
ABSTRACT
Regulatory T cells are integral to the regulation of autoimmune and anti-tumor immune responses. However, several studies have suggested that changes in T cell signaling networks can result in T cells that are resistant to the suppressive effects of regulatory T cells. Here, we investigated the role of Cbl-b, an E3 ubiquitin ligase, in establishing resistance to Treg-mediated suppression. We found that the absence of Cbl-b, a negative regulator of multiple TCR signaling pathways, rendered T cells impartial to Treg suppression by regulating cytokine networks leading to improved anti-tumor immunity despite the presence of Treg cells in the tumor. Specifically, Cbl-b KO CD4+FoxP3− T cells hyper-produced IL-2 and together with IL-2 Rα upregulation served as an essential mechanism to escape suppression by Treg cells. Furthermore, we report that IL-2 serves as the central molecule required for cytokine-induced Treg resistance. Collectively our data emphasize the role of IL-2 as a key mechanism that renders CD4+ T cells resistant to the inhibitory effects of Treg cells.
KEYWORDS:
Introduction
Regulatory T cells (Tregs) play a pivotal role in the pathogenesis of human diseases.Citation1,Citation2 Loss of Treg function promotes a breakdown in immune tolerance, and facilitates early onset multi-organ autoimmunity in humans with IPEX syndrome and in scurfy mice.Citation3–Citation7 In contrast, Treg cells also constrain the beneficial anti-tumor immune responses thereby serving as a negative prognostic indicator.Citation8–Citation13 Tregs exploit diverse contact-dependent and/or cytokine-mediated mechanisms, including TGF-β secretion, IL-2 consumption, and downregulation of co-stimulatory molecules on antigen presenting cells, to restrict T cell function in a context-dependent manner.Citation1,Citation2,Citation14,Citation15 Thus, numerous strategies have been proposed to reverse the suppressive effects of Treg cells in the context of tumor immunity.Citation15 One particular approach involves rendering T cells refractory to the suppressive effects of Treg cells.
Studies have demonstrated that there are a variety of molecular pathways and cellular mechanisms which render T cells resistant to Treg suppression.Citation16–Citation25 First, TLR4 and TLR9-mediated stimulation of dendritic cells and the subsequent increase in IL-6 production by DCs have been shown to render T cells resistant to the suppressive effects of CD4+CD25+ Tregs in vitro.Citation16 This is consistent with other studies demonstrating that IL-6 inhibited the suppressive ability of Treg cells in lupus-prone and graft-versus-host disease (GVHD) murine models, through mechanisms that have not been elucidated.Citation23,Citation25 Furthermore, cognate ligand-induced stimulation of glucocorticoid-induced TNFR-related protein (GITR) on activated CD4+ T cells and subsequent production of IL-2 by the CD4+ T cells have been shown to potentially mediate Treg resistance.Citation17,Citation18 Similarly, several other T cell stimulating cytokines such as IL-1β, IL-4, IL-7, IL-15, and IL-21, have also been shown to render T cells resistant to Treg-mediated suppression.Citation19–Citation22,Citation24 Although the specific mechanism utilized by these cytokines to render T cells resistant to Treg cells has not been defined, evidence suggests that the phosphatidylinositol 3-kinase (PI3 K)/Akt signaling pathway may play a role.Citation26,Citation27
Beyond the role of cytokines in T cell resistance to Treg cells, intracellular molecules such as Casitas B- Lineage Lymphoma-b (Cbl-b), p105 (NFкB precursor protein), miR-155, Akt1, MyD88, TRAF6 and SHP-1 have also been reported to render effector T cells resistant or sensitive to Treg-mediated suppression.Citation28–Citation35 In particular, studies involving Cbl-b deficient T cells have been one of the first to demonstrate the role of intracellular molecules in rendering T cells resistant to the suppressive effects of Treg cells in vitro and in vivo.Citation28,Citation30,Citation33
Cbl-b is an E3 ubiquitin ligase and directly ubiquitinates PKCθ, PLCγ1, Vav1, Nedd4 along with several other TCR signaling molecules.Citation36,Citation37 Thus, Cbl-b serves as a powerful negative regulator of T cell activation.Citation38 Studies have shown that Cbl-b KO T cells utilize multiple molecular mechanisms which may render T cells resistant to the suppressive effects of Treg cells. Cbl-b directly ubiquitinates SMAD7, a negative regulator of TGF-β signaling pathway,Citation39,Citation40 which has also been implicated in generating CD4+ T cells that are refractory to Treg-mediated suppression.Citation41 This is consistent with findings demonstrating that the lack of Cbl-b results in a robust T cell activation even in the presence of TGF-β.Citation30,Citation40 Furthermore, regulatory T cells utilize several other context-dependent suppressive mechanisms, one of which includes downregulation of CD80/86 expressed on antigen presenting cells.Citation42,Citation43 Cbl-b KO T cells can be stimulated in a CD28-independent manner and demonstrate impaired induction of T cell anergy,Citation44,Citation45 therefore Cbl-b KO T cells potentially bypass the requirement for CD80/86-induced co-stimulation. In summary, Cbl-b KO CD4+FoxP3− T cells display context-dependent insensitivity to TGF-β signaling, and CD28 independent stimulation, all of which contributes to a robust T cell activation.Citation30,Citation33,Citation38,Citation44 However, the precise mechanism which renders Cbl-b KO T cells resistant to Treg cells is still under debate.
Despite the emergence of multiple studies characterizing the role of immunoregulatory molecules and cytokines in mediating T cell resistance against Tregs, no consensus was achieved in identifying the specific biological mechanism. This study investigated the role of defined cytokines in rendering Cbl-b deficient T cells resistant to suppression by regulatory T cells.
Materials and methods
Mice
C57BL/6 Wild-type (WT), CD25 KO and CD45.1 mice were purchased from The Jackson Laboratory. Cbl-b KO mice in C57BL/6 background were a kind gift from Dr, J.M. Penninger, and DEREG mice in C57BL/6 background were a kind gift from Dr. T. Sparwasser. All mice were maintained and bred under the guidelines and policies set by UHN Animal Resource Center. Mice used for experiments were between 2 to 4 months old.
In vivo tumor studies
E.G7-Ova tumor cells, previously cultured in complete RPMI 1640 media (10% FCS, 50 μM 2-mercaptoethanol, 2 mM glutamine, and 0.1% pen/strep.), were subcutaneously injected into the shaved left flanks of Cbl-b deficient and sufficient mice on day 10 post inoculation. Tumor growth was tracked every 3 days using a caliper. Mice bearing tumors over 225 mm2 were euthanized and recorded as deceased. For in vivo depletion of Treg cells in the tumor study, FoxP3+ cells were transiently ablated in DEREG mice by administering 10ng per g body weight diphtheria toxin (Merck) intraperitoneally on day 10, 11, 19, and 20 post tumor inoculation.
Cell isolation
CD4+ T cells were negatively selected from spleens and lymph nodes of mice using the magnetic purification kit (CAT# 130-104-454 & 130-104-075, Miltenyi Biotec). BD Fluorescence-activated cell sorting (FACS) Aria was used to further separate un-stimulated naïve T cells (CD4+CD25−) and Treg cells (CD4+CD25+), using FoxP3 expression to confirm the purity of the populations. For purification of APCs, CD5 (Ly-1) MicroBeads were used to deplete CD5+ T and B cells (CAT# 130-049-301, Miltenyi Biotec).
In vitro T cell stimulation
All in vitro T cell stimulation was performed using anti-CD3 and irradiated APCs. Prior to co-culture, APCs were irradiated with a dose of 2500cGy using X-RAD 320 (PXi Precision X-Ray). Unless noted otherwise, purified CD4+CD25− T cells were stained with 10 µM cell proliferation dye eFluor® 450 (CAT# 65-0842-90, eBioscience) in PBS for 20 min. at 4°C. After three washes, 5 × 104 T cells were co-cultured with 2 × 105 irradiated APCs and 1 µg/ml anti-CD3 Ab (Clone 145–2 C11, CAT# 14-0031-85, eBioscience) in complete RPMI 1640 media (CAT# 11875119, Invitrogen) containing 10% FCS, 1% L-glutamine, 1% Penicillin-Streptomycin, and 0.0004% 2-mercaptoethanol. Cells were incubated in Thermo ScientificTM NuncTM MicroWellTM 96-well polystyrene microplates (CAT# 12-565-66, Fisher Scientific) in 5% CO2 and 37°C incubation. Supernatants were harvested on day 1 or 3 post-stimulation for cytokine analysis and flow analysis was also performed either on day 1 or 3 post-stimulation.
Treg suppression assay
Upon FACS sorting of naïve CD4+ T cells (CD4+CD25−) and Treg cells (CD4+CD25+), purified CD4+CD25− T cells were stained with 10 µM cell proliferation dye eFluor® 450 (CAT# 65-0842-90, eBioscience) in PBS for 20 min, followed by three washes in complete RPMI 1640 media (CAT# 11875119, Invitrogen). 5 × 104 CD4+CD25− T cells were subsequently cultured with 2 × 105 irradiated APCs, anti-CD3 Ab (1 µg/ml), and 5 × 104 CD4+CD25+ Treg cells. For Treg suppression assays involving 1:1 to 8:1 Teff: Treg ratios, the quantity of Treg cells was adjusted from 5 × 104 to 6.25 × 103 cells, respectively. For exogenous supplementation of IL-2 signaling inhibitors/agonist, recombinant IL-2 (CAT# 575404, BioLegend), recombinant IL-15 (CAT# 34-8153-82, eBioscience), anti–IL-2 (S4B6, CAT# 16-7020-85, eBioscience) and anti-CD25 (PC61) were used. For cytokine screening, all cytokines (IFN-α, IFN-γ, IL-1β, IL-2, IL-4, IL-6, IL-7, IL-9, IL-10, IL-12, IL-17A, IL-18, IL-23 and TNF-α) were purchased from BioLegend except for IFN-γ purchased from eBioscience. Cells were incubated in Thermo ScientificTM NuncTM MicroWellTM 96-well polystyrene microplates (CAT# 12-565-66, Fisher Scientific) in 5% CO2 and 37°C incubation. Unless noted otherwise, supernatants were harvested on day 1 or 3 post stimulation for cytokine analysis and flow analysis was performed either on day 1 or 3 post-stimulation.
Cytokine analysis
Co-culture supernatants, stored in −80°C, were analyzed using IL-2 and IFN-γ ELISA kits (CAT# 88-7024-88, 88-7314-86, eBioscience). LEGENDplexTM Mouse Th Cytokine Panel cytometric bead array (CAT# 740005, BioLegend) was used for T cell cytokine secretion profiling.
Surface/Intracellular staining & flow cytometry
Individual cell suspensions were washed twice in FACS buffer (PBS supplemented with 2% FCS and 0.05% sodium azide), followed by FcR blocking (30 min.) using anti-CD16/32 (CAT# 14-0161-85, eBioscience). For surface marker analyses, cells were subsequently stained with Abs for 30 min. on ice followed by two washes. The following antibodies were used for experiments: anti-CD25-PE (PC61), anti-CD122-PE (5H4), anti-CD4-APC (GK1.5), anti-GITR-APC (DTA-1) or anti-MHCII-APC-Cy7/FITC (M5/114.15.2), all purchased from eBioscience; and anti-MHCII-AmCyan (M5/114.15.2) was purchased from BioLegend. Anti-CD132-PE (TUGm2) was purchased from BD Biosciences. For all surface marker staining, cells were fixed using 4% paraformaldehyde after washes.
Intracellular transcription factor staining was performed using FoxP3 Transcription Factor Staining Buffer Set (CAT# 00-5523-00, eBioscience) with the following antibodies: anti-CTLA-4-PE (UC10-489), or anti-FoxP3-PE-Cy7 (FKJ-16 s), from eBioscience. All data were acquired using BD FACSCantoTM II Flow Cytometer (BD Biosciences), and were analyzed on FlowJo software 7.6.1 (FlowJo LLC).
Statistical analysis
Two-tailed paired Student’s t-test or 1-way ANOVA with Tukey’s Post-Hoc test was performed for comparisons. All data are presented as mean with standard error (n = 3) using GraphPad Prism 5 (GraphPad Software Inc.). All experiments are representative of 2 or more biological replicates.
Results
Cbl-b deficiency enhances anti-tumor immunity despite the presence of regulatory T cells
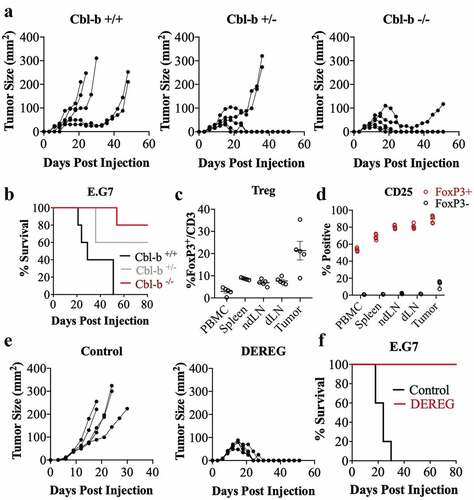
Previous studies have demonstrated that Cbl-b deficient mice have improved anti-tumor immunity using transplantable tumor models.Citation46,Citation47 In order to evaluate whether resistance to immune regulation may play a role in the improved response in the Cbl-b KO mice, we first compared the ability of Cbl-b sufficient and deficient mice in controlling tumor growth using the E.G7 thymoma model. Consistent with a study conducted by Chiang et al. (2007), Cbl-b deficient mice were capable of spontaneous tumor rejection as demonstrated through controlled tumor size () and increased overall survival (). Cbl-b expression is generally found in the lymphocytic lineages, and T cells have been shown to play a particularly important role in this tumor model. To examine the role of Tregs in this model, we evaluated the proportion and markers expressed by this population of cells. Regulatory T cells were present in higher frequency in comparison to other lymphoid organs with a mean of 21% of CD3+ cells expressing FoxP3 (). Furthermore, Treg cells from the tumor expressed highest percentage of CD25+ compared to Tregs in the peripheral blood, spleen, draining and non-draining LN suggestive of enhanced Treg activation (). To evaluate whether Treg cells play a functional role in regulating anti-tumor immunity against the E.G7 tumor, the transgenic DEREG mouse model was used which expresses the diphtheria toxin receptor under the control of FoxP3 promoter. Ablation of Treg cells by treatment with diphtheria toxin resulted in impaired tumor growth (), and 100% survival in mice (). In summary, we demonstrate that Tregs play an important role in the E.G7 tumor model, and that the absence of Cbl-b can lead to the control of tumor growth.
Figure 1. Cbl-b deficiency enhances anti-tumor immunity against E.G7 tumor
CD4+T cells that are deficient in Cbl-b are resistant to inhibition by regulatory T cells
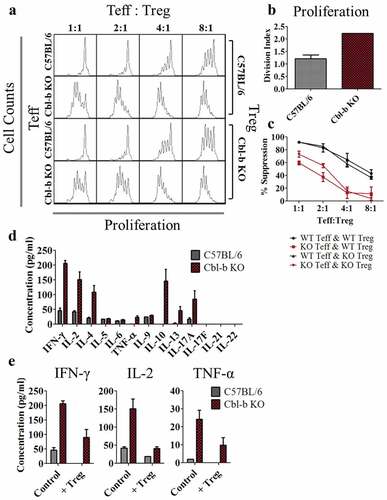
Numerous studies have suggested that the absence of Cbl-b in T cells results in resistance to the inhibitory effects of regulatory T cells.Citation28,Citation30,Citation47 Due to the limitations of models available to precisely evaluate the impact of Treg cells on CD4+ effector T cells, we examined the importance of Cbl-b in Treg-resistance through in vitro Treg suppression assays. The proliferative capacity and the ability of T cells to be suppressed by Tregs were quantified in Cbl-b KO CD4+FoxP3− T cells. Consistent with previous literature, Cbl-b KO CD4+ effector T cells rapidly proliferated despite the presence of Treg cells ().Citation28,Citation30 In contrast, the absence of Cbl-b in Treg cells did not impact the ability of Treg cells to suppress T cell proliferation (). As T cell proliferation may not necessarily indicate Treg resistance, we calculated a % suppression score. Division index (D.I) was quantified by averaging the number of cell divisions using FlowJo software, and % suppression was calculated by normalizing T cell proliferation in the presence of Tregs to intrinsic proliferative capacity of the T cells (% suppression = 100 – (D.I. with Tregs/D.I. without Tregs) x 100).Citation48 While Cbl-b KO CD4+FoxP3− T cells intrinsically possessed enhanced proliferative capacity, as quantified by an approximate 2-fold increase in its division index (), Cbl-b KO CD4+FoxP3− T cells still demonstrated significantly lower % suppression in any given ratio of Teff to Treg cells in comparison to C57BL/6 CD4+FoxP3− T cells (). Thus, CD4+FoxP3− T cells from Cbl-b KO mice were refractory to the inhibitory effects of Treg cells.
Figure 2. Cbl-b KO CD4+FoxP3− T cells display resistance to Treg-mediated suppression and enhanced cytokine secretion
Potential role for cytokines in mediating T cell resistance to regulatory cells
Despite the inability of Tregs to suppress proliferation of Cbl-b KO CD4+FoxP3− T cells, the precise mechanism remains to be elucidated. Previous studies have highlighted the ability of Cbl-b deficient T cells to secrete higher levels of cytokines including IFN-γ and IL-2.Citation33,Citation44,Citation46,Citation47,Citation49 Therefore, we extensively characterized soluble factors secreted by Cbl-b KO CD4+ T cells in the context of Treg resistance. Cytokine production by Cbl-b KO CD4+FoxP3− T cells was characterized by cytometric bead array (CBA) in cultures that did not include Treg cells and was compared with C57BL/6 control T cells. The hyperactive Cbl-b KO CD4+FoxP3− T cells secreted increased levels of Th1 and Th2 cytokines including IFN-γ, IL-2, IL-4 and TNF-α, as well as IL-10, IL-13 and IL-17A (). To examine which cytokines were elevated in the context of Treg suppression, we performed Treg suppression assays and analyzed the supernatant by CBA. Cbl-b KO CD4+ effector T cells cultured with C57BL/6 Treg cells secreted similar or higher levels of IFN-γ, IL-2 and TNF-α compared to C57BL/6 CD4+ effector T cells stimulated without Treg cells (), indicating the availability of these cytokines during Treg-mediated suppression. Therefore, Cbl-b KO CD4+ T cells secrete a diverse array of cytokines including IFN-γ, IL-2, and TNF-α that may potentially play a role in mediating Treg resistance.
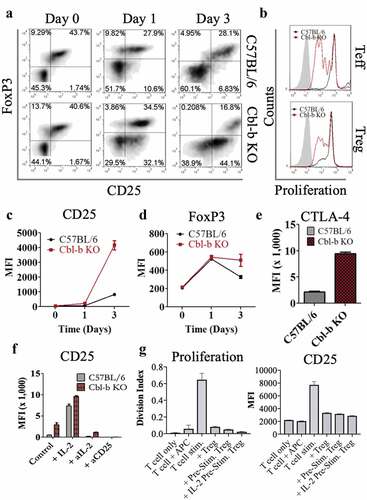
IL-2 hyper-secretion is correlated to CD25 overexpression in Cbl-b KO CD4+ T cells
One way that regulatory T cells have been reported to suppress effector T cells is via IL-2 consumption, leading to cytokine deprivation-mediated cell death or blockade of differentiation.Citation50–Citation52 We speculated that elevated secretion of IL-2 by Cbl-b KO CD4+ T cells may be responsible for sustaining T cell proliferation and that upregulation of the IL-2 receptor by Cbl-b deficient effector cells may enhance the ability to outcompete Treg cells for IL-2, thereby providing resistance to suppression. Therefore, we further characterized the effect of Cbl-b on IL-2 secretion by performing an ELISA on the supernatant collected from Treg suppression assays. In addition, we evaluated IL-2Rα expression on C57BL/6 or Cbl-b KO CD4+CD25− T cells in the absence of Treg cells on day 1 and 3 post-stimulation. In agreement with previous findings, Cbl-b KO CD4+ T cells hyper-secreted IL-2 in the absence () or presence of Treg cells (), and displayed strikingly enhanced CD25 expression even at day 1 post-stimulation further suggesting that the T cells may have increased IL-2 uptake ().
Figure 3. Cbl-b deficient CD4+ T cells upregulate CD25 levels and IL-2 production
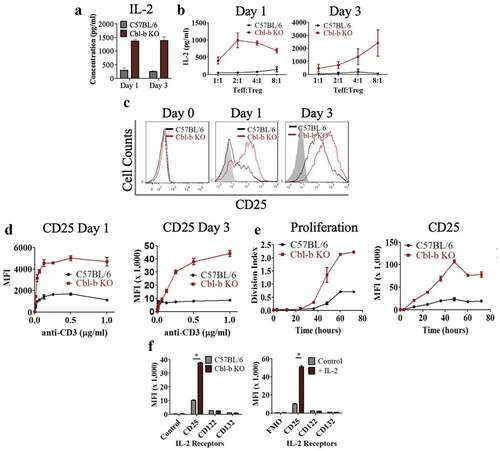
Previous reports suggested that TCR signaling is enhanced in the absence of Cbl-b, and that T cell activation is independent of CD28 signaling.Citation38,Citation44,Citation45,Citation53 This augmented TCR signaling may explain the higher expression of CD25 on T cells. Therefore, we examined whether the upregulation of CD25 expression was relative to the strength of the TCR signal. Overall, a substantial increase in CD25 expression is observed by increasing TCR signaling using graded anti-CD3 concentrations or increased duration of stimulation (,). However, Cbl-b KO CD4+ T cells required less TCR signal to induce a high level of CD25 expression, suggesting a link with TCR sensitivity and upregulation of CD25 (,). Lastly, when stimulated without Treg cells, Cbl-b deficiency or exogenous IL-2 supplementation specifically enhanced CD25 expression while CD122 and CD132 surface expression were kept at basal expression (). This observation can be explained by previous studies demonstrating that high-affinity IL-2 receptors (consisting of CD25, CD122 and CD132) are internalized upon IL-2 ligation, whereas CD122 and CD132 are degraded while CD25 is recycled back to the plasma membrane.Citation54–Citation56 In summary, hyper-secretion of IL-2 is correlated with the upregulation of IL-2 receptors on Cbl-b KO CD4+ T cells and may play an important role in rendering T cells refractory to Treg suppression.
IL-2 signaling is critical for the ability of Cbl-b KO CD4+FoxP3− T cells to counteract the suppressive effects of Treg cells
To further explore the possibility that Cbl-b KO CD4+ T cells overcome Treg suppression through increased IL-2 production and CD25 expression, we directly examined CD25 expression on T cells and Treg cells at different time-points throughout the suppression assay. First, the levels of CD25 expression on CD4+FoxP3+ and CD4+FoxP3− T cells were compared on day 0, 1 and 3 post-stimulation. Even in the presence of Treg cells, Cbl-b KO CD4+ T cells expressed a markedly higher level of CD25 on day 1 and 3 post-stimulation, but were still lower in comparison to Treg cells (). It was worth noting that on day 3, CD25 expression of Cbl-b KO CD4+ effector T cells suppressed by Treg cells were still higher than that of C57BL/6 T cells stimulated without Tregs (), demonstrating an enhanced potential for Cbl-b KO CD4+ T cells to capture IL-2 even in the presence of Tregs.
Figure 4. IL-2 signaling in Cbl-b KO CD4+FoxP3− T cells mediate Treg cell resistance
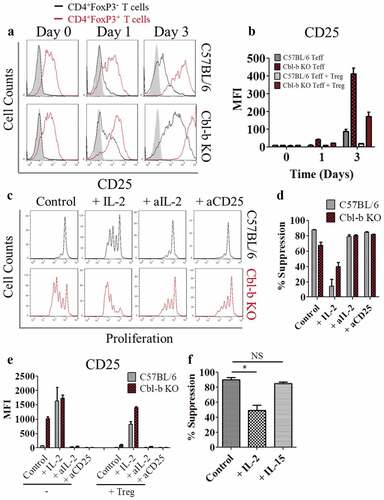
To examine the direct role of IL-2 on the Treg-resistant phenotype observed in Cbl-b KO CD4+ T cells, a Treg suppression assay was performed with the addition of exogenous IL-2, anti–IL-2 or anti-CD25. The addition of IL-2 induced C57BL/6 CD4+ T cell proliferation in the presence of Treg cells, and the addition of anti–IL-2 or anti-CD25 on Cbl-b KO CD4+ effector T cells reversed its hyper-proliferative state (). When quantified for Treg cell resistance, addition of IL-2 to C57BL/6 T cells induced an approximately 5-fold decrease in % suppression. In contrast, the addition of anti–IL-2 or anti-CD25 on Cbl-b KO CD4+ T cells resulted in an increase in % suppression (). As expected, the addition of IL-2 led to an increased CD25 expression on T cells despite the presence of Tregs, whereas blocking IL-2 or CD25 showed the opposite effect (), strengthening the link between the presence of IL-2 and CD25 expression.
To demonstrate the importance of CD25 on the Treg-resistant phenotype, Treg suppression assays were performed on C57BL/6 T cells with either exogenous IL-2 or IL-15 (interacting with CD122 and CD132, but lacking CD25 interaction). IL-15 did not decrease % suppression of C57BL/6 CD4+FoxP3− T cells co-cultured with Treg cells (). In summary, elevated secretion of IL-2 by Cbl-b KO CD4+FoxP3− T cells and the increased CD25 surface-expression by Cbl-b KO T cells serves as an important mechanism that alters the ability of CD4+ T cells to be suppressed by Tregs.
Cbl-b KO CD4+FoxP3− T cells mediate Treg resistance despite Treg expansion
While the overproduction of IL-2 and increased CD25 expression by Cbl-b KO CD4+ T cells played an important role in rendering these T cells refractory to regulation, the overall effect on Treg cells remained to be explored. As such, Treg suppression assays were performed using either C57BL/6 or Cbl-b KO CD4+CD25− T cells with C57BL/6 Treg cells. By gating on CD4+ cells, a higher proportion of CD25+FoxP3+ population was present on day 1 post-stimulation and only a 2-fold decrease in % FoxP3+ T cells were observed with Cbl-b KO CD4+ T cells on day 3 post-stimulation (). Despite the inability for C57BL/6 Tregs to proliferate in vitro, Cbl-b KO CD4+ T cells were able to promote the expansion of Treg cells in the suppression assay (). Three days post-stimulation, co-culture of C57BL/6 Treg cells with Cbl-b KO CD4+ T cells led to increased levels of CD25, FoxP3 and CTLA-4 on Treg cells () which are correlated with enhanced Treg function. Finally, we performed Treg suppression assays with either IL-2 or IL-2 signaling blockers (anti–IL-2 or anti-CD25) and monitored the phenotype of Treg cells. The addition of IL-2 also increased CD25 expression on FoxP3+ T cells, and the use of IL-2 blockers decreased CD25 expression on Treg cells (). Treg suppression assay using IL-2 pre-stimulated wild-type Treg cells demonstrated comparable suppressive function to pre-stimulated Treg cells without IL-2 (). Despite an IL-2-induced Treg expansion during the co-culture, Cbl-b KO CD4+ T cells were still capable of hyper-proliferating. These assays showed that the effect of Cbl-b deficient CD4+ T cells was primarily exerted on T cells without disrupting the function of Treg cells.
Figure 5. IL-2 secretion by Cbl-b KO CD4+FoxP3− T cells positively contribute to Treg activities
IL-2 serves as the central cytokine in mediating Treg cell-resistance
In addition to IL-2, Cbl-b KO CD4+ T cells secreted other cytokines including IFN-γ, IL-4, IL-17A, and IL-10 (). Since several reports have highlighted the role of cytokines such as IL-6 in rendering T cells refractory to the suppressive function of Tregs,Citation16,Citation57–Citation59 we explored whether other cytokines could also modulate Treg resistance. First, co-cultures were set up including C57BL/6 CD4+ T cells and C57BL/6 Treg cells, supplemented with 14 independent cytokines to determine whether these cytokines could inhibit Treg cell mediated suppression. The cytokines used for this Treg suppression assay were selected from those that were hyper-secreted by Cbl-b KO CD4+ T cells, or were previously implicated in Treg resistance. Along with % suppression, CD4+FoxP3+ and CD4+FoxP3− cells from each condition were analyzed for expression of activation markers (). The addition of IL-2 or IL-4 to the co-culture demonstrated the strongest capacity to block the ability of Tregs to inhibit proliferation, as well as the highest expression of CD25 and GITR on effector T cells (). Beyond IL-2 and IL-4, other cytokines that reduced % suppression by approximately 2-fold or more included: IFN-α, IL-6, IL-7, IL-10 and IL-18 (). It is important to note that most of these cytokines had a stimulatory effect on the CD4+ effector T cells (), potentially inducing higher IL-2 secretion for subsequent Treg resistance. To test whether these cytokines mediated Treg resistance through indirect IL-2 upregulation by T cells, we collected supernatants from the Treg suppression assay performed in . We then correlated the influence of each exogenous cytokine on % suppression with either IL-2 secretion or CD25 surface expression by effector T cells. Percent suppression achieved by individual cytokines negatively correlated with IL-2 secretion by T cells, where cytokines such as IL-6 induced high levels of IL-2 secretion (). Similarly, % suppression quantified for individual cytokines also negatively correlated with CD25 expression on effector T cells (). These data suggest that cytokine-induced Treg cell resistance may likely converge on the amplification of IL-2 production and signaling for its in vitro phenotype.
Figure 6. IL-2 serves as a central cytokine responsible for CD4+FoxP3- T cell resistance against Tregs
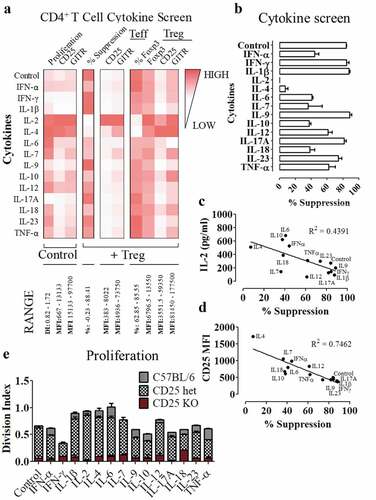
To directly confirm whether these cytokines depend on IL-2 signaling to mediate the observed phenotype, naïve C57BL/6 or CD25 KO CD4+ T cells were stimulated with the cytokines used in . In the absence of CD25 expression, majority of the cytokines were insufficient to induce CD4+ T cell proliferation (). When stained with viability dye on day 3 post-stimulation without exogenous cytokines, C57BL/6 CD4+ effector T cells had a higher viability of 78%, while approximately 48% of CD25 KO CD4+ T cells were still viable (data not shown). Altogether, these data suggest that while other cytokines could be used to confer resistance against regulatory T cells, the cytokine networks tested here relied on IL-2 as the central molecule to mediate Treg resistance.
Discussion
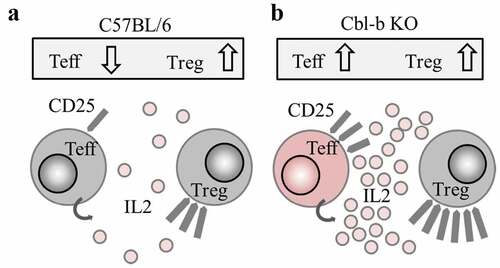
Recently, a genome-wide CRISPR screen of primary human T cells has identified Cbl-b as the top candidate to enhance T cell activation, proliferation and tumor cell killing in vitro.Citation60 This is consistent with previous findings which identify Cbl-b as an important gene regulating autoimmunity and anti-tumor immunity in mice.Citation46,Citation47,Citation53 In this study, we focused on CD4+ T cells and explored the mechanism of Cbl-b in mediating T cell resistance to Treg cells. We show that hyper-secretion of IL-2 by Cbl-b KO CD4+ T cells and upregulation of CD25 increase effector T cell exposure and sensitivity to IL-2 (,). Increased exposure to IL-2 signaling then counteracts the previously established ability of Treg cells in sequestering IL-2 required for effector T cell proliferation and survival.Citation15,Citation61 Furthermore, while other cytokines can mediate Treg cell resistance, we found that IL-2 served as the central molecule essential for cytokine-driven Treg resistance.
Figure 7. Proposed mechanism: Cbl-b KO CD4+ T cells promote Treg resistance through enhanced IL-2 production
Previous studies have highlighted the importance of other T cell activating cytokines including IL-4, IL-6, and IL-7 in generating resistance to Treg-mediated suppression.Citation16,Citation19,Citation21,Citation23 It is possible that many of these cytokines can induce resistance to the suppressive effects of Treg cells (), because these cytokines also share overlapping signaling pathways with IL-2 such the JAK/STAT and PI3 K/Akt pathways. While the additional exposure to T cell activating cytokines such as IL-6 was sufficient in mediating a robust T cell response, as well as Treg resistance, they also triggered the release of IL-2 and were unable to induce proliferation in the absence of CD25 (,). Although these cytokines will likely exert distinct effects on T cell activation, differentiation and survival, their mechanism of resistance to Tregs depend on the ability of CD4+ T cells to process IL-2 signaling. The precise signaling pathway responsible for this phenotype is currently unknown, but it has been proposed that the PI3 K/Akt signaling could serve as a convergence point of numerous cytokine signaling pathways to mediate Treg resistance.Citation27 However, the cytokines implicated in Treg resistance such as IL-4, IL-6, and IL-7, which were also capable of inducing PI3 K signaling, were heavily dependent on IL-2 (). Thus, the PI3 K/Akt signaling pathway itself may not be sufficient to explain the effects of IL-2, and may act in conjunction with other components of the IL-2 signaling pathway to mediate Treg resistance. Here, we highlight the importance of IL-2 as a crucial cytokine required to induce CD4+ T cell resistance to Treg cells.
While Cbl-b deficiency renders T cells resistant to Treg-mediated suppression, we found that Treg activities were also enhanced (). Our observation that Treg expansion occurred under conditions for Treg resistance is novel, but perhaps not unexpected. The role of IL-2 in Treg expansion has been reported in numerous studies, where systemic administration of IL-2 increased Treg activities.Citation62–Citation64 Because IL-2R signaling is crucial for STAT5-driven Treg cell function,Citation52,Citation65 and Cbl-b KO CD4+ T cells confer resistance through overproduction of IL-2, Treg cell expansion is indeed inevitable. Consistent with our model, when TC1 or EL4 exogenous tumor cells were transplanted into the flank of C57BL/6 or Cbl-b KO mice, Cbl-b deficient mice displayed higher tumor-infiltrating Tregs despite the increased anti-tumor immune response and overall survival.Citation46,Citation47 Although enhanced quality and quantity of Treg cells are worth noting, the overall immunological phenotype is dependent on the balance between inhibitory and activating mechanisms, not strictly based on the number of cells.
Clinically, IL-2 served as the first effective immune therapy against human cancer.Citation66 Earlier studies involving systemic administration of IL-2 demonstrated promising clinical responses in patients with metastatic melanoma and renal cancer, resulting in durable, complete and curative regressions.Citation66–Citation68 Some of these initial studies stimulated substantial activity evaluating the therapeutic potential of IL-2, despite the known toxicity.Citation66 To date, IL-2 has been investigated for diverse applications including monotherapy, combination therapy, and ex vivo expansion of T cells for cell therapy.Citation66,Citation69,Citation70
However, the role of IL-2 as therapy in disease has been controversial. Numerous studies have shown that low dose IL-2 preferentially expand Treg cells for the treatment of autoimmune diseases,Citation70–Citation72 whereas high dose IL-2 expands Tregs while also enabling anti-tumor immunity.Citation69,Citation73,Citation74 Sim et al. (2014) demonstrated that patients with metastatic melanoma treated with high-dose recombinant IL-2 (720,000 IU/kg; Proleukin, Novartis) elevates the number of ICOS+ Treg cells for both responders and non-responders. In the study, those with higher frequency of ICOS+ Treg cells remaining in blood post high-dose IL-2 therapy had worse clinical outcomes than those with lower percentage of ICOS+ Treg cells.Citation75 Similarly, Yao et al. (2012) performed nonmyeloablative chemotherapy followed by tumor infiltrating lymphocyte (TIL) transfer and IL-2 administration, and suggest that the proportion of reconstituted CD4+FoxP3+ Treg cells in blood after the treatment negatively correlated to responsiveness to the therapy.Citation76 Adoptive T cell therapies using TILs routinely include IL-2.Citation77–Citation83 However, controversies surrounding its ability to promote T cell survival versus the detrimental effects of Treg cell expansion has been unclear. Our studies suggest that if patients are treated with TIL and high dose IL-2, it may lead to the expansion of Treg cells due to the presence of high dose IL-2 but this may not necessarily be detrimental if it occurs together with the development of resistance to the inhibitory effects of Treg cells.
Alternatively, recently proposed strategies involve preferential expansion of T cells over Treg cells using IL-2-antibody complexes or modified IL-2 biologics.Citation15,Citation84–Citation88 For example, Bempegaldesleukin (NKTR-214), which consists of an IL-2 core conjugated to 6 releasable polyethylene glycol chains, preferentially bind to IL-2Rβ over IL-2Rα which induce greater T cell to Treg cell ratio and a more robust anti-tumor immunity in comparison to Aldesleukin.Citation87 Similarly, conjugating anti–IL-2 with IL-2 selectively enhance T cell performance to induce stronger anti-tumor immunity.Citation86,Citation88 These strategies may be more effective in comparison to conventional IL-2 usage, however most of these still require validation in a clinical setting.
In summary, our studies have defined a mechanism to render T cells refractory to the inhibitory effects of Tregs. We also highlight the potential in promoting an immune response by IL-2 overproduction through targeting Cbl-b or other related signaling pathways within CD4+ T cells. Such a strategy could potentially be incorporated in many treatment modalities including the Chimeric Antigen Receptor (CAR) -T cell therapy or TCR-engineering based immunotherapy to generate robust effector T cell responses and subsequent anti-tumor immunity.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Author Contributions
SH conceived and performed experiments, managed the research project and wrote the manuscript. DCC, MSP, ZQL and CGB performed experiments. ARE provided reagents. CWT, LC and PSO provided expertise and feedback. PSO secured funding and supervised the project.
Acknowledgments
This research was supported by a Foundation Grant from Canadian Institute for Health Research (CIHR) to PSO. We thank the Animal Resource Centre as well as Princess Margaret Cancer Centre flow facility for technical support. Special acknowledgements are made for Dr. Tak Mak and Dr. Naoto Hirano for their suggestions and insights for this project. The authors declare no competing financial interests.
Additional information
Funding
References
- Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. 2008;133(5):775–15. doi:10.1016/j.cell.2008.05.009.
- Sakaguchi S, Miyara M, Costantino CM, Hafler DA. FOXP3+ regulatory T cells in the human immune system. Nat Rev Immunol. 2010;10(7):490–500. doi:10.1038/nri2785.
- Lyon MF, Peters J, Glenister PH, Ball S, Wright E. The scurfy mouse mutant has previously unrecognized hematological abnormalities and resembles Wiskott-Aldrich syndrome. Proc Natl Acad Sci U S A. 1990;87:2433–2437.
- Clark LB, Appleby MW, Brunkow ME, Wilkinson JE, Ziegler SF, Ramsdell F. Cellular and molecular characterization of the scurfy mouse mutant. J Immunol. 1999;162:2546–2554.
- Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, Kelly TE, Saulsbury FT, Chance PF, Ochs HD. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet. 2001;27(1):20–21. doi:10.1038/83713.
- Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko S-A, Wilkinson JE, Galas D, Ziegler SF, Ramsdell F. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet. 2001;27(1):68–73. doi:10.1038/83784.
- Wildin RS, Ramsdell F, Peake J, Faravelli F, Casanova J-L, Buist N, Levy-Lahad E, Mazzella M, Goulet O, Perroni L, et al. X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat Genet. 2001;27(1):18–20. doi:10.1038/83707.
- Onizuka S, Tawara I, Shimizu J, Sakaguchi S, Fujita T, Nakayama E. Tumor rejection by in vivo administration of anti-CD25 (interleukin-2 receptor alpha) monoclonal antibody. Cancer Res. 1999;59:3128–3133.
- Shimizu J, Yamazaki S, Sakaguchi S. Induction of Tumor Immunity by Removing CD25+CD4+ T Cells: A Common Basis Between Tumor Immunity and Autoimmunity. J Immunol. 1999;163:5211–5218.
- Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10(9):942–949. doi:10.1038/nm1093.
- Klages K, Mayer CT, Lahl K, Loddenkemper C, Teng MWL, Ngiow SF, Smyth MJ, Hamann A, Huehn J, Sparwasser T. Selective Depletion of Foxp3+ Regulatory T Cells Improves Effective Therapeutic Vaccination against Established Melanoma. Cancer Res. 2010;70(20):7788–7799. doi:10.1158/0008-5472.CAN-10-1736.
- Nishikawa H, Sakaguchi S. Regulatory T cells in tumor immunity. Int J Cancer. 2010;127(4):759–767. doi:10.1002/ijc.25429.
- Arce Vargas F, Furness AJS, Solomon I, Joshi K, Mekkaoui L, Lesko MH, Miranda Rota E, Dahan R, Georgiou A, Sledzinska A, et al. Fc-Optimized Anti-CD25 Depletes Tumor-Infiltrating Regulatory T Cells and Synergizes with PD-1 Blockade to Eradicate Established Tumors. Immunity. 2017;46(4):577–586. doi:10.1016/j.immuni.2017.03.013.
- Josefowicz SZ, Lu L-F, Rudensky AY. Regulatory T Cells: mechanisms of Differentiation and Function. Annu Rev Immunol. 2012;30(1):531–564. doi:10.1146/annurev.immunol.25.022106.141623.
- Han S, Toker A, Liu ZQ, Ohashi PS. Turning the Tide Against Regulatory T Cells. Front Oncol. 2019;9:279. doi:10.3389/fonc.2019.00279.
- Pasare C. Toll Pathway-Dependent Blockade of CD4+CD25+ T Cell-Mediated Suppression by Dendritic Cells. Science. 2003;299(5609):1033–1036. doi:10.1126/science.1078231.
- Ji H, Liao G, Faubion WA, Abadía-Molina AC, Cozzo C, Laroux FS, Caton A, Terhorst C. Cutting edge: the natural ligand for glucocorticoid-induced TNF receptor-related protein abrogates regulatory T cell suppression. J Immunol. 2004;172(10):5823–5827. doi:10.4049/JIMMUNOL.172.10.5823.
- Stephens GL, McHugh RS, Whitters MJ, Young DA, Luxenberg D, Carreno BM, Collins M, Shevach EM. Engagement of glucocorticoid-induced TNFR family-related receptor on effector T cells by its ligand mediates resistance to suppression by CD4+CD25+ T cells. J Immunol. 2004;173(8):5008–5020. doi:10.4049/jimmunol.173.8.5008.
- Ruprecht CR, Gattorno M, Ferlito F, Gregorio A, Martini A, Lanzavecchia A, Sallusto F. Coexpression of CD25 and CD27 identifies FoxP3+ regulatory T cells in inflamed synovia. J Exp. 2005;201(11):1793–1803. doi:10.1084/jem.20050085.
- O’Sullivan BJ, Thomas HE, Pai S, Santamaria P, Iwakura Y, Steptoe RJ, Kay TWH, Thomas R. IL-1β Breaks Tolerance through Expansion of CD25 + Effector T Cells. J Immunol. 2006;176(12):7278–7287. doi:10.4049/JIMMUNOL.176.12.7278.
- Pace L, Rizzo S, Palombi C, Brombacher F, Doria G. Cutting Edge: IL-4-Induced Protection of CD4+CD25 Th Cells from CD4+CD25+Regulatory T Cell-Mediated Suppression. J Immunol. 2006;176(7):3900–3904. doi:10.4049/JIMMUNOL.176.7.3900.
- Peluso I, Fantini MC, Fina D, Caruso R, Boirivant M, MacDonald TT, Pallone F, Monteleone G. IL-21 counteracts the regulatory T cell-mediated suppression of human CD4+ T lymphocytes. J Immunol. 2007;178(2):732–739. doi:10.4049/JIMMUNOL.178.2.732.
- Wan S, Xia C, Morel L. IL-6 produced by dendritic cells from lupus-prone mice inhibits CD4+CD25+ T cell regulatory functions. J Immunol. 2007;178(1):271–279. doi:10.4049/jimmunol.178.1.271.
- Ben Ahmed M, Belhadj Hmida N, Moes N, Buyse S, Abdeladhim M, Louzir H, Cerf-Bensussan N. IL-15 renders conventional lymphocytes resistant to suppressive functions of regulatory T cells through activation of the phosphatidylinositol 3-kinase pathway. J Immunol. 2009;182(11):6763–6770. doi:10.4049/jimmunol.0801792.
- Chen X, Das R, Komorowski R, Beres A, Hessner MJ, Mihara M, Drobyski WR. Blockade of interleukin-6 signaling augments regulatory T-cell reconstitution and attenuates the severity of graft-versus-host disease. Blood. 2009;114(4):891–900. doi:10.1182/blood-2009-01-197178.
- Wohlfert EA, Clark RB. ‘Vive la Résistance!’ – the PI3K–Akt pathway can determine target sensitivity to regulatory T cell suppression. Trends Immunol. 2007;28(4):154–160. doi:10.1016/j.it.2007.02.003.
- Mercadante ER, Lorenz UM. Breaking free of control: how conventional T cells overcome regulatory T cell suppression. Front Immunol. 2016;193(7):1–16. doi:10.3389/fimmu.2016.00193.
- Wohlfert EA, Callahan MK, Clark RB. Resistance to CD4+CD25+ regulatory T cells and TGF-beta in Cbl-b-/- mice. J Immunol. 2004;173(2):1059–1065. doi:10.4049/jimmunol.173.2.1059.
- King CG, Kobayashi T, Cejas PJ, Kim T, Yoon K, Kim GK, Chiffoleau E, Hickman SP, Walsh PT, Turka LA, et al. TRAF6 is a T cell–intrinsic negative regulator required for the maintenance of immune homeostasis. Nat Med. 2006;12(9):1088–1092. doi:10.1038/nm1449.
- Wohlfert EA, Gorelik L, Mittler R, Flavell RA, Clark RB. Cutting edge: deficiency in the E3 Ubiquitin Ligase Cbl-b results in a multifunctional defect in T cell TGF-beta sensitivity In Vitro and In Vivo. J Immunol. 2006;176(3):1316–1320. doi:10.4049/jimmunol.176.3.1316.
- Chang M, Lee AJ, Fitzpatrick L, Zhang M, Sun S-C. NF- B1 p105 regulates T cell homeostasis and prevents chronic inflammation. J Immunol. 2009;182(5):3131–3138. doi:10.4049/jimmunol.0803637.
- Stahl HF, Fauti T, Ullrich N, Bopp T, Kubach J, Rust W, Labhart P, Alexiadis V, Becker C, Hafner M, et al. miR-155 inhibition sensitizes CD4+ Th cells for TREG mediated suppression Unutmaz D, editor. PLoS One. 2009;4(9):e7158. doi:10.1371/journal.pone.0007158.
- Adams CO, Housley WJ, Bhowmick S, Cone RE, Rajan TV, Forouhar F, Clark RB. Cbl-b(-/-) T cells demonstrate in vivo resistance to regulatory T cells but a context-dependent resistance to TGF-beta. J Immunol. 2010;185(4):2051–2058. doi:10.4049/jimmunol.1001171.
- Schenten D, Nish SA, Yu S, Yan X, Lee HK, Brodsky I, Pasman L, Yordy B, Wunderlich FT, Brüning JC, et al. Signaling through the adaptor molecule MyD88 in CD4+ T cells is required to overcome suppression by regulatory T cells. Immunity. 2014;40(1):78–90. doi:10.1016/j.immuni.2013.10.023.
- Mercadante ER, Lorenz UM. T cells deficient in the tyrosine phosphatase SHP-1 resist suppression by regulatory T cells. J Immunol. 2017;199(1):129–137. doi:10.4049/jimmunol.1602171.
- Liu Y-C. Ubiquitin ligases and the immune response. Annu Rev Immunol. 2004;22(1):81–127. doi:10.1146/annurev.immunol.22.012703.104813.
- Paolino M, Penninger JM. Cbl-b in T-cell activation. Semin Immunopathol. 2010;32(2):137–148. doi:10.1007/s00281-010-0197-9.
- Bachmaier K, Krawczyk C, Kozieradzki I, Kong -Y-Y, Sasaki T, Oliveira-dos-santos A, Mariathasan S, Bouchard D, Wakeham A, Itie A, et al. Negative regulation of lymphocyte activation and autoimmunity by the molecular adaptor Cbl-b. Nature. 2000;403(6766):211–216. doi:10.1038/35003228.
- Nakao A, Afrakhte M, Morn A, Nakayama T, Christian JL, Heuchel R, Itoh S, Kawabata M, Heldin N-E, Heldin C-H, et al. Identification of Smad7, a TGFβ-inducible antagonist of TGF-β signalling. Nature. 1997;389(6651):631–635. doi:10.1038/39369.
- Gruber T, Hinterleitner R, Hermann-Kleiter N, Meisel M, Kleiter I, Wang CM, Viola A, Pfeifhofer-Obermair C, Baier G. Cbl-b mediates TGFβ sensitivity by downregulating inhibitory SMAD7 in primary T cells. J Mol Cell Biol. 2013;5(6):358–368. doi:10.1093/jmcb/mjt017.
- Fantini MC, Rizzo A, Fina D, Caruso R, Sarra M, Stolfi C, Becker C, MacDonald TT, Pallone F, Neurath MF, et al. Smad7 controls resistance of colitogenic T cells to regulatory T cell-mediated suppression. Gastroenterology. 2009;136(4):1308–1316.e3. doi:10.1053/j.gastro.2008.12.053.
- Oderup C, Cederbom L, Makowska A, Cilio CM, Ivars F. Cytotoxic T lymphocyte antigen-4-dependent down-modulation of costimulatory molecules on dendritic cells in CD4+ CD25+ regulatory T-cell-mediated suppression. Immunology. 2006;118(2):240–249. doi:10.1111/j.1365-2567.2006.02362.x.
- Onishi Y, Fehervari Z, Yamaguchi T, Sakaguchi S. Foxp3+ natural regulatory T cells preferentially form aggregates on dendritic cells in vitro and actively inhibit their maturation. Proc Natl Acad Sci U S A. 2008;105(29):10113–10118. doi:10.1073/pnas.0711106105.
- Chiang YJ, Kole HK, Brown K, Naramura M, Fukuhara S, Hu R-J, Jang IK, Gutkind JS, Shevach E, Gu H. Cbl-b regulates the CD28 dependence of T-cell activation. Nature. 2000;403(6766):216–220. doi:10.1038/35003235.
- Jeon M-S, Atfield A, Venuprasad K, Krawczyk C, Sarao R, Elly C, Yang C, Arya S, Bachmaier K, Su L, et al. Essential role of the E3 ubiquitin ligase Cbl-b in T cell anergy induction. Immunity. 2004;21(2):167–177. doi:10.1016/j.immuni.2004.07.013.
- Chiang JY, Jang IK, Hodes R, Gu H, Boon T, Houghton AN, Gold JS, Blachere NE, Rosenberg SA, Rosenberg SA, et al. Ablation of Cbl-b provides protection against transplanted and spontaneous tumors. J Clin Invest. 2007;117(4):1029–1036. doi:10.1172/JCI29472.
- Loeser S, Loser K, Bijker MS, Rangachari M, van der Burg SH, Wada T, Beissert S, Melief CJM, Penninger JM. Spontaneous tumor rejection by cbl-b–deficient CD8+ T cells. J Exp Med. 2007;204(4):879–891. doi:10.1084/jem.20061699.
- McMurchy AN, Levings MK. Suppression assays with human T regulatory cells: A technical guide. Eur J Immunol. 2012;42(1):27–34. doi:10.1002/eji.201141651.
- Stromnes IM, Blattman JN, Tan X, Jeevanjee S, Gu H, Greenberg PD. Abrogating Cbl-b in effector CD8(+) T cells improves the efficacy of adoptive therapy of leukemia in mice. J Clin Invest. 2010;120(10):3722–3734. doi:10.1172/JCI41991.
- Pandiyan P, Zheng L, Ishihara S, Reed J, Lenardo MJ. CD4+CD25+Foxp3+ regulatory T cells induce cytokine deprivation–mediated apoptosis of effector CD4+ T cells. Nat Immunol. 2007;8(12):1353–1362. doi:10.1038/ni1536.
- McNally A, Hill GR, Sparwasser T, Thomas R, Steptoe RJ. CD4+CD25+ regulatory T cells control CD8+ T-cell effector differentiation by modulating IL-2 homeostasis. Proc Natl Acad Sci U S A. 2011;108(18):7529–7534. doi:10.1073/pnas.1103782108.
- Chinen T, Kannan AK, Levine AG, Fan X, Klein U, Zheng Y, Gasteiger G, Feng Y, Fontenot JD, Rudensky AY. An essential role for the IL-2 receptor in Treg cell function. Nat Immunol. 2016;17(11):1322–1333. doi:10.1038/ni.3540.
- Gronski MA, Boulter JM, Moskophidis D, Nguyen LT, Holmberg K, Elford AR, Deenick EK, Kim HO, Penninger JM, Odermatt B, et al. TCR affinity and negative regulation limit autoimmunity. Nat Med. 2004;10(11):1234–1239. doi:10.1038/nm1114.
- Hémar A, Subtil A, Lieb M, Morelon E, Hellio R, Dautry-Varsat A. Endocytosis of interleukin 2 receptors in human T lymphocytes: distinct intracellular localization and fate of the receptor alpha, beta, and gamma chains. J Cell Biol. 1995;129(1):55–64. doi:10.1083/JCB.129.1.55.
- Yu A, Malek TR. The proteasome regulates receptor-mediated endocytosis of interleukin-2. J Biol Chem. 2001;276(1):381–385. doi:10.1074/jbc.M007991200.
- Su EW, Moore CJ, Suriano S, Johnson CB, Songalia N, Patterson A, Neitzke DJ, Andrijauskaite K, Garrett-Mayer E, Mehrotra S, et al. IL-2Rα mediates temporal regulation of IL-2 signaling and enhances immunotherapy. Sci Transl Med. 2015;7(311):1–19. doi:10.1126/scitranslmed.aac8155.
- Goodman WA, Levine AD, Massari JV, Sugiyama H, McCormick TS, Cooper KD. IL-6 signaling in psoriasis prevents immune suppression by regulatory T cells. J Immunol. 2009;183(5):3170–3176. doi:10.4049/jimmunol.0803721.
- Goodman WA, Young AB, McCormick TS, Cooper KD, Levine AD. Stat3 phosphorylation mediates resistance of primary human T cells to regulatory T cell suppression. J Immunol. 2011;186(6):3336–3345. doi:10.4049/jimmunol.1001455.
- Schneider A, Long SA, Cerosaletti K, Ni CT, Samuels P, Kita M, Buckner JH. In active relapsing-remitting multiple sclerosis, effector T cell resistance to adaptive T(regs) involves IL-6-mediated signaling. Sci Transl Med. 2013;5(170):1–15. doi:10.1126/scitranslmed.3004970.
- Shifrut E, Carnevale J, Tobin V, Diolaiti ME, Ashworth A, Marson Correspondence A. Genome-wide CRISPR screens in primary human T cells reveal key regulators of immune function. Cell. 2018;175(1958–1971.e15). doi:10.1016/j.cell.2018.10.024.
- Thornton AM, Donovan EE, Piccirillo CA, Shevach EM. Cutting edge: IL-2 is critically required for the in vitro activation of CD4+CD25+ T cell suppressor function. J Immunol. 2004;172(11):6519–6523. doi:10.4049/JIMMUNOL.172.11.6519.
- Zhang H, Chua KS, Guimond M, Kapoor V, Brown MV, Fleisher TA, Long LM, Bernstein D, Hill BJ, Douek DC, et al. Lymphopenia and interleukin-2 therapy alter homeostasis of CD4+CD25+ regulatory T cells. Nat Med. 2005;11(11):1238–1243. doi:10.1038/nm1312.
- Zorn E, Nelson EA, Mohseni M, Porcheray F, Kim H, Litsa D, Bellucci R, Raderschall E, Canning C, Soiffer RJ, et al. IL-2 regulates FOXP3 expression in human CD4+CD25+ regulatory T cells through a STAT-dependent mechanism and induces the expansion of these cells in vivo. Blood. 2006;108(5):1571–1579. doi:10.1182/blood-2006-02-004747.
- Lemoine FM, Cherai M, Giverne C, Dimitri D, Rosenzwajg M, Trebeden-Negre H, Chaput N, Barrou B, Thioun N, Gattegnio B, et al. Massive expansion of regulatory T-cells following interleukin 2 treatment during a phase I-II dendritic cell-based immunotherapy of metastatic renal cancer. Int J Oncol. 2009;35(3):569–581.
- Antov A, Yang L, Vig M, Baltimore D, Van Parijs L. Essential role for STAT5 signaling in CD25+CD4+ regulatory T cell homeostasis and the maintenance of self-tolerance. J Immunol. 2003;171:3435–3441.
- Rosenberg SA. IL-2: the first effective immunotherapy for human cancer. J Immunol. 2014;192(12):5451–5458. doi:10.4049/jimmunol.1490019.
- Rosenberg SA, Yang JC, Topalian SL, Schwartzentruber DJ, Weber JS, Parkinson DR, Seipp CA, Einhorn JH, White DE. Treatment of 283 consecutive patients with metastatic melanoma or renal cell cancer using high-dose bolus interleukin 2. JAMA. 1994;271:907–913.
- Lotze MT, Chang AE, Seipp CA, Simpson C, Vetto JT, Rosenberg SA. High-dose recombinant interleukin 2 in the treatment of patients with disseminated cancer. JAMA. 1986;256(22):3117. doi:10.1001/jama.1986.03380220083027.
- Spolski R, Li P, Leonard WJ. Biology and regulation of IL-2: from molecular mechanisms to human therapy. Nat Rev Immunol. 2018;18(10):648–659. doi:10.1038/s41577-018-0046-y.
- Abbas AK, Trotta E, Simeonov DR, Marson A, Bluestone JA. Revisiting IL-2: biology and therapeutic prospects. Sci Immunol. 2018;3(25). doi:10.1126/sciimmunol.aat1482.
- Klatzmann D, Abbas AK. The promise of low-dose interleukin-2 therapy for autoimmune and inflammatory diseases. Nat Rev Immunol. 2015;15(5):283–294. doi:10.1038/nri3823.
- Hirakawa M, Matos TR, Liu H, Koreth J, Kim HT, Paul NE, Murase K, Whangbo J, Alho AC, Nikiforow S, et al. Low-dose IL-2 selectively activates subsets of CD4+ Tregs and NK cells. JCI Insight. 2016;1(18):e89278. doi:10.1172/jci.insight.89278.
- Ahmadzadeh M, Rosenberg SA. IL-2 administration increases CD4+ CD25(hi) Foxp3+ regulatory T cells in cancer patients. Blood. 2006;107(6):2409–2414. doi:10.1182/blood-2005-06-2399.
- Cesana GC, DeRaffele G, Cohen S, Moroziewicz D, Mitcham J, Stoutenburg J, Cheung K, Hesdorffer C, Kim-Schulze S, Kaufman HL. Characterization of CD4+CD25+ regulatory T cells in patients treated with high-dose interleukin-2 for metastatic melanoma or renal cell carcinoma. J Clin Oncol. 2006;24(7):1169–1177. doi:10.1200/JCO.2005.03.6830.
- Sim GC, Martin-Orozco N, Jin L, Yang Y, Wu S, Washington E, Sanders D, Lacey C, Wang Y, Vence L, et al. IL-2 therapy promotes suppressive ICOS+ Treg expansion in melanoma patients. J Clin Invest. 2014;124(1):99–110. doi:10.1172/JCI46266.
- Yao X, Ahmadzadeh M, Lu Y-C, Liewehr DJ, Dudley ME, Liu F, Schrump DS, Steinberg SM, Rosenberg SA, Robbins PF, et al. Levels of peripheral CD4(+)FoxP3(+) regulatory T cells are negatively associated with clinical response to adoptive immunotherapy of human cancer. Blood. 2012;119(24):5688–5696. doi:10.1182/blood-2011-10-386482.
- Dudley ME, Yang JC, Sherry R, Hughes MS, Royal R, Kammula U, Robbins PF, Huang J, Citrin DE, Leitman SF, et al. Adoptive cell therapy for patients with metastatic melanoma: evaluation of intensive myeloablative chemoradiation preparative regimens. J Clin Oncol. 2008;26(32):5233–5239. doi:10.1200/JCO.2008.16.5449.
- Besser MJ, Shapira-Frommer R, Treves AJ, Zippel D, Itzhaki O, Hershkovitz L, Levy D, Kubi A, Hovav E, Chermoshniuk N, et al. Clinical responses in a phase II study using adoptive transfer of short-term cultured tumor infiltration lymphocytes in metastatic melanoma patients. Clin Cancer Res. 2010;16(9):2646–2655. doi:10.1158/1078-0432.CCR-10-0041.
- Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, Citrin DE, Restifo NP, Robbins PF, Wunderlich JR, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res. 2011;17(13):4550–4557. doi:10.1158/1078-0432.CCR-11-0116.
- Pilon-Thomas S, Kuhn L, Ellwanger S, Janssen W, Royster E, Marzban S, Kudchadkar R, Zager J, Gibney G, Sondak VK, et al. Efficacy of adoptive cell transfer of tumor-infiltrating lymphocytes after lymphopenia induction for metastatic melanoma. J Immunother. 2012;35(8):615–620. doi:10.1097/CJI.0b013e31826e8f5f.
- Radvanyi LG, Bernatchez C, Zhang M, Fox PS, Miller P, Chacon J, Wu R, Lizee G, Mahoney S, Alvarado G, et al. Specific lymphocyte subsets predict response to adoptive cell therapy using expanded autologous tumor-infiltrating lymphocytes in metastatic melanoma patients. Clin Cancer Res. 2012;18(24):6758–6770. doi:10.1158/1078-0432.CCR-12-1177.
- Andersen R, Donia M, Ellebaek E, Borch TH, Kongsted P, Iversen TZ, Hölmich LR, Hendel HW, Met Ö, Andersen MH, et al. Long-lasting complete responses in patients with metastatic melanoma after adoptive cell therapy with tumor-infiltrating lymphocytes and an attenuated IL2 regimen. Clin Cancer Res. 2016;22(15):3734–3745. doi:10.1158/1078-0432.CCR-15-1879.
- Nguyen LT, Saibil SD, Sotov V, Le MX, Khoja L, Ghazarian D, Bonilla L, Majeed H, Hogg D, Joshua AM, et al. Phase II clinical trial of adoptive cell therapy for patients with metastatic melanoma with autologous tumor-infiltrating lymphocytes and low-dose interleukin-2. Cancer Immunol Immunother. 2019 Feb;11:1–13. doi:10.1007/s00262-019-02307-x.
- Levin AM, Bates DL, Ring AM, Krieg C, Lin JT, Su L, Moraga I, Raeber ME, Bowman GR, Novick P, et al. Exploiting a natural conformational switch to engineer an interleukin-2 “superkine”. Nature. 2012;484(7395):529–533. doi:10.1038/nature10975.
- Colbeck EJ, Hindley JP, Smart K, Jones E, Bloom A, Bridgeman H, McPherson RC, Turner DG, Ladell K, Price DA, et al. Eliminating roles for T-bet and IL-2 but revealing superior activation and proliferation as mechanisms underpinning dominance of regulatory T cells in tumors. Oncotarget. 2015;6(28):24649–24659. doi:10.18632/oncotarget.5584.
- Klevorn LE, Berrien-Elliott MM, Yuan J, Kuehm LM, Felock GD, Crowe SA, Teague RM. Rescue of tolerant CD8+ T cells during cancer immunotherapy with IL2: antibodyComplexes. Cancer Immunol Res. 2016;4(12):1016–1026. doi:10.1158/2326-6066.CIR-16-0159.
- Charych DH, Hoch U, Langowski JL, Lee SR, Addepalli MK, Kirk PB, Sheng D, Liu X, Sims PW, VanderVeen LA, et al. NKTR-214, an engineered cytokine with biased IL2 receptor binding, increased tumor exposure, and marked efficacy in mouse tumor models. Clin Cancer Res. 2016;22(3):680–690. doi:10.1158/1078-0432.CCR-15-1631.
- Sockolosky JT, Trotta E, Parisi G, Picton L, Su LL, Le AC, Chhabra A, Silveria SL, George BM, King IC, et al. Selective targeting of engineered T cells using orthogonal IL-2 cytokine-receptor complexes. Science. 2018;359(6379):1037–1042. doi:10.1126/science.aar3246.