blast vaccination in well-established leukemia. 104 APL blasts were inoculated i.v. into FVB/N mice at day 0. Mice were then vaccinated with RT53-treated APL blast 3 or 10 days after leukemia engraftment (n = 5 per group). Survival curves were analyzed with the Mantel–Cox test. The schematic protocol used is illustrated (right panel)

Concluding remarks
Acute leukemia such as AML is known to generate an immunosuppressive environment that circumvent immunosurveillance mechanisms.Citation21–Citation27 Therefore, immunotherapy has great potential as a more effective and less toxic alternative to current treatments for leukemia patients. We report here a simple, highly efficient and practical strategy for immunotherapy of leukemia. By using a well-established, aggressive APL model, we showed that single vaccination with a vaccine comprising whole leukemic cells exposed to RT53, shown here to induce immunogenic cell death, protected against the development of leukemia in vivo both prophylactically and therapeutically. Cure of the tumor was observed and the vaccinated animals were protected against subsequent leukemia challenge in the absence of any further boosts, through the generation of long-lasting, protective tumor-specific responses involving CD4 + T cells. Such vaccine-based approach is practical, as it does not require the knowledge of specific tumor antigens, is not limited by the HLA phenotype and is safe because the antitumor effect is obtained without the use of potentially toxic immunostimulatory adjuvants in the vaccine. Because leukemic blasts can be easily obtained from the blood or bone marrow of patients at diagnosis, yielding sufficient material for clinical use, RT53-treated leukemic cells vaccines may be a workable and effective strategy for immunotherapy of leukemia. Further, we demonstrated the single-agent efficacy of RT53 for the treatment of established leukemia, even at late stages, suggesting that RT53 might constitute a therapeutic option for patients who experienced multiple relapses because of resistance to approved therapies. Along with the absence of toxicity, these preclinical efficacy evidences support further development and clinical testing of RT53-based therapies in the context of acute leukemia and maybe other hematological malignancies.
Conflict of interest
The authors declare that they have no conflict of interest.
Supplemental Material
Download (){kind=link}
Acknowledgments
We thank the coworkers of the Animal Experimental Facilities of the Institut de Recherche Saint-Louis as well as the staff of the Institut Cochin imaging platform. We are grateful to Michael Bishop and Scott Kogan (University of California San Francisco (UCSF)) for the APL cells. This study was supported in part by grants from Association Laurette Fugain to T.B., H.D. and J-L.P. and La Ligue Nationale contre le Cancer (Grant No SR17/75-38) and Cancéropole Ile de France to J-L.P.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
References
- Ellis LM, Hicklin DJ. Resistance to targeted therapies: refining anticancer therapy in the era of molecular oncology. Clin Cancer Res. 2009;15:7471–9. doi:10.1158/1078-0432.CCR-09-1070.
- Neel DS, Bivona TG. Resistance is futile: overcoming resistance to targeted therapies in lung adenocarcinoma. NPJ Precis Oncol. 2017;1(3).
- Martin SD, Coukos G, Holt RA, Nelson BH. Targeting the undruggable: immunotherapy meets personalized oncology in the genomic era. Ann Oncol. 2015;26:2367–2374. doi:10.1093/annonc/mdv382.
- Thallinger C, Fureder T, Preusser M, Heller G, Mullauer L, Holler C, Prosch H, Frank N, Swierzewski R, Berger W, et al. Review of cancer treatment with immune checkpoint inhibitors: current concepts, expectations, limitations and pitfalls. Wien Klin Wochenschr. 2018;130:85–91. doi:10.1007/s00508-017-1285-9.
- De Kouchkovsky I, Abdul-Hay M. Acute myeloid leukemia: a comprehensive review and 2016 update. Blood Cancer J. 2016;6:e441. doi:10.1038/bcj.2016.50.
- Jagot-Lacoussiere L, Kotula E, Villoutreix BO, Bruzzoni-Giovanelli H, Poyet JL. A cell-penetrating peptide targeting AAC-11 specifically induces cancer cells death. Cancer Res. 2016;76:5479–5490. doi:10.1158/0008-5472.CAN-16-0302.
- Rigou P, Piddubnyak V, Faye A, Rain JC, Michel L, Calvo F, Poyet JL. The antiapoptotic protein AAC-11 interacts with and regulates Acinus-mediated DNA fragmentation. Embo J. 2009;28:1576–1588. doi:10.1038/emboj.2009.106.
- Pasquereau-Kotula E, Habault J, Kroemer G, Poyet JL. The anticancer peptide RT53 induces immunogenic cell death. PLoS One. 2018;13:e0201220. doi:10.1371/journal.pone.0201220.
- Brown D, Kogan S, Lagasse E, Weissman I, Alcalay M, Pelicci PG, Atwater S, Bishop JM. A PMLRARalpha transgene initiates murine acute promyelocytic leukemia. Proc Natl Acad Sci U S A. 1997;94:2551–2556. doi:10.1073/pnas.94.6.2551.
- Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature. 2015;517:311–320. doi:10.1038/nature14191.
- Dodo K, Katoh M, Shimizu T, Takahashi M, Sodeoka M. Inhibition of hydrogen peroxide-induced necrotic cell death with 3-amino-2-indolylmaleimide derivatives. Bioorg Med Chem Lett. 2005;15:3114–3118. doi:10.1016/j.bmcl.2005.04.016.
- Lallemand-Breitenbach V, Guillemin MC, Janin A, Daniel MT, Degos L, Kogan SC, Bishop JM, de The H. Retinoic acid and arsenic synergize to eradicate leukemic cells in a mouse model of acute promyelocytic leukemia. J Exp Med. 1999;189:1043–1052. doi:10.1084/jem.189.7.1043.
- Pokorna K, Le Pogam C, Chopin M, Balitrand N, Reboul M, Cassinat B, Chomienne C, Padua RA, Pla M. Tracking the extramedullary PML-RARalpha-positive cell reservoirs in a preclinical model: biomarker of long-term drug efficacy. Mol Cell Probes. 2013;27:1–5. doi:10.1016/j.mcp.2012.08.001.
- Casares N, Pequignot MO, Tesniere A, Ghiringhelli F, Roux S, Chaput N, Schmitt E, Hamai A, Hervas-Stubbs S, Obeid M, et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J Exp Med. 2005;202:1691–1701. doi:10.1084/jem.20050915.
- Kepp O, Senovilla L, Vitale I, Vacchelli E, Adjemian S, Agostinis P, Apetoh L, Aranda F, Barnaba V, Bloy N, et al. Consensus guidelines for the detection of immunogenic cell death. Oncoimmunol. 2014;3:e955691.
- Chen SA, Tsai MH, Wu FT, Hsiang A, Chen YL, Lei HY, Tzai TS, Leung HW, Jin YT, Hsieh CL, et al. Induction of antitumor immunity with combination of HER2/neu DNA vaccine and interleukin 2 gene-modified tumor vaccine. Clin Cancer Res. 2000;6:4381–4388.
- Luckheeram RV, Zhou R, Verma AD, Xia B. CD4(+)T cells: differentiation and functions. Clin Dev Immunol. 2012;2012:925135. doi:10.1155/2012/925135.
- Quezada SA, Simpson TR, Peggs KS, Merghoub T, Vider J, Fan X, Blasberg R, Yagita H, Muranski P, Antony PA, et al. Tumor-reactive CD4(+) T cells develop cytotoxic activity and eradicate large established melanoma after transfer into lymphopenic hosts. J Exp Med. 2010;207:637–650. doi:10.1084/jem.20091918.
- Fu J, Zhang Z, Zhou L, Qi Z, Xing S, Lv J, Shi J, Fu B, Liu Z, Zhang JY, et al. Impairment of CD4+ cytotoxic T cells predicts poor survival and high recurrence rates in patients with hepatocellular carcinoma. Hepatol. 2013;58:139–149. doi:10.1002/hep.26054.
- Sharma RK, Yolcu ES, Srivastava AK, Shirwan H. CD4+ T cells play a critical role in the generation of primary and memory antitumor immune responses elicited by SA-4-1BBL and TAA-based vaccines in mouse tumor models. PLoS One. 2013;8:e73145. doi:10.1371/journal.pone.0073145.
- Barrett AJ, Le Blanc K. Immunotherapy prospects for acute myeloid leukaemia. Clin Exp Immunol. 2010;161:223–232. doi:10.1111/j.1365-2249.2010.04197.x.
- Gibbins JD, Ancelet LR, Weinkove R, Compton BJ, Painter GF, Petersen TR, Hermans IF. An autologous leukemia cell vaccine prevents murine acute leukemia relapse after cytarabine treatment. Blood. 2014;124:2953–2963. doi:10.1182/blood-2014-04-568956.
- Ustun C, Miller JS, Munn DH, Weisdorf DJ, Blazar BR. Regulatory T cells in acute myelogenous leukemia: is it time for immunomodulation? Blood. 2011;118:5084–5095. doi:10.1182/blood-2011-07-365817.
- Mohty M, Jarrossay D, Lafage-Pochitaloff M, Zandotti C, Briere F, de Lamballeri XN, Isnardon D, Sainty D, Olive D, Gaugler B. Circulating blood dendritic cells from myeloid leukemia patients display quantitative and cytogenetic abnormalities as well as functional impairment. Blood. 2001;98:3750–3756. doi:10.1182/blood.V98.13.3750.
- Szczepanski MJ, Szajnik M, Czystowska M, Mandapathil M, Strauss L, Welsh A, Foon KA, Whiteside TL, Boyiadzis M. Increased frequency and suppression by regulatory T cells in patients with acute myelogenous leukemia. Clin Cancer Res. 2009;15:3325–3332. doi:10.1158/1078-0432.CCR-08-3010.
- Zhou Q, Bucher C, Munger ME, Highfill SL, Tolar J, Munn DH, Levine BL, Riddle M, June CH, Vallera DA, et al. Depletion of endogenous tumor-associated regulatory T cells improves the efficacy of adoptive cytotoxic T-cell immunotherapy in murine acute myeloid leukemia. Blood. 2009;114:3793–3802. doi:10.1182/blood-2009-03-208181.
- Shenghui Z, Yixiang H, Jianbo W, Kang Y, Laixi B, Yan Z, Xi X. Elevated frequencies of CD4(+) CD25(+) CD127lo regulatory T cells is associated to poor prognosis in patients with acute myeloid leukemia. Int J Cancer J Int Du Cancer. 2011;129:1373–1381. doi:10.1002/ijc.25791.