
ABSTRACT
Myeloid-derived suppressor cells (MDSCs) are immunosuppressive cells that promote tumor progression by inhibiting anti-tumor immunity and may be the cause of patient resistance to immune checkpoint inhibitors (ICIs). Therefore, MDSCs are a promising target for cancer immunotherapy, especially in combination with ICIs. Previous studies have shown that the anticonvulsant drug valproic acid (VPA) has additional anti-cancer and immunoregulatory activities due to its inhibition of histone deacetylases. We have previously shown that VPA can attenuate the immunosuppressive function of differentiated MDSCs in vitro. In the present study, we utilized anti-PD-1-sensitive EL4 and anti-PD-1-resistant B16-F10 tumor-bearing mouse models and investigated the effects of VPA on MDSCs with the aim of enhancing the anti-cancer activity of an anti-PD-1 antibody. We showed that VPA could inhibit EL4 and B16-F10 tumor progression, which was dependent on the immune system. We further demonstrated that VPA down-regulated the expression of CCR2 on monocytic (M)-MDSCs, leading to the reduced infiltration of M-MDSCs into tumors. Importantly, we demonstrated that VPA could relieve the immunosuppressive action of MDSCs on CD8+ T-cell and NK cell proliferation and enhance their activation in tumors. We also observed that the combination of VPA plus an anti-PD-1 antibody was more effective than either agent alone in both the EL4 and B16-F10 tumor models. These results suggest that VPA can effectively relieve the immunosuppressive tumor microenvironment by reducing tumor infiltration of M-MDSCs, resulting in tumor regression. Our findings also show that VPA in combination with an immunotherapeutic agent could be a potential new anti-cancer therapy.
Introduction
In recent decades, immune checkpoint blockade therapy has shown tremendous promise for the treatment of multiple cancers by boosting the existing immune response.Citation1,Citation2 Immune checkpoint inhibitors (ICIs) targeting programmed cell death 1 (PD-1), programmed cell death-ligand 1 (PD-L1), and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) have been approved for treating patients with multiple cancer types. However, most patients still show limited or no clinical response, which is probably due to the immunosuppressive machinery in the tumor microenvironment (TME), including the presence of immunosuppressive cells, such as myeloid-derived suppressor cells (MDSCs), tumor associated macrophages (TAMs), and regulatory T-cells (Tregs).Citation3,Citation4
Numerous studies have revealed that MDSCs play important roles in the suppression of anti-tumor immunity, which can lead to tumor evasion from immune surveillance.Citation5,Citation6 Remarkably, several recent studies have shown that MDSCs cause patient resistance to ICI.Citation7,Citation8 Therefore, it is important to identify an effective therapy that depletes or modulates MDSCs. Phenotypically and morphologically, MDSCs can be classified into two subpopulations, namely monocytic (M)-MDSCs with the phenotype CD11b+Ly-6G−Ly-6Chi and polymorphonuclear (PMN)-MDSCs with the phenotype CD11b+Ly-6G+Ly-6C.int Citation9 An ex vivo study has shown that valproic acid (VPA) inhibits the differentiation of M-MDSCs to PMN-MDSCs by inhibiting histone deacetylases (HDACs).Citation10 However, many questions remain about the differences between these MDSC subsets.
Accumulating evidence indicates that epigenetic control mediated by HDAC inhibitor(s) (HDACi) plays an important role in tumor progression and even in immune regulation.Citation11,Citation12 Recent research has shown that in vitro, a pan-HDACi, trichostatin A, or suberoylanilide hydroxamic acid, can robustly expand the M-MDSC population.Citation13 Another study has shown that the class I HDACi entinostat can neutralize MDSCs and enhance the anti-tumor effect of PD-1 antibody in murine models of lung and renal cell carcinoma.Citation14 In addition, Kim et al. demonstrated a similar HDACi effect in colon and breast cancer models.Citation15 We have previously shown that VPA attenuates the immunosuppressive function of differentiated MDSCs in vitro.Citation16 Overall, although HDACi have emerged as an important therapeutic strategy for the treatment of malignancies, the effect and mechanisms underlying the regulation of MDSCs by HDACi still remain largely unexplored.
VPA has been widely used as an antiepileptic drug in the clinic for more than fifty years and is considered to be a safe drug. Repurposing of VPA as an anti-cancer drug is now expected.Citation17 In this study, we investigate the effects of VPA on MDSCs with the aim of enhancing the anti-tumor activity of ICIs. We believe that our data support the importance of targeting MDSCs, especially in combination with ICIs, and our findings further support the use of VPA in the clinic as an anti-cancer drug.
Materials and methods
Cell lines
The EL4 and B16-F10 cell lines were purchased from American Type Culture Collection (ATCC). The EL4 cell line was maintained in RPMI-1640 medium (FUJIFILM Wako) supplemented with 10% fetal bovine serum (FBS, Gibco, Carlsbad) and 1% Antibiotic-Antimycotic Mixed Stock Solution (100 ×) (Nacalai Tesque). The B16-F10 cell line was maintained in DMEM (FUJIFILM Wako) supplemented with 10% FBS and 1% Antibiotic-Antimycotic Mixed Stock Solution (100 ×). Cells were resuscitated and cultured according to ATCC guidelines. The B16-F10 cell line was passaged for less than 6 months after resuscitation. All cell lines were used within 1 month of thawing from early-passage (<3 passages of original vial) lots.
Mice
C57BL/6J mice were purchased from Japan SLC (Shizuoka, Japan) and were used at 6–8 weeks of age. All animals were bred and maintained under specific-pathogen-free conditions. All animal experimental procedures in this study were performed in accordance with the institutional guidelines for animal experiments of Osaka University (Douyaku28-8-8).
Murine tumor studies
EL4 cells (4 × 105 cells/mouse) or B16-F10 cells (1 × 106 cells/mouse) were injected subcutaneously into the lower right flank of C57BL/6 J mice. Five days after inoculation with EL4 cells, mice received daily intraperitoneal (i.p.) injection of PBS or VPA (500 mg/kg, Sigma-Aldrich) until day 13. Anti-Ly-6G (clone: 1A8, 400 µg/mouse), anti-CD8α (clone: 2.43), and anti-NK1.1 (clone: PK136) antibodies (Abs) (200 µg/mouse, Bio X Cell) were injected i.p. on days 4, 7, and 10. The anti-PD-1 (clone: J43) Ab or IgG (both 200 µg/mouse, Bio X Cell) were injected i.p on days 5, 8, and 11. Eight days after inoculation with B16-F10 cells, mice received daily i.p. injection of PBS or VPA (500 mg/kg) until day 15. Anti-Ly-6G (400 µg/mouse), anti-CD8α and anti-NK1.1 Abs (200 µg/mouse) were injected i.p. on days 7, 10 and 13. The anti-PD-1 Ab or IgG (both 200 µg/mouse, Bio X Cell) were injected i.p. on days 8, 11, and 14. The tumor volume was calculated periodically using the following formula: Tumor volume (cm3) = 0.5 × length (cm) × width (cm)2.
Bone marrow, spleen, and tumor cell isolation and blood sample preparation
EL4 tumor-bearing mice were sacrificed on 14 days post tumor inoculation. Whole bone marrow (BM) and spleens were harvested from mice and processed into single cell suspensions. Cell suspensions were then treated with ammonium-chloride-potassium (ACK) lysis buffer and used for either flow cytometry analysis or FACS sorting. Tumors were harvested and cut into small pieces. Tumor pieces were digested with 0.1% collagenase type I (Funakoshi) for 30 minutes at 37ºC with agitation, then ground to a single cell suspension. Dissociated tumors were strained through a 70 µm cell strainer and used for flow cytometry analysis. To prepare blood samples, mouse retro-orbital blood (75 µL) was collected and incubated in 1 mL of ACK lysis buffer on ice for 10 min, after which the cells were pelleted by centrifugation at 400 × g for 5 min. Cells were then resuspended in 0.5 mL ACK lysis buffer and incubated on ice for 5 min. After centrifugation at 400 × g for 3 min, cells were resuspended in 2% FBS/PBS and used for flow cytometry analysis.
In vitro MDSC differentiation
The in vitro differentiation of BM cells into MDSCs was performed as described previously.Citation16 Briefly, BM cells from C57BL/6J mice were stimulated with 40 ng/mL recombinant GM-CSF (Peprotech) for 4 days in the absence or presence of VPA or valpromide (VPM, Sigma-Aldrich) (0.25, 0.5, or 1 mM) to examine its effects on MDSC differentiation.
Flow cytometry analysis
Cells were washed and suspended in 2% FBS/PBS, blocked with a TruStain FcX (anti-mouse CD16/32) Ab (Clone 93, BioLegend), and then stained with the following Abs: allophycocyanin (APC)-labeled anti-mouse CD11b (Clone M1/70, eBiosicence), Pacific Blue-labeled anti-mouse Gr-1 (Clone RB6-8C5), fluorescein isothiocyanate (FITC)-labeled anti-mouse Ly-6G (Clone 1A8), APC-Cy7-labeled anti-mouse Ly-6C (Clone HK1.4), PE or PE-Cy7-labeled anti-mouse F4/80 (Clone BM8), PE-labeled anti-mouse CD25 (Clone PC61.5), Pacific Blue-labeled anti-mouse CD45 (Clone 30-F11), Pacific Blue-labeled anti-mouse CD4 (Clone RM4-4), FITC-labeled anti-mouse CD8α (Clone 53–6.7), FITC-labeled anti-mouse CD3ɛ (Clone 145-2C11), APC-labeled anti-mouse NK1.1 (Clone PK136), PE-labeled anti-mouse CCR2 (Clone SA203G11), PerCP/Cy5.5-labeled anti-mouse CD69 (Clone H1.2F3, BioLegend). The cells were then washed and resuspended in 2% FBS/PBS containing 7-aminoactinomycin D as a viability stain (BioLegend) or ZombieAqua (BioLegend) as a stain for dead cells. For intracellular cytokine staining of PE-Cy7-labeled anti-mouse IFN-γ (Clone XMG1.2, BioLegend), a BD Cytofix/Cytoperm kit (BD Biosciences) was used according to the manufacturer’s instructions. Flow cytometry analysis was performed using a BD FACSCanto II device (BD Biosciences), and the acquired data were analyzed using FlowJo software (BD Biosciences).
MDSC suppression assay
Spleens were harvested from C57BL/6J mice, ground to release splenocytes, and then treated with ACK lysis buffer to eliminate any contaminating red blood cells. Next, the splenocytes were subjected to CD4+ T-cell isolation using the MojoSort magnetic cell separation system and Mouse CD4 Nanobeads (BioLegend). Isolated cells were then labeled with the proliferation dye eFluor 670 (eBioscience) and seeded into 96-well plates at a density of 1 × 105 cells/200 µL per well. All wells had been pre-coated with anti-CD3ɛ Ab (BioLegend) diluted with PBS to a concentration of 1 µg/mL and stored at 4°C overnight before use. MDSCs purified from the spleen of EL4 tumor-bearing mice (M-MDSC: CD11b+Ly-6Chi; PMN-MDSC: CD11b+Ly-6Cint; purity > 90%; JSAN, Bay bioscience Co., Ltd., Kobe, Japan) were added at different ratios to T-cells. The anti-CD28 Ab (BioLegend) was then added to each well at a final concentration of 0.5 µg/mL. After 3 days of incubation at 37°C in an atmosphere of 5% CO2, the proliferation of CD4+ and CD8+ T-cells was analyzed using flow cytometry.
CD8+ T-cell and NK cell proliferation assays
CD8+ T-cells or NK cells were purified from murine splenocytes by FACS sorting (JSAN). Isolated CD8+ T-cells or NK cells were loaded with eFluor 670 and then seeded into 96-well plates at a density of 5 × 104 cells/200 µL per well in the presence of different concentrations of VPA (0, 0.1, and 1 mM). For CD8+ T-cell stimulation, wells were pre-coated with 1 µg/mL anti-CD3ɛ overnight, after which CD8+ T-cells immobilized on the anti-CD3ɛ antibody were stimulated in the presence of 0.5 µg/mL soluble anti-CD28. NK cells were stimulated with 500 U/mL recombinant IL-2 (BioLegend). After 4 days of incubation at 37°C in an atmosphere of 5% CO2, proliferation was quantified by analyzing eFluor 670 dilution by flow cytometry.
In vitro chemotaxis assay
The cell migration assay was performed in 24-well plates with Transwell polycarbonate-permeable supports (8 mm; Corning). BM cells harvested from PBS- or VPA-treated EL4 tumor-bearing mice were stained with PE-labeled anti-mouse CD11b, FITC-labeled anti-mouse Ly-6G and APC-Cy7-labeled anti-mouse Ly-6C. Following this, 2 × 106 BM cells were plated in the upper chambers, and RPMI-1640 medium (10% FBS) supplemented with CCL2/CCL7 (100 ng/mL, BioLegend) was placed in the lower chamber. After incubation for 5 hours at 37ºC, the number of PMN-MDSCs (CD11b+Ly-6G+Ly-6Cint) and M-MDSCs (CD11b+Ly-6G−Ly-6Chi) that had migrated into the lower chamber were counted by flow cytometry. The migration index was calculated as a ratio of the number of migrated cells treated with the chemokines CCL2/CCL7 to that in non-treated wells.
Statistical analyses
Significant differences were assessed using a Student’s t test, or a one- or two-way analysis of variance (ANOVA) using GraphPad Prism (GraphPad Software). p < .05 was considered to be statistically significant.
Results
VPA delays EL4 and B16-F10 tumor progression and reactivates tumor infiltrating immune cells
We tested whether VPA alone had an anti-tumor effect in the anti-PD-1-sensitive EL4 lymphoma and anti-PD-1-resistant B16-F10 melanoma-bearing mouse models. The induction of MDSCs and infiltration into tumors has previously been reported in both of these models. These processes play a key role in the suppression of anti-tumor T-cell activity.Citation18,Citation19 VPA alone delayed tumor growth in the EL4 lymphoma-bearing mice and prolonged the survival () and )). In the B16-F10 melanoma-bearing mouse model, mice that received VPA also showed a slower tumor progression and had a longer survival than those that received PBS () and )).
Figure 1. VPA delays the progression of EL4 and B16-F10 tumors
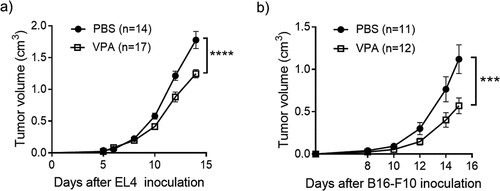
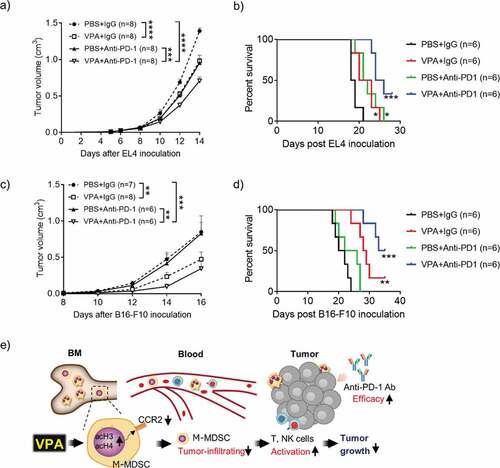
Figure 6. VPA enhances response to anti-PD-1 immunotherapy
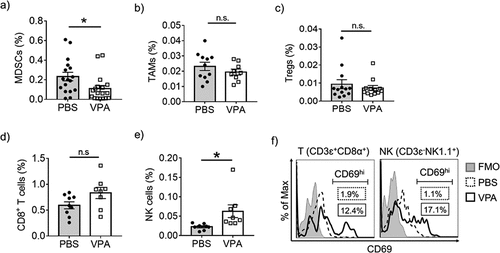
To determine whether the inhibition of tumor growth resulting from VPA was associated with an enhanced immune response, we examined the EL4 tumor infiltrating lymphoid and myeloid populations. MDSCs, TAMs, and Tregs are believed to be major cell components of the immune suppressive TME. VPA caused a reduction in the level of MDSCs (CD11b+Gr-1+) in the tumor site, whereas there were no significant changes in the levels of TAMs and Tregs (–), Supplementary Figure 1a). Furthermore, we found that the administration of VPA resulted in an increase in both NK cells (CD3ɛ−NK1.1+) and CD8+ T-cells (CD3ɛ+CD4−CD8α+) in tumors, as well as in peripheral blood (,), Supplementary Figure 1a), although the increase in CD8+ T-cells was not significant. We evaluated the expression of the early activation marker CD69 in NK and CD8+ T cells. The number of CD69hi cells had increased in both cases (). These results indicate that reducing MDSC levels through VPA may relieve the immunosuppressive action of MDSCs on the proliferation of CD8+ T-cells and NK cells and enhance their re-activation in the TME.
Figure 2. VPA reactivates tumor-infiltrating immune cells
Impaired tumor progression by VPA is anti-tumor immune cell-dependent
Next, to investigate whether the mechanism of the in vivo efficacy of VPA is anti-tumor immunity-mediated, tumor growth was assessed in mice lacking specific anti-tumor immune cell subsets. The anti-tumor effect of VPA was completely absent in mice depleted of NK cells in the EL4 tumor model, whereas CD8+ T-cell depletion alone had a lesser effect, indicating that NK cells are required for the full anti-tumor effect of VPA (). Although VPA could inhibit cell proliferation (Supplementary Figure 2a, c) and enhance apoptosis (Supplementary Figure 2b, d) in EL4 and B16-F10 cells in vitro, the results also suggested that the existence of anti-tumor immune cells was more important for the anti-tumor effect of VPA in vivo.
Figure 3. VPA impairs EL4 tumor progression via anti-tumor immune cells
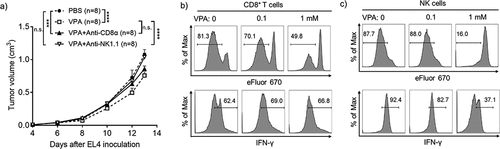
Next, to determine whether VPA could directly stimulate anti-tumor immune cells activity, CD8+ T-cells or NK cells were stimulated with anti-CD3ɛ/anti-CD28 Abs or IL-2, respectively, in the presence or absence of VPA in the media. The proliferation of CD8+ T-cells and NK cells was not stimulated in the presence of VPA, and in fact a high concentration of VPA inhibited proliferation in both cell types (,). In addition, VPA did not show any ability to enhance interferon-gamma (IFN-γ) expression in both CD8+ T-cells and NK cells (,). These results indicate that VPA does not have a direct effect of the proliferation or IFN-γ production by CD8+ T-cells and NK cells. Taken together, these results suggest that immune cell reactivation in the TME is not directly mediated by VPA, but likely occurs through other mechanisms such as a decrease in MDSCs, thus relieving immunosuppression in the TME.
VPA reduces infiltration of M-MDSCs in EL4 tumors
To clarify the mechanism of the reduction of MDSCs in the TME, we examined the levels of the two subsets of MDSCs present in bone marrow (BM), spleen, blood, and tumors from PBS- or VPA-treated EL4 tumor-bearing mice. Although there were no significant decreases in M-MDSCs in the BM and spleen, the levels of M-MDSCs in the blood and tumors were significantly reduced after administration of VPA (). On the other hand, PMN-MDSCs were significantly increased in BM, but no changes were seen in spleen, blood, and tumors comparing PBS- and VPA-treated mice (). Based on these data, we conclude that VPA probably selectively inhibits the migration of M-MDSCs from the BM to the tumor site. We could not find any evidence that VPA had an effect on CD8+ T-cells and NK cells in the spleen, including their relative proportions and their CD69 expression levels (Supplementary Figure 1b-d). These data are consistent with the lack of effect of VPA on the level of MDSCs in the spleen, supporting our hypothesis that VPA relieves immunosuppression by reducing the levels of M-MDSCs in tumors.
Figure 4. VPA reduces the infiltration of M-MDSCs in EL4 tumors
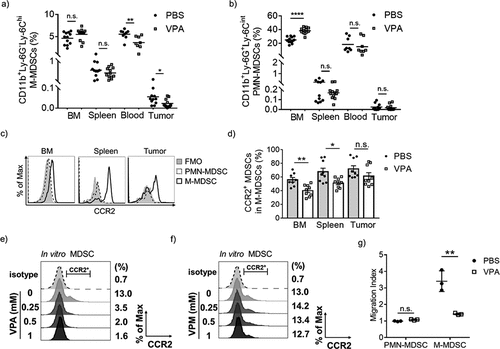
Previous work has shown that the CCR2/CCR2 ligand axis is crucial in the migration and accumulation of monocytic cells including M-MDSCs.Citation20 To examine whether CCR2 is involved in the migration of M-MDSCs into tumors, we first assessed the expression levels of CCR2 on MDSCs. As shown in ), CCR2 could barely be detected on PMN-MDSCs but was highly expressed in M-MDSCs. In addition, VPA down-regulated CCR2 expression on M-MDSCs in the BM, spleen, and in tumors on days 11 and 14 post EL4 inoculation () and Supplementary Figure 3a). Decreased CCR2 expression was also observed in the TAMs, although it is not statistically significant (Supplementary Figure 3b). Previous studies have shown that GM-CSF stimulates BM cells to differentiate into MDSCs (hereafter referred to as in vitro MDSCs).Citation14,Citation21 We demonstrated that VPA also decreased the proportion of CCR2+ in vitro MDSCs in a dose-dependent manner and also down-regulated Ccr2 gene expression at high concentrations, which suggests that VPA directly regulates CCR2 expression levels in MDSCs () and Supplementary Figure 4a). In addition, VPA caused an increase in histone H3 and histone H4 acetylation in MDSCs with an approximately 2.5-fold increase in vivo (Supplementary Figure 5a, b). To clarify whether VPA regulates CCR2 via HDAC modification, we examined the effect of valpromide (VPM), an amide derivative of VPA, on CCR2 expression in MDSCs. VPM does not possess HDAC inhibitory activity (Supplementary Figure 5c, d), and thus, it is often used as a control to explore whether VPA functions through the HDAC mechanism.Citation22 We demonstrated that VPM could not down-regulate the CCR2 expression in in vitro MDSCs similar to VPA (), which suggested increased histone acetylation by VPA is likely responsible for the down-regulation of CCR2 expression. Both EL4 and B16-F10 tumors expressed CCL2 and CCL7, the two major CCR2 ligands (Supplementary Figure 4e). As chemotaxis is likely to be a key driver in cell mobilization, we next examined in vitro whether CCR2 ligands are chemoattractants for M-MDSCs. It came as no surprise to us to find that PMN-MDSCs were not responsive to either CCL2 or CCL7. On the other hand, CCL2 and CCL7 were much stronger chemoattractants for M-MDSCs. Since the levels of tumor migration of M-MDSCs were found to be much higher in PBS-treated mice than in VPA-treated mice (), these data suggest that VPA induces a down-regulation of CCR2 expression which leads to an attenuation of the migration of M-MDSCs into tumors.
PMN-MDSCs are not vital for the anti-tumor effect of VPA
Next, we investigated whether VPA directly affects the immunosuppressive activity of MDSCs. We isolated both M-MDSCs and PMN-MDSCs from the spleens of treated mice ()) and then combined them in a 1:1 ratio with eFluor 670-labeled CD4+ T-cells, which were then stimulated with anti-CD3ɛ and anti-CD28 Abs. T-cell proliferation was much higher when T-cells were co-cultured with PMN-MDSCs derived from VPA-treated mice but there was no difference in T-cell proliferation following co-culture with M-MDSCs from VPA-treated mice, suggesting that VPA can attenuate the immunosuppressive activity of PMN-MDSCs, but has little effect on that of M-MDSCs (). Several genes that are responsible for the immunosuppressive function of PMN-MDSCs have been reported, including Arg1, Cox2, Nos2, Cebpb, and Ptges.Citation23 We have previously shown that VPA significantly down-regulates Arg1 expression but had no effect of Nos2 expression levels in in vitro MDSCs.Citation16 In this study, we further found that the levels of Ptges were significantly down-regulated by VPA but there were no significant changes in the levels of Cox2 and Cebpb (Supplementary Figure 4b-d). Those results suggest that VPA not only impairs the migration of M-MDSCs to tumors but also attenuate the immunosuppressive function of PMN-MDSCs, contributing to the reactivation of CD8+ T-cells and NK cells.
Figure 5. VPA attenuates the immunosuppressive activity of MDSCs in vivo.
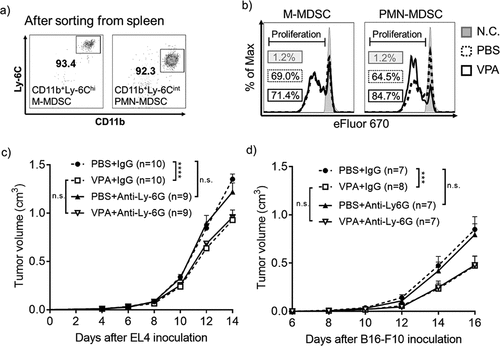
Next, we depleted PMN-MDSCs in vivo using anti-Ly-6G Ab to assess which subset of MDSCs is more important in the anti-tumor effect of VPA in the EL4 tumor-bearing mouse model. The data in supplementary figure 6 show that PMN-MDSCs could be totally depleted from the blood and tumor after administration of the anti-Ly-6G Ab. In the PBS groups, depletion of PMN-MDSCs failed to inhibit tumor progression, similar to the IgG control. In addition, the anti-tumor effect of VPA was the same in both the anti-Ly-6G Ab and IgG groups ()). Similar results were obtained in the B16-F10 tumor-bearing mouse model (Supplementary Figure 6b and )). These results demonstrate that PMN-MDSCs are not vital for enhancing tumor growth in these two tumor models, suggesting that the anti-tumor effect of VPA is dependent on the impaired tumor infiltration of M-MDSCs rather than on PMN-MDSCs.
VPA enhances the response to anti-PD-1 immunotherapy
Growing evidence suggests that resistance to ICI is directly mediated by the suppressive activity of infiltrating myeloid cells in various tumors.Citation24 Therefore, we reasoned that combining VPA with an ICI could further reduce tumor growth and may overcome resistance to ICIs. In anti-PD-1-sensitive EL4 tumor-bearing mice, both VPA and an anti-PD-1 Ab impaired tumor progression equally, whereas the combination of VPA plus anti-PD-1 therapy showed an enhanced anti-tumor effect compared to either VPA or the anti-PD-1 Ab alone (). In line with previous reports,Citation25,Citation26 the anti-PD-1 Ab alone exerted a poor anti-tumor effect in the anti-PD-1-resistant B16-F10 tumor-bearing mice. In contrast, a significant inhibition of tumor growth was seen in mice with the combination of VPA plus anti-PD-1 Ab compared to mice treated with anti-PD-1 Ab ()). We further examined the survival outcome for the VPA and anti-PD-1 antibody combination. We observed a significant increase in survival for the combination treatment in both EL4 and B16-F10 tumor models (,)). Furthermore, we found that the combination effect of VPA and anti-PD-1 immunotherapy resulted in a significant increase in tumor infiltrating CD8+ T cells (Supplementary Figure 7a, c). These results suggest that the anti-tumor effect induced by anti-PD-1 is improved by combination with VPA, which is associated with the increased antitumor immune response. Moreover, VPA and the anti-PD-1 Ab alone, or in combination, were well-tolerated, with no significant decreases in body weight being observed (Supplementary Figure 7b, d). These results suggest that VPA could be an attractive combination agent for ICI treatments.
Discussion
Among the four classes of HDACs, VPA preferentially inhibits class I HDACs (HDAC1, 2, 3, and 8).Citation27 Although VPA has been reported to directly inhibit tumor cell proliferation and attenuate tumor immunogenicity via HDAC inhibition,Citation28,Citation29 the immune regulation ability of VPA in the TME has rarely been addressed. In this study, we clearly showed that VPA could effectively relieve the immunosuppressive TME through a reduction in tumor-infiltrating M-MDSCs, leading to a reduction of tumor burden ().
It has been reported that MDSCs are recruited to tumor tissues from the BM or blood by chemokines produced by the tumor tissue, playing pivotal roles in inhibiting anti-tumor immunity.Citation30,Citation31 It appears that the therapeutic strategy of inhibiting recruitment of MDSCs into tumor tissues could be effective. Distinct chemokine axes regulate the trafficking of M-MDSCs and PMN-MDSCs into the tumor site. In our EL4 tumor model, VPA down-regulated CCR2 expression in the M-MDSCs, leading to reduced tumor-infiltration of M-MDSCs, which suggests the possible use of VPA in types of cancers that secrete high levels of CCR2 ligands such as CCL2 and CCL7. At this point, VPA showed a similar effect to CCR2 inhibitors. A recent study showed CCR2 antagonist CCX872 could impede invasion of MDSCs into tumors and enhance an anti-PD-1 effect to improve the survival of resistant murine gliomas.Citation32 However, VPA have targets other than CCR2, hence VPA might be better than CCR2 inhibitors for cancer therapy.
It has been well established that gene expression can be modulated by chromatin remodeling.Citation33 Although histone acetylation usually increases gene transcription, previous research has also shown that in some cases histone acetylation can suppress gene transcription.Citation34 In our study, down-regulation of CCR2 expression could be correlated with the acetylation status of histone. Hence, direct activation of a CCR2 repressor protein through its acetylation may result in down-regulation of CCR2 expression. Alternatively, previous studies have shown that VPA has the ability to induce apoptosis in a number of different cell types, including tumor cells and immune cells, by regulating apoptosis related genes through HDAC inhibition.Citation35,Citation36 Thus, the possibility that VPA is more cytotoxic to CCR2+ M-MDSCs still remains, and further research is warranted. On the other hand, although CCR2 expression on the TAMs also decreased slightly, there were no significant changes in TAMs found in the tumor site. In addition to the CCL2/CCR2 axis, several other tumor derived factors attract circulating monocytes to the tumor site and differentiate into TAMs, such as chemokines CSF1, CCL5, CXCL12, and CX3CL1.Citation37 VPA might up-regulate these factors, resulting in no significant change in the levels of TAMs at the tumor site.
In other tumors such as breast cancer HM1Citation38 and prostate cancer TRAMP-C1 models,Citation30 PMN-MDSCs have been reported to play a key role in tumor growth. In our EL4 and B16-F10 tumor models, the depletion of PMN-MDSCs failed to inhibit tumor growth, which may be related to the relatively small number of PMN-MDSCs in these two tumor models. In a previous study, we found that VPA could inhibit HDAC2 expression and thereby prevent differentiation of M-MDSCs into PMN-MDSCs. Therefore, although the cancer-promoting effect of PMN-MDSCs are not the same in different tumor-bearing mouse models, VPA could also exert its anti-cancer effect by inhibiting the differentiation of MDSCs even in those tumors rich in PMN-MDSCs.
Even though VPA is currently being tested alone, or in combination, with other agents in clinical studies in cancer,Citation39,Citation40 the effects and molecular mechanism of VPA, especially in immune cells, are largely unknown. Therefore, it is essential to understand the mechanism of action of VPA in order to improve its effectiveness and ensure its safety. Our results have uncovered that the molecular mechanism of tumor growth inhibition by VPA is immune cell-mediated, arising from reduced tumor-infiltration of M-MDSCs. Furthermore, our findings clearly show that VPA in combination with an immunotherapy could be a potential new anti-tumor therapy that acts by neutralizing MDSC function, although further research is required to verify this hypothesis. Compared with combination therapies such as anti-CTLA-4/anti-PD-1 Abs that target T-cells alone, a strategy that targets both T-cells and immunosuppressive myeloid cells could further improve response rates and therapeutic effects. Depletion of MDSCs using Gr-1 or Ly-6G/C antibodies, although successful in mice models, has limited utility in the clinic. To the best of our knowledge, there are currently no clinically applicable approaches to mitigate MDSC-mediated immune escape. Our findings are notable in suggesting the safety of VPA and its combination with ICI might target patient MDSCs through a different mechanism attenuating M-MDSC recruitment into tumors. These results have direct potential clinical implications in designing rational combination treatments for clinical use.
Abbreviations
| CTLA-4 | = | Cytotoxic T-lymphocyte-associated protein 4 |
| HDACs | = | Histone deacetylases |
| HDACi | = | HDAC inhibitor(s) |
| ICIs | = | Immune checkpoint inhibitors |
| M | = | Monocytic |
| MDSCs | = | Myeloid-derived suppressor cells |
| PD-1 | = | Programmed cell death 1 |
| PD-L1 | = | Programmed cell death-ligand 1 |
| PMN | = | Polymorphonuclear |
| TAMs | = | Tumor associated macrophages |
| TME | = | Tumor microenvironment |
| Tregs | = | Regulatory T-cells |
| VPA | = | Valproic acid |
| VPM | = | Valpromide |
Disclosure of Potential Conflicts of Interest
The authors declare that they have no conflict of interest.
Supplemental Material
Download ()Supplemental Material
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
References
- Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell. 2015;161(2):205–10. doi:10.1016/j.cell.2015.03.030.
- Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348(6230):56–61. doi:10.1126/science.aaa8172.
- Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature. 2011;480(7378):480–489. doi:10.1038/nature10673.
- Weber R, Fleming V, Hu X, Nagibin V, Groth C, Altevogt P, Utikal J, Umansky V. Myeloid-derived suppressor cells hinder the anti-cancer activity of immune checkpoint inhibitors. Front Immunol. 2018;9:1310. doi:10.3389/fimmu.2018.01310.
- Ramachandran IR, Martner A, Pisklakova A, Condamine T, Chase T, Vogl T, Roth J, Gabrilovich D, Nefedova Y. Myeloid-derived suppressor cells regulate growth of multiple myeloma by inhibiting T cells in bone marrow. J Immunol. 2013;190(7):3815–3823. doi:10.4049/jimmunol.1203373.
- Finke J, Ko J, Rini B, Rayman P, Ireland J, Cohen P. MDSC as a mechanism of tumor escape from sunitinib mediated anti-angiogenic therapy. Int Immunopharmacol. 2011;11(7):856–861. doi:10.1016/j.intimp.2011.01.030.
- Limagne E, Euvrard R, Thibaudin M, Rebe C, Derangere V, Chevriaux A, Boidot R, Vegran F, Bonnefoy N, Vincent J, et al. Accumulation of MDSC and Th17 cells in patients with metastatic colorectal cancer predicts the efficacy of a FOLFOX-bevacizumab drug treatment regimen. Cancer Res. 2016;76(18):5241–5252. doi:10.1158/0008-5472.CAN-15-3164.
- Gebhardt C, Sevko A, Jiang H, Lichtenberger R, Reith M, Tarnanidis K, Holland-Letz T, Umansky L, Beckhove P, Sucker A, et al. Myeloid cells and related chronic inflammatory factors as novel predictive markers in melanoma treatment with ipilimumab. Clin Cancer Res. 2015;21(24):5453–5459. doi:10.1158/1078-0432.CCR-15-0676.
- Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, Mandruzzato S, Murray PJ, Ochoa A, Ostrand-Rosenberg S, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun. 2016;7:12150. doi:10.1038/ncomms12150.
- Youn JI, Kumar V, Collazo M, Nefedova Y, Condamine T, Cheng P, Villagra A, Antonia S, McCaffrey JC, Fishman M, et al. Epigenetic silencing of retinoblastoma gene regulates pathologic differentiation of myeloid cells in cancer. Nat Immunol. 2013;14(3):211–220. doi:10.1038/ni.2526.
- Shah RR. Safety and tolerability of histone deacetylase (HDAC) inhibitors in oncology. Drug Saf. 2019;42(2):235–245. doi:10.1007/s40264-018-0773-9.
- Llopiz D, Ruiz M, Villanueva L, Iglesias T, Silva L, Egea J, Lasarte JJ, Pivette P, Trochon-Joseph V, Vasseur B, et al. Enhanced anti-tumor efficacy of checkpoint inhibitors in combination with the histone deacetylase inhibitor Belinostat in a murine hepatocellular carcinoma model. Cancer Immunol Immunother. 2019;68(3):379–393. doi:10.1007/s00262-018-2283-0.
- Rosborough BR, Castellaneta A, Natarajan S, Thomson AW, Turnquist HR. Histone deacetylase inhibition facilitates GM-CSF-mediated expansion of myeloid-derived suppressor cells in vitro and in vivo. J Leukoc Biol. 2012;91(5):701–709. doi:10.1189/jlb.0311119.
- Orillion A, Hashimoto A, Damayanti N, Shen L, Adelaiye-Ogala R, Arisa S, Chintala S, Ordentlich P, Kao C, Elzey B, et al. Entinostat neutralizes myeloid-derived suppressor cells and enhances the antitumor effect of PD-1 inhibition in murine models of lung and renal cell carcinoma. Clin Cancer Res. 2017;23(17):5187–5201. doi:10.1158/1078-0432.CCR-17-0741.
- Kim K, Skora AD, Li Z, Liu Q, Tam AJ, Blosser RL, Diaz LA Jr., Papadopoulos N, Kinzler KW, Vogelstein B, et al. Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. Proc Natl Acad Sci USA. 2014;111(32):11774–11779. doi:10.1073/pnas.1410626111.
- Xie Z, Ago Y, Okada N, Tachibana M. Valproic acid attenuates immunosuppressive function of myeloid-derived suppressor cells. J Pharmacol Sci. 2018;137(4):359–365. doi:10.1016/j.jphs.2018.06.014.
- Soria-Castro R, Schcolnik-Cabrera A, Rodriguez-Lopez G, Campillo-Navarro M, Puebla-Osorio N, Estrada-Parra S, Estrada-Garcia I, Chacon-Salinas R, Chavez-Blanco AD. Exploring the drug repurposing versatility of valproic acid as a multifunctional regulator of innate and adaptive immune cells. J Immunol Res. 2019;2019:9678098. doi:10.1155/2019/9678098.
- Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, Bronte V, Chouaib S. PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med. 2014;211(5):781–790. doi:10.1084/jem.20131916.
- Corzo CA, Condamine T, Lu L, Cotter MJ, Youn JI, Cheng P, Cho HI, Celis E, Quiceno DG, Padhya T, et al. HIF-1alpha regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J Exp Med. 2010;207(11):2439–2453. doi:10.1084/jem.20100587.
- Huang B, Lei Z, Zhao J, Gong W, Liu J, Chen Z, Liu Y, Li D, Yuan Y, Zhang GM, et al. CCL2/CCR2 pathway mediates recruitment of myeloid suppressor cells to cancers. Cancer Lett. 2007;252(1):86–92. doi:10.1016/j.canlet.2006.12.012.
- Highfill SL, Rodriguez PC, Zhou Q, Goetz CA, Koehn BH, Veenstra R, Taylor PA, Panoskaltsis-Mortari A, Serody JS, Munn DH, et al. Bone marrow myeloid-derived suppressor cells (MDSCs) inhibit graft-versus-host disease (GVHD) via an arginase-1-dependent mechanism that is up-regulated by interleukin-13. Blood. 2010;116(25):5738–5747. doi:10.1182/blood-2010-06-287839.
- Zhang C, Zhang E, Yang L, Tu W, Lin J, Yuan C, Bunpetch V, Chen X, Ouyang H. Histone deacetylase inhibitor treated cell sheet from mouse tendon stem/progenitor cells promotes tendon repair. Biomaterials. 2018;172:66–82. doi:10.1016/j.biomaterials.2018.03.043.
- Veglia F, Perego M, Gabrilovich D. Myeloid-derived suppressor cells coming of age. Nat Immunol. 2018;19(2):108–119. doi:10.1038/s41590-017-0022-x.
- De Henau O, Rausch M, Winkler D, Campesato LF, Liu C, Cymerman DH, Budhu S, Ghosh A, Pink M, Tchaicha J, et al. Overcoming resistance to checkpoint blockade therapy by targeting PI3Kgamma in myeloid cells. Nature. 2016;539(7629):443–447. doi:10.1038/nature20554.
- Kleffel S, Posch C, Barthel SR, Mueller H, Schlapbach C, Guenova E, Elco CP, Lee N, Juneja VR, Zhan Q, et al. Melanoma cell-intrinsic PD-1 receptor functions promote tumor growth. Cell. 2015;162(6):1242–1256. doi:10.1016/j.cell.2015.08.052.
- Pan D, Kobayashi A, Jiang P, Ferrari de Andrade L, Tay RE, Luoma AM, Tsoucas D, Qiu X, Lim K, Rao P, et al. A major chromatin regulator determines resistance of tumor cells to T cell-mediated killing. Science. 2018;359(6377):770–775. doi:10.1126/science.aao1710.
- Gottlicher M, Minucci S, Zhu P, Kramer OH, Schimpf A, Giavara S, Sleeman JP, Lo Coco F, Nervi C, Pelicci PG, et al. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. Embo J. 2001;20(24):6969–6978. doi:10.1093/emboj/20.24.6969.
- Nebbioso A, Clarke N, Voltz E, Germain E, Ambrosino C, Bontempo P, Alvarez R, Schiavone EM, Ferrara F, Bresciani F, et al. Tumor-selective action of HDAC inhibitors involves TRAIL induction in acute myeloid leukemia cells. Nat Med. 2005;11(1):77–84. doi:10.1038/nm1161.
- Roulois D, Blanquart C, Panterne C, Gueugnon F, Gregoire M, Fonteneau JF. Downregulation of MUC1 expression and its recognition by CD8(+) T cells on the surface of malignant pleural mesothelioma cells treated with HDACi. Eur J Immunol. 2012;42(3):783–789. doi:10.1002/eji.201141800.
- Hawila E, Razon H, Wildbaum G, Blattner C, Sapir Y, Shaked Y, Umansky V, Karin N. CCR5 directs the mobilization of CD11b(+)Gr1(+)Ly6C(low) polymorphonuclear myeloid cells from the bone marrow to the blood to support tumor development. Cell Rep. 2017;21(8):2212–2222. doi:10.1016/j.celrep.2017.10.104.
- Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol. 2016;37(3):208–220. doi:10.1016/j.it.2016.01.004.
- Flores-Toro JA, Luo D, Gopinath A, Sarkisian MR, Campbell JJ, Charo IF, Singh R, Schall TJ, Datta M, Jain RK, et al. CCR2 inhibition reduces tumor myeloid cells and unmasks a checkpoint inhibitor effect to slow progression of resistant murine gliomas. Proc Natl Acad Sci USA. 2019. doi:10.1073/pnas.1910856117.
- Wu C. Chromatin remodeling and the control of gene expression. J Biol Chem. 1997;272(45):28171–28174. doi:10.1074/jbc.272.45.28171.
- Bug G, Gul H, Schwarz K, Pfeifer H, Kampfmann M, Zheng X, Beissert T, Boehrer S, Hoelzer D, Ottmann OG, et al. Valproic acid stimulates proliferation and self-renewal of hematopoietic stem cells. Cancer Res. 2005;65(7):2537–2541. doi:10.1158/0008-5472.CAN-04-3011.
- Zhu MM, Li HL, Shi LH, Chen XP, Luo J, Zhang ZL. The pharmacogenomics of valproic acid. J Hum Genet. 2017;62(12):1009–1014. doi:10.1038/jhg.2017.91.
- Lv J, Du C, Wei W, Wu Z, Zhao G, Li Z, Xie X. The antiepileptic drug valproic acid restores T cell homeostasis and ameliorates pathogenesis of experimental autoimmune encephalomyelitis. J Biol Chem. 2012;287(34):28656–28665. doi:10.1074/jbc.M112.356584.
- Ugel S, De Sanctis F, Mandruzzato S, Bronte V. Tumor-induced myeloid deviation: when myeloid-derived suppressor cells meet tumor-associated macrophages. J Clin Invest. 2015;125(9):3365–3376. doi:10.1172/JCI80006.
- Taki M, Abiko K, Baba T, Hamanishi J, Yamaguchi K, Murakami R, Yamanoi K, Horikawa N, Hosoe Y, Nakamura E, et al. Snail promotes ovarian cancer progression by recruiting myeloid-derived suppressor cells via CXCR2 ligand upregulation. Nat Commun. 2018;9(1):1685. doi:10.1038/s41467-018-03966-7.
- Nilubol N, Merkel R, Yang L, Patel D, Reynolds JC, Sadowski SM, Neychev V, Kebebew E. A phase II trial of valproic acid in patients with advanced, radioiodine-resistant thyroid cancers of follicular cell origin. Clin Endocrinol (Oxf). 2017;86(1):128–133. doi:10.1111/cen.13154.
- Catalano MG, Pugliese M, Gallo M, Brignardello E, Milla P, Orlandi F, Limone PP, Arvat E, Boccuzzi G, Piovesan A. Valproic acid, a histone deacetylase inhibitor, in combination with paclitaxel for anaplastic thyroid cancer: results of a multicenter randomized controlled phase II/III trial. Int J Endocrinol. 2016;2016:2930414. doi:10.1155/2016/2930414.