
ABSTRACT
UBR5 is a nuclear phosphoprotein of obscure functions. Clinical analyses reveal that UBR5 amplifications and overexpression occur in over 20% cases of human breast cancers. Breast cancer patients carrying UBR5 genetic lesions with overexpression have significantly reduced survival. Experimental work in vitro and in vivo demonstrates that UBR5, functioning as an oncoprotein, plays a profound role in breast cancer growth and metastasis. UBR5 drives tumor growth largely through paracrine interactions with the immune system, particularly through inhibiting the cytotoxic response mediated by CD8+ T lymphocytes, whereas it facilitates metastasis in a tumor cell-autonomous manner via its transcriptional control of key regulators of the epithelial–mesenchymal transition, ID1 and ID3. Furthermore, simultaneous targeting of UBR5 and PD-L1 yields strong therapeutic benefit to tumor-bearing hosts. This work significantly expands our scarce understanding of the pathophysiology and immunobiology of a fundamentally important molecule and has strong implications for the development of novel immunotherapy to treat highly aggressive breast cancers that resist conventional treatment.
Introduction
Breast cancer is the most common malignancy among women in the United States and the second most common cause of mortality among women ages 45 to 55 y.Citation1 Estimated 249,260 new breast cancer cases were identified in the United States in 2016 and the predicted number of deaths is that year was 40,890.Citation1 Despite the development of newer diagnostic methods, selective as well as targeted chemotherapies and their combinations, surgery, hormonal therapy, radiotherapy, breast cancer recurrence, metastasis, and drug resistance are still the major problems for breast cancer. Clearly, more therapeutic modalities are needed for this most common form of cancer diagnosed in women worldwide. In an effort to identify potential “driver” genetic alterations for human breast cancer development and/or pathogenesis, we analyzed the whole-exon sequence data from the surgical tumor samples of a cohort of triple-negative breast cancer (TNBC) patients in comparison to the surrounding normal tissues.Citation2 One of the most strikingly altered genes in over 30% of the specimens that we examined was ubiquitin protein ligase E3 component n-recognin 5 (UBR5, a.k.a., EDD), a member of the rare HECT-domain E3 ubiquitin ligase family.Citation3
The biology of UBR5 is poorly understood. UBR5 is highly conserved in metazoans and is essential for early embryonic development in mice,Citation4,Citation5 indicating its fundamental importance. A recent report demonstrates that UBR5 ubiquitylates CSPP1, a centrosomal and ciliary protein involved in cilia formation, and is required for cytoplasmic organization of CSPP1-comprising centriolar satellites in the centrosomal periphery, suggesting that UBR5-mediated ubiquitylation of CSPP1 or associated centriolar satellite constituents is one underlying requirement for cilia expression.Citation6 Another report indicates that as a modulator of super-vigilant proteostasis UBR5 suppresses proteostasis collapse in pluripotent stem cells from Huntington’s disease patients.Citation7 Human UBR5 was originally identified in a screen for progestin-regulated genes in breast cancer cells.Citation3 It is rarely mutated in healthy somatic tissues but is mutated and overexpressed in many major cancers.Citation8 However, the causality, activities, and mechanisms of UBR5’s tumorigenic activities and the associated genetic lesions had not been established. Through genetic, cellular, and molecular manipulations in mouse models, we first uncovered a distinctive and profound role of UBR5 in the aggression of an experimental TNBC model.Citation2 Further, we found that UBR5’s tumorigenic activities are exerted paracrine mainly through its interaction with the adaptive immune apparatus, whereas its metastasis-promoting property is purely “cell-intrinsic”, independent of the immune system and even of UBR5’s E3 ubiquitin ligase activity.Citation2 In the present study, we further explored the cellular and molecular mechanisms whereby UBR5 drives tumor growth and metastasis, and the potential of UBR5 as a novel immunotherapeutic target for aggressive breast cancer.
Materials and methods
Cell cultures
4T1 cell line (CRL-2539) was obtained from ATCC in 2012. 4T1 cells were cultured in RPMI-1640 supplemented with 10% fetal bovine serum, 2 mM glutamine, 100 U/mL Penicillin and 100 μg/mL Streptomycin at 37°C in a humidified atmosphere of 5% CO2. Cells were split when they reached 80%-90% confluence. For all experiments, cells were grown to 80%-90% confluence before experimentation.
Human breast cancer cell MCF-7 was cultured in DMEM with 10% fetal bovine serum, 2 mM glutamine, 100 U/mL Penicillin, and 100 μg/mL Streptomycin. For RNAi-mediated UBR5/EDD expression silencing, cells were transfected with 20 μmol/L of Ubr5-siRNA duplex (5ʹ-CAACUUAGAUCUCCUGAAA-3ʹ) and Lipofectamine RNAiMAX (Invitrogen, 13778075) as per the manufacturer’s instructions. Nonspecific siRNA oligo (Sigma, SIC002) was used as a negative control.
To generate UBR5-reconstituted 4T1/Ubr5−/- cell line, cells were transfected with pCMV-Tag2B EDD1 (Addgene, #37188) using lipofectamine 3000 (Invitrogen, L3000008) as per the manufacturer’s protocol. To generate Id1 or Id1/Id3 reconstituted cell lines in 4T1/Ubr5−/-, cells were transfected with pCMV3-Id1 (provided by Dr. Robert Benezra), pEF1α-IRES-Id1/Id3 using lipofectamine 3000, respectively. All stable cell lines were selected with G418 and confirmed by q-PCR and western blot.
To knockdown Raet1e expression in 4T1/Ubr5−/-, cells were transfected with 20 μmol/L of Raet1e-siRNA duplex (ThermoFisher, AM16708) and Lipofectamine RNAiMAX (Invitrogen, 13778075) as per the manufacturer’s instructions. Nonspecific siRNA oligo (Sigma, SIC002) was used as a negative control. Forty-eight hour post-siRNA transfection, cells were harvested for knocking down efficiency evaluation and mice injection.
Mice and tumor model
Female BALB/c mice aged 6–8 weeks were purchased from Jackson Laboratory. To induce the 4T1 orthotopic mammary tumor, 5 × 105 4T1 cells were injected subcutaneously (s.c.) in the abdominal mammary gland and mice were euthanized at indicated times for analysis or monitored for survival. The volume of tumors was calculated as V = L× W2/2, where L and W stand for tumor length and width, respectively. For the lung metastasis model of 4T1, 2 × 105 cells were intravenously infused through the tail vein and mice were sacrificed on d 18 for analysis. For T cell depletion, 4T1 tumor-bearing mice were given anti-mouse CD8α antibody (100 μg/mouse, Clone 2.43, BioXcell) or anti-mouse CD4 antibody (250 μg/mouse, Clone GK1.5, BioXcell) at indicated times. All animal experiments were performed in accordance with National Institutes of Health guidelines for housing and care of laboratory animals after protocol (protocol Number 0701-569A) approved by IACUC at Weill Cornell Medicine.
Cell proliferation assay
Cell proliferation was evaluated by Sulforhodamine B (SRB) assay. Briefly, cells (1*104/well) were seeded into 96 well plates and cultured for 24 h, 48 h, and 72 h, then fixed with 10% trichloroacetic acid (Sigma, T8657) for 30 min at 4°C and stained with 0.4% (w/v) SRB (Sigma, 230162) in 1% acetic acid solution for 30 min. After washing with 1% acetic acid, bound SRB was solubilized with 10 mmol/L Tris buffer, and absorbance (OD) was measured at 510 nm using 96 well plate reader.
RNA isolation and qRT-PCR
Total RNAs were extracted with the RNeasy Plus Mini Kit (QIAGEN, 74134), and cDNA was synthesized using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystem, 4368814). Quantitative PCR was performed on QuantStudioTM 6 Flex System (Applied Biosystem) using PowerUp SYBR Green Master Mix (Thermo Fisher). RT-PCR primers for Id1: CCCAGAACCGCAAAGTGAGC (forward), CGGTGGTCCCGACTTCAGAC (reverse); for Id3: CGACCGAGGAGCCTCTTAGC (forward), ACGCTGCAGGATTTCCACCT (reverse); for Gapdh: GTCCTTCCTGGCCTAGAGGT (forward), GATGCAGGGATGATGTTCTG (reverse). The expression levels of target genes were normalized with Gapdh abundance. Data were presented as the average of triplicates ± SD.
Western blot
Cells were lysed in RIPA buffer (Thermal Scientific) and the lysates were centrifuged at 12,000× rpm for 30 min at 4°C. Supernatants were collected and protein concentration was quantified by Bio-rad protein assay (Bio-rad, 5000006). Cell lysates were subjected to SDS-PAGE and transected to the PVDF membrane, followed by immunoblotting with antibodies against UBR5 (NBP2-1591, Novus Biologicals), E-cadherin (NBP2-19051, Novus Biologicals), ID1 (195–14, CalBioreagents), ID3 (16–1, CalBioreagents), Raet1e (ab95202, Abcam), and GAPDH (sc-FL335, Santa Cruz).
Clonogenic assays
4T1 cells were seeded in 6-well plates (100 cells/well) and cultured at 37°C in a humidified atmosphere of 5% CO2. After 10 d, cells were washed with PBS and fixed with 4% paraformaldehyde and then stained with 0.5% crystal violet. The number of colonies formed in each well was counted and photographed under the microscope. All assays were performed in triplicate.
Flow cytometry
Primary tumor tissues were harvested, weighed, and digested with tissue dissociation buffer [~280 U/mL Collagenase Type3, 4ug/mL DNase in HBSS] for 1 h in 37°C water bath with periodic vortexing and mashed through 70 μm filters, layered on a 44% and 66% Percoll gradient (GE), and centrifuged at 3000 rpm for 30 min without brake. After 20 min incubation with Zombie UV TM Fixable stain at room temperature, all samples were washed with BD FACS buffer and stained with the appropriate surface antibodies. CD3 (17A2), CD8α (53–6.7), NK1.1 (PK136), CD11b (M1/70), F4/80 (BM8), Gr-1 (RB6-8C5), Ly6 G-PE (Clone 1A8), Ly6 C-PEcy7 (Clone HK1.4), MHCⅡ (Clone CD11 c (N418), MHCI (M5/114.15.2) were purchased from Biolegend. CD45 (30-F11), CD4 (GK1.5), CD25 (PC61.5), and Foxp3 (FJK-16 s) were purchased from eBioscience. All antibodies were tested with their isotype controls. Intracellular staining for Foxp3 and Granzyme B was performed according to Foxp3/Transcription Factor Staining buffer set (eBioscience). Data acquisition was performed on FACSCabibur (BC Biosciences) and analyzed via FlowJo.
Invasion transwell assay
The Transwell assay was performed as previously described.Citation2 Briefly, 2 × 104 4T1 cells overnight cultured in serum-free medium were seeded onto Matrigel bedding Chambers (24-well, 356234, Corning) with serum-free medium. After 24 h incubation, migrated cells determined and quantified.
Bioinformatic analysis of UBR5 mRNA expression in breast cancer patients’ specimens and survival
The bioinformatic analysis of breast cancer patients was based on the data from The Cancer Genome Atlas (TCGA) project and MET500 dataset.Citation9 UBR5 mRNA expression data was retrieved based on RNA-seq assay from Firehose (http://gdac.broadinstitute.org/). Analysis of UBR5 mRNA expression was based on UALCAN.Citation10 The clinical survival data of breast cancer patients in TCGA was retrieved according to the pipeline of https://portal.gdc.cancer.gov and Liu.Citation11 Kaplan–Meier analysis of percent survival was performed with R version 3.4.4 (https://www.r-project.org/about.html).
RNA-seq analysis
cDNA libraries were made using the Illumina TruSeq Stranded mRNA Library Prep kit and were sequenced with single-end 51 bps on Illumina HiSeq4000 instrument. STARCitation12 was used to align raw sequencing reads to the mouse GRCm38 reference genome. Raw read counts were calculated using HTseq-count.Citation13 Differential expression analysis was performed using the DEseq2 package.Citation14 We selected 1269 differentially expressed genes with adjusted p-value <1e-10 and log2 FoldChange greater than 2.0 (up-regulated) or smaller than −2.0 (down-regulated), for pathway analysis using the Ingenuity Pathway Analysis (IPA).Citation15
LC-MS and data analysis
A Thermo Fisher Scientific EASY-nLC 1000 coupled on-line to a Fusion Lumos mass spectrometer (Thermo Fisher Scientific) was used. Buffer A (0.1% FA in water) and buffer B (0.1% FA in 100% ACN) were used as mobile phases for gradient separation. A 75 µm x 15 cm chromatography column (ReproSil-Pur C18-AQ, 3 µm, Dr. Maisch GmbH, German) was packed in-house for peptides separation. Peptides were separated with a gradient of 5–30% buffer B over 220 min, 30%-80% B over 20 min at a flow rate of 300 nL/min. The Fusion Lumos mass spectrometer was operated in data-dependent mode. Full MS scans were acquired in the Orbitrap mass analyzer over a range of 300–1500 m/z with resolution 120,000 at m/z 200. Up to the top 20, most abundant precursors were selected with an isolation window of 0.4 Thomsons and fragmented by higher-energy collisional dissociation with normalized collision energy of 40. MS/MS scans were acquired in the Orbitrap mass analyzer with resolution 30000 at m/z 200. The automatic gain control target value was 1ECitation6 for full scans and 1ECitation5 for MS/MS scans, respectively, and the maximum ion injection time is 60 ms for both.
The raw files were processed by using the MaxQuant computational proteomics platform (version 1.5.5.1). The fragmentation spectra were searched against the UniProt mouse protein database and allowed up to two missed tryptic cleavages. Oxidation of methionine and protein N-terminal acetylation were used as variable modifications for database searching. Carbamidomethyl cysteine was used as fixed modification. The precursor and fragment mass tolerances were set to 7 and 20 ppm, respectively. Both peptide and protein identifications were filtered at 1% false discovery rate (FDR).
Lentivirus generation and transduction
Lentiviruses were produced by co-transfection of 293 T cells with psPAX2, pMDG2, and UBR5 MISSION shRNA (TRCN0000238583, sigma) with lipofectamine 3000 (Life Technology). Virus supernatants collected 48-h and 72-h post-transfection were combined, centrifuged at 2000rpm for 10 min to remove cellular debris. Lentiviral supernatants were sterilized with 0.45 μm pore filters. 4T1 cells were transduced with UBR5-shRNA containing lentivirus using 10ug/ml polybrene (Santa Cruz) and treated with 2 μg/ml puromycin after 48 h.
Histological analysis
For paraffin-embedded tissue, organs were fixed in 4% formaldehyde overnight at 4°C. Fixed sections were then made from paraffin-embedded tissue blocks. To score tumor nodules in lung, tissue blocks were sectioned and stained with hematoxylin/eosin (H&E) according to the manufacturer’s instruction. Four to Five fields were selected from whole lung sections for five mice and all histological quantifications are presented as the mean of different tumor nodules for each individual mouse. Anti-UBR5 (Santa Cruz, sc-9562) and anti-PD-L1 (Thermo Fisher, PA5-20343) antibodies were used for immunohistochemistry, and tumor tissue sections were dewaxed in xylene and rehydrated with distilled water. After antigen unmasking with citric antigen retrieval buffer, blocking step was performed, followed by 1 h incubation at 37°C with the primary antibodies. Slides were counterstained with hematoxylin, dehydration, and mounting.
Statistics
Data are shown as mean ± S.E.M. All experiments were repeated at least two times and results were similar between repeats. Animal experiments used between three to six mice per group. All statistical analyses were performed using Graph Pad Prism 5.0 software. Differences between experimental groups were calculated using a two-tailed unpaired Student’s t-test. Survival rates were compared using the log-rank test.
Results
UBR5 gene amplification and overexpression occur frequently in human breast cancers
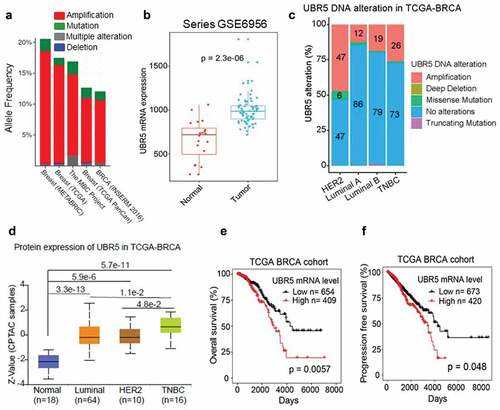
Ubiquitin protein ligase E3 component n-recognin 5 (UBR5, a.k.a., EDD) is encodes a 309 nuclear protein. Using a pooled shRNA screen to interrogate death receptor signaling, Dompe et al. identified UBR5 along with 15 others as having the property to modulate the sensitivity to ligand-induced apoptosis, and also exhibiting frequent overexpression and/or copy number gain in human breast, pancreas, and lung cancers.Citation16 In a TCGA-based meta-analysis involving five independent studies and 5,151 breast cancer samples, we found that human UBR5 gene alterations (predominantly amplifications) occur in 1221% of breast cancer cases (). Consistent with the gene amplification, UBR5 mRNA expression levels in breast cancer tissues were significantly higher than in normal breast tissues (). Further analysis revealed that UBR5 alterations, principally amplifications, occur in all subtypes of breast cancer to varying degrees with greatest frequency in Her2+ subtype (). Consistent with this observation, UBR5 protein expression was significantly higher in all subtypes of breast cancer than that in normal breast tissues (). In addition, high UBR5 expression positively correlates with the overall survival of breast cancer patients (P = .0057) (), as well as progression-free survival (P = .048) (). Nevertheless, very little was known about the causality, the activities, and mechanisms of UBR5 in promoting tumor growth and metastasis, particularly in vivo. We undertook a series of experimental approaches through genetic, cellular, and molecular manipulations in a highly aggressive 4T1 experimental murine mammary TNBC model.
Figure 1. Meta-analysis of UBR5’s involvement in human breast cancers. (a) Analysis of UBR5 gene alterations in human breast cancer studies based on the data of The Cancer Genome Atlas (TCGA) project. (b) UBR5 mRNA level in breast cancer specimens compared to normal breast tissues (TCGA). (c) UBR5 gene alterations in breast cancer subtypes as percentages. (d) UBR5 protein expression in subtypes of breast cancer. (e) Correlation of UBR5 expression with overall survival of breast cancer patients. (f) Correlation of UBR5 expression with progression-free survival
Ubr5-deletion in tumors triggers a more robust T-cell response
We explored UBR5 functionally in the syngeneic and highly metastatic murine TNBC model, 4T1, which expresses high levels of UBR5. We “knocked out” UBR5 expression in tumor cells by CRISPR/Cas9, designated 4T1/Ubr5−/-. The efficiency of the knock out was confirmed at genomic, mRNA and protein levels.Citation2 We randomly selected three independent Ubr5−/- clones, which displayed very similar in vitro propagation capacities to that of control 4T1 (Figure S1A-B). To understand the broad impact of Ubr5 deletion on gene expression in 4T1 cells, we performed transcriptome analysis by RNA-seq comparing the three Ubr5−/- clones with three WT GFP clones. The reproducibility of the clones was validated (Fig S2A). Hierarchical cluster analysis revealed two strikingly opposite gene expression groups between WT and Ubr5−/- 4T1 cells (Figure S2B). Ingenuity Pathway analysis (IPA) of the combined data showed that compared to WT controls, signaling pathways involved in epithelial to mesenchymal transition (EMT) and extracellular matrix degradation as well as immune co-stimulation were up-regulated (positive z-score) in Ubr5−/- cells, whereas STAT3-, TGF-β-, P38MAPK- and endothelin signaling pathways were downregulated (negative z-score), implying lower malignant potential and tumorigenicity of 4T1/Ubr5−/- compared to the WT control ().
Figure 2. Immunological alterations in mice carrying Ubr5−/- tumor. 4T1 tumor cells were s.c. injected into the mammary pad of BALB/c mice and on d 11, tumor-bearing mice were sacrificed and tumor organs were harvested for FACS analysis. (a) 4T1 tumor growth curve. Around d11, 4T1/Ubr5−/- tumor started to shrink. (b) RNA-seq analysis of WT vs Ubr5−/ – tumors isolated on d 11. The activation states of various biological pathways are indicated. The dotted line represents the statistical significance threshold (p < .05). (c,d) Increased tumor-infiltrating CD4+/CD8+ T cells in 4T1/Ubr5−/- tumor-bearing mice. (c) Representative FACS images and quantification of intratumoral CD4+ T cells. (d) Representative FACS images and quantification of intratumoral CD8+ T cells. (e) Representative FACS histogram and quantification of granzyme B+ cells gated on intratumoral CD8+ T cells. (f) Decreased proportion of Tregs in TDLNs of 4T1/Ubr5−/- tumor-bearing mice. Representative FACS images of Tregs in TDLNs and the quantification of Tregs in TDLN. In all cases, data are representative of at least three independent experiments using three to five mice per group. Data represent mean ± SEM; *P < .05, **P < .01
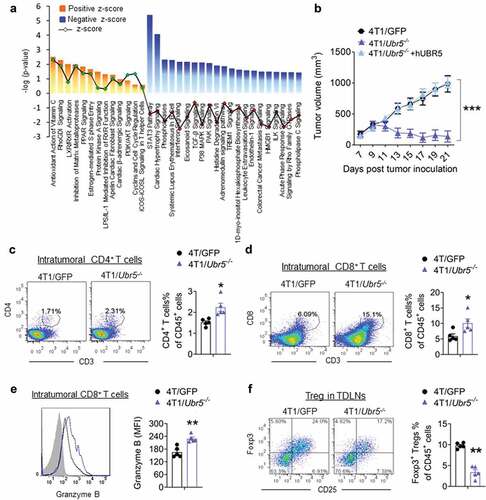
To assess the effects of Ubr5 deletion on tumor growth and metastasis, female BALB/c mice (syngeneic with 4T1 tumor) were inoculated in the abdominal mammary gland with control GFP+ or Ubr5−/- tumors. However, Ubr5−/- tumor growth in vivo was very much arrested from d 10 onwards, and the tumors shrank gradually over time and some even disappeared completely 3 weeks after tumor cell inoculation (, Figure S1 C).
To further elucidate the immune mechanisms involved in the antitumor effect of Ubr5 depletion, we analyzed the major subsets of leukocytes in the tumor microenvironment (TME), spleen, and tumor-draining lymph nodes (TDLNs) of 4T1 bearing mice on d 11 post-tumor inoculation at which time the 4T1/Ubr5 −/- tumor started to shrink (). The proportion of tumor-infiltrating CD4+ () and CD8+ () T cells was increased by approximately twofold in 4T1/Ubr5−/- bearing mice, compared with mice bearing control tumors. Furthermore, infiltrating CD8+ T cells exhibited enhanced granzyme B expression (), indicating a more active cytolytic state of the CD8+ T cells in mice carrying the Ubr5-deleted tumor. Although there was no significant difference in tumor-infiltrating Foxp3+ CD25+ Tregs in both types of hosts (data not shown), decreased Tregs were observed in TDLNs of 4T1/Ubr5−/- bearing mice (). Populations of intra-tumoral CD3−NKp46+ NK cells (Figure S2A), CD11b+Ly6 C+/Ly6 G+ MDSCs (Figure S2B), and CD11 c+MHCⅡ+ DCs (Figure S2 C) were not significantly altered between these two types of 4T1 bearing mice. Similar results were obtained in spleens, with a higher frequency of both CD4+ and CD8+ T cells in 4T1/Ubr5−/- bearing mice (Figure S2D-G). Together, these results demonstrate that targeting UBR5 in tumors induces a heightened immune active state of the T lymphocytes.
CD8 + T cell-dependent immunity is essential for controlling Ubr5−/- tumor growth
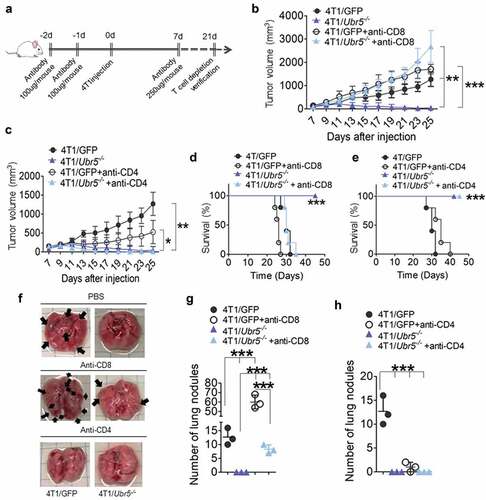
To determine whether and which type of T cells control 4T1/Ubr5−/- tumor growth, we devised a scheme to deplete CD4+ and CD8+ T cells in 4T1 tumor-bearing mice with antibodies (). Although both CD4+ and CD8+ T cells were markedly depleted in spleens, peritoneal cavity, and tumors as evaluated on d 21 post-tumor inoculation (Figure S4 and S5), only depletion of CD8+ T, not CD4+ T cells, caused a complete reversal of the tumor growth of 4T1/Ubr5−/- to and above the WT tumor (), and animal survival (). In addition, lung metastasis of 4T1/Ubr5−/- tumors was partially rescued by CD8+ T cell depletion but not by CD4+ T cell depletion (). Taken together, these data suggest that UBR5 promotes tumor growth in a manner that is solely dependent on restricting the activities of CD8+ T cells.
Figure 3. Effects of T cell depletion on tumor growth and metastasis. (a) Schematic for T cell depletion in 4T1 tumor-bearing mice. (bc) 4T1 tumor growth curves with CD8 T cell depletion (b) or CD4 T cell depletion (c) (n = 5 per group). (d,e) The survival rates of 4T1 tumor-bearing mice with CD8 T cell depletion (d) or CD4 T cell depletion (e) were monitored and quantified (n = 5 per group). (f,h) Lung metastasis in mice bearing 4T1 on d 24 post-tumor cells s.c. injection. (f) Representative images for lung nodules. Lung nodules in CD8 T cell-depleted tumor-bearing mice (g) and CD4 T cell-depleted tumor-bearing mice (h) were quantified. Data are representative of three independent experiments with similar results. Data represent mean ± SEM; *P < .05, **P < .01, ***P < .001
Ubr5 deletion causes dysregulated epithelial–mesenchymal transition and impaired tumor metastasis
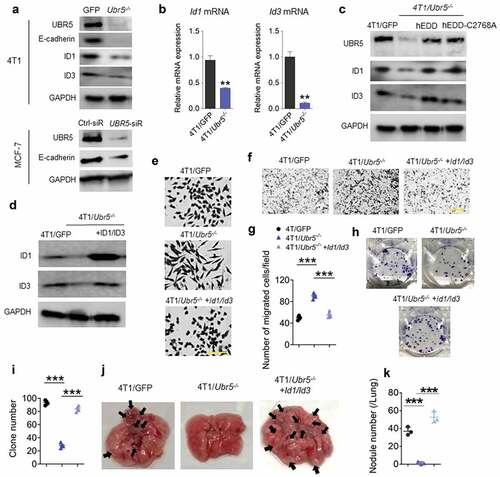
We previously observed that the morphology of 4T1/Ubr5−/- was altered from a cuboidal epithelial shape, to a more elongated “mesenchymal” shape, characteristic of epithelial–mesenchymal transition (EMT).Citation2 4T1/Ubr5−/- cells displayed strongly reduced levels of E-cadherin, ID1 and ID3 proteins (), the latter two of which are critical regulators of mesenchymal–epithelial transition (MET) in metastatic colonization.Citation17 The defects in ID1 and ID3 protein expression were also observed at the mRNA level (), indicating that UBR5 regulates directly or indirectly the transcription of Id1 and Id3 genes. Notably, the protein expression of ID1/ID3 was completely restored to the control (4T1/GFP) level in both human EDD-reconstituted- and catalytic mutant EDD-C2768A-reconstituted- 4T1/Ubr5−/- cells, suggesting that UBR5 regulates ID1/ID3 expression independent of its E3 ubiquitin ligase activity (). In addition, the defective E-cadherin expression was also observed in UBR5-deficient human breast cancer cell line MCF-7 (). Reconstitution of 4T1/Ubr5−/- cells with ID1 and ID3 together (), but not with ID1 alone (Figure. S7) restored their epithelial phenotype to that of the wild type 4T1 cells () and reduced 4T1/Ubr5−/- tumor cells’ ability to migrate in vitro, measured by transwell assay, to that of the WT cells (). The reconstitution also restored 4T1/Ubr5−/- tumor cells’ clonogenicity to the WT level (,i), and completely rescued the impaired lung metastasis of 4T1/Ubr5−/- tumors in vivo without any difference in in vitro propagation capacity (, Figure S1D). These data demonstrate that UBR5 promotes tumor metastasis primarily through its transcriptional control of the major EMT regulators ID1 and ID3 in a cell-intrinsic manner.
Figure 4. Effects of Ubr5 deletion on tumor metastasis. (a) Protein expression of murine UBR5, E-cadherin, ID1 and ID3 in 4T1 cells, and human E-cadherin in MCF-7 after UBR5 depletion by siRNA was evaluated by western blot. (b) Messenger RNA expression of Id1 and Id3 in 4T1 cells was assessed by qRT-PCR. (c) ID1 and ID3 protein expression in human EDD-reconstituted and catalytic mutant EDD-C2768A-reconstituted 4T1/Ubr5-/- cells was evaluated by western blot. (d) Murine ID1 and ID3 reconstitution in 4T1/Ubr5 -/- cells were verified by western blot. (e) Representative micrographs of cell morphology of 4T1/GFP, 4T1/Ubr5-/-, and 4T1/Ubr5-/- + ID1/ID3 reconstituted cells in culture. Scale bars: 100 μm. (f,g) Representative micrographs (f) and the quantification (g) of a Transwell invasion assay. (hi) Representative images (h) and quantified values (i) of clonogenic assay with 100 cells/well. (j–k) 5 × 105 cells were i.v. injected into BALB/c mice and the lungs of the recipient mice were harvested on d 18. Representative images (j) and quantified values of metastatic nodules in lung (k) (n = 3 per group). Data are representative of two independent experiments with similar results. Data represent mean ± SEM, **P < .01, ***P < .001
Targeting UBR5 may lead to induction of immunogens
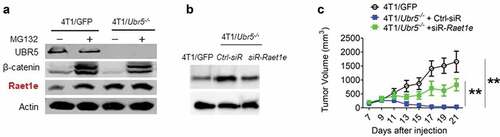
We were interested in identifying UBR5’s substrates and direct targets. To begin such a search effort, we compared protein expression between WT and Ubr5−/- 4T1 tumor cells by LC/MS. Out of more than 4,300 proteins identified and analyzed, 30 of them were significantly upregulated in Ubr5−/- cells over WT cells (), whereas 33 proteins were downregulated (Figure S8). One of the upregulated proteins, Raet1e, belong to a family of ligands for NKG2D activating receptor of human natural killer (NK) cells. It has been shown to function as a ligand for both TCRγ9δ2 and NKG2D through which Raet1e induces T cell-mediated cytotoxicity to tumor cells.Citation18 Decreased Raet1e expression is a poor prognostic marker in nasopharyngeal carcinoma,Citation19 whereas high expression of Raet1e is an indicator of good prognosis in cervical cancer.Citation20 We first confirmed the MS finding of Raet1e’s overexpression by Western blot in WT and in Ubr5−/- cells treated with the proteasome inhibitor MG132 (), which also indicated that Raet1e protein level was indeed regulated by a proteasome-mediated mechanism. We then transiently knocked down the expression of Raet1e via siRNA in Ubr5−/- 4T1 tumor (), which, when implanted in mice, caused a partial reversal of the inhibited growth of the parental tumor (), but did not alter the in vitro growth rates (Figure S1E), suggesting that higher Raet1e expression may mediate an adverse response against the tumor and that UBR5 deficiency in tumor cells may generate immunogens that attract anti-tumor responses.
Table 1. Upregulated proteins in Ubr5−/- 4T1 tumor
Figure 5. Potential immunogens and interacting partners controlled by UBR5. (a) Protein expression of Raet1e in WT vs. Ubr5−/- 4T1 cells treated with MG132 or not, by Western blot analysis. (b,c) Knocking down Raet1e using siRNA in 4T1/Ubr5−/- cells partially restored its tumor growth in vivo. (b) 48 h after siRNA transfection, Raet1e protein levels in 4T1 cells were measured by western blot. (c) After Raet1e-siRNA transfection, 4T1/Ubr5−/- tumor cells were s.c. injected in the abdominal mammary gland. Tumor growth was monitored for 3 weeks. Data are representative of two independent experiments with similar results. Data represent mean ± SEM, **P < .01
Dual targeting of UBR5 and PD-L1 yields superior therapeutic benefits
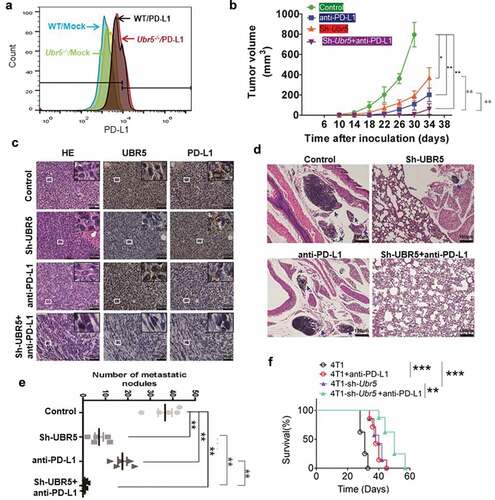
UBR5 is an important regulator of genome stability through its modulation of the activity of the DNA damage checkpoint kinase, CHK2,Citation21 its control of p53 levelsCitation22 and ATM-mediated phosphorylation of p53.Citation23 We hypothesize that UBR5 deletion may result in genome instability and mutagenesis, inducing potential neoantigens which will attract T cell-mediated tumor-specific responses. We observed that Ubr5-deletion did not affect PD-L1 surface expression in the tumor (), which suggested to us that combinational therapy targeting both UBR5 and PD-L1 may produce an additive or synergistic therapeutic benefit compared to either treatment alone. We tested the idea by targeting UBR5 expression with an shRNA and PD-L1 activity by a PD-L1 blocking antibody. The dual targeting yielded superior therapeutic benefits than either treatment alone with respect to tumor size (). The targeting efficiency was evidenced via immunohistochemical (IHC) analysis by the diminished protein expression levels of UBR5 and PD-L1 in treated tumors, and the lowest level of both UBR5 and PD-L1 was presented in the tumor tissues with the interference of both UBR5 and PD-L1 compared to the control group (). Consistently with the drastically impaired tumor growth, dual targeting of UBR5 and PD-L1 resulted in dramatically reduced lung metastasis than mono-targeting (,e), and significantly extended survival (). These data demonstrate the therapeutic superiority of the combinational targeting strategy.
Figure 6. Combinational therapy targeting UBR5 and PD-L1. (a) FACS analysis of surface expression of PD-L1 on WT and Ubr5−/- 4T1 cells. (b) Combination therapy targeting UBR5 (via shRNA) and PD-L1 (via antibody) in 4T1 tumors showing tumor growth. PD-L1 antibody (Bioxcell, clone 10 F.9G2) or isotype IgG (Rat IgG2b, κ) was injected intraperitoneally at 200 μg per mouse weekly since d 7 after tumor inoculation for 3 times in total. (c) Representative H&E staining and IHC staining of UBR5 and PD-L1 in WT 4T1 tumor tissues of indicated groups (scale bar = 50 μm). (de) Metastasis of the four treated groups illustrated by H&E staining of tumor tissues (d) and by quantifying metastatic nodules in the lungs (e). (f) Survival rate of mice with WT or Ubr5-deficient tumors treated with anti-PD-L1. Three different replicates were performed for all experiments. Data represent mean ± SEM; *P < .05; **P < .01
Discussion
An ingenuity pathway analysis (IPA)-based study that we performed (unpublished data) reveals that UBR5 may represent a major signaling hub connecting with many important molecules in various ways such as TP53, argonaute 1 (AGO1), Bcl-2-associated X protein (BAX), calcium and integrin binding 1 (CIB1), growth arrest and DNA damage-inducible 45 (GADD45) proto-oncogene MDM2, poly(A)-binding and interacting protein 2 (PAIP2), progesterone receptor (PGR), protein phosphatase 2 regulatory subunit Balpha (PPP2R2A), and AGTRI, etc., suggesting a broad network of biological pathways regulated by UBR5 through direct or indirect physical interactions. In this study, we present evidence that UBR5 gene amplifications are common occurrence in human breast cancers and that high UBR5 expression is adversely associated with disease progression and patient survival. Targeting UBR5 in the mammary tumor through gene editing or expression silencing causes paracrine effects that trigger the activation of CD8+ T cells, which help control the tumor growth. The antigenic specificities of this response are presently unknown. We have shown that Raet1e expression was upregulated in Ubr5−/- tumor, which may stimulate an anti-tumor response. This response is unlikely to be of NK cell origin since depletion of NK cells did not have an impact on the anti-tumor response (data not shown). It remains to be determined if γδ T cells are involved.Citation18 Given UBR5’s demonstrated essential role for G(1)/S and intra S phase DNA damage checkpoint activation and for the maintenance of G(2)/M arrest and genome integrity after double-strand DNA breaks,Citation21 it is conceivable that deletion of Ubr5 may cause genomic instability and the induction of “neoantigens” that attract T cell-mediated immune responses. Further studies in this area may lead to the identification of these neoantigens for potential applications as novel cancer vaccine and immunotherapy targets.
In contrast to the strong paracrine involvement of CD8+ T-mediated immunity in UBR5-regulated tumor growth, the metastatic process driven by UBR5 appears to be primarily cell-intrinsic. Our data demonstrate that the annulling of Ubr5 in 4T1 cells is causative for the loss of E-cadherin expression and impairs the tumor cells’ mesenchymal to epithelial transition (MET) and their ability to colonize in secondary organs. This effect is controlled by UBR5 principally through transcriptional regulation of the key EMT regulators ID1 and ID3. The result is the maintenance of Ubr5−/- tumor cells in the mesenchymal state lacking E-cadherin expression, thus unable to complete MET and take roots in the lungs. It is thus of great importance to further understand how UBR5 loss leads to ID1/ID3 downregulation. Given the mechanism of UBR5’s action, it is possible that loss of UBR5 may lead to the stabilization of a repressor which inhibits ID1/ID3 expression. ATF3 is a well-characterized, known repressor of ID1 expression.Citation24 It will be interesting to determine if UBR5 destabilizes ATF3. It is equally possible that loss of UBR5 leads indirectly to the loss of a positively acting transcription factor that controls ID1/ID3 expression. A variety of factors that control ID1 expression in TNBC cells have been identified. The basic helix-loop-helix (bHLH) transcription factor Lyl1 and CREB1, a widely expressed transcription factor, and a suspected oncogene, interact and form a molecular complex. The histone acetyltransferases p300 and CBP are recruited to this complex. Together they activate CREB1 target promoters such as Id1, Id3, cyclin D3, Brca1, Btg2, and Egr1.Citation25
In summary, our findings may lead to the development of groundbreaking therapeutic modalities that, when combined with conventional treatment, will bring us significantly closer to ameliorating the immense casualty caused by highly aggressive breast cancer because targeting UBR5-mediated signaling pathway will not only bring immediate clinical benefits to patients by abrogating cancer growth and metastasis but may also lead to immune activation that will likely result in the generation of T cell memories and delicate immunosurveillance against cancer recurrence and drug resistance.
Highlights
We identify a profound role of UBR5 in breast cancer aggression.
Loss of UBR5 in cancer cells causes the appearance of certain putative immunogens.
Loss of UBR5 in cancer cells causes a strong CD8+ T cell-mediated anti-tumor response in a paracrine manner.
Loss of UBR5 in cancer cells severely impairs metastasis in a cell-intrinsic manner that is dependent on EMT regulators ID1 and ID3.
Combinational targeting of UBR5 and PD-L1 yields strong therapeutic benefit to tumor-bearing hosts.
The work significantly expands our scarce understanding of the pathophysiology and immunobiology of a fundamental molecule and has strong implications for the development of novel immunotherapy to treat aggressive breast cancers that resist conventional treatment.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Author contributions
M.S., C.W., X.C., and X.M.: contributed to study concept and design, analysis, and interpretation of data, writing of the manuscript, statistical analysis, obtained funding, study supervision. H.W.: performed meta-analysis of clinical data; T.Z.: performed bioinformatic analysis of the RNA-seq data; J.L. made the Raet1e mutants; R.B.: guided the metastasis experiments; L.C.: contributed human clinical samples. All authors read and approved the final manuscript.
Supplemental Material
Download ()Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
References
- R L S, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65:5–12. doi:10.3322/caac.21254.
- Liao L, Song M, Li X, Tang L, Zhang T, Zhang L, Pan Y, Chouchane L, Ma X. E3 ubiquitin ligase UBR5 drives the growth and metastasis of triple negative breast cancer. Cancer Res. 2017;77:2090–2101. doi:10.1158/0008-5472.CAN-16-2409.
- M J C, Russell AJ, Woollatt E, Sutherland GR, Sutherland RL, Watts CK. Identification of a human HECT family protein with homology to the Drosophila tumor suppressor gene hyperplastic discs. Oncogene. 1998;17:3479–3491. doi:10.1038/sj.onc.1202249.
- D N S, Hird SL, Withington SL, Dunwoodie SL, Henderson MJ, Biben C, Sutherland RL, Ormandy CJ, Watts CK. Edd, the murine hyperplastic disc gene, is essential for yolk sac vascularization and chorioallantoic fusion. Mol Cell Biol. 2004;24:7225–7234. doi:10.1128/MCB.24.16.7225-7234.2004.
- Kinsella E, Dora N, Mellis D, Lettice L, Deveney P, Hill R, Ditzel M. Use of a conditional Ubr5 mutant allele to investigate the role of an N-end rule ubiquitin-protein ligase in hedgehog signalling and embryonic limb development. PLoS One. 2016;11:e0157079. doi:10.1371/journal.pone.0157079.
- R F S, Frikstad KM, McKenna J, McCloy RA, Deng N, Burgess A, Stokke T, Patzke S, Saunders DN. The E3 ubiquitin ligase UBR5 regulates centriolar satellite stability and primary cilia. Mol Biol Cell. 2018:mbcE17040248. doi:10.1091/mbc.E17-04-0248.
- Koyuncu S, Saez I, Lee HJ, Gutierrez-Garcia R, Pokrzywa W, Fatima A, Hoppe T, Vilchez D. The ubiquitin ligase UBR5 suppresses proteostasis collapse in pluripotent stem cells from Huntington’s disease patients. Nat Commun. 2018;9:2886. doi:10.1038/s41467-018-05320-3.
- J L C, Henderson MJ, Russell AJ, Anderson DW, Bova RJ, Campbell IG, Choong DY, Macdonald GA, Mann GJ, Nolan T, et al. EDD, the human orthologue of the hyperplastic discs tumour suppressor gene, is amplified and overexpressed in cancer. Oncogene. 2003;22:5070–5081. doi:10.1038/sj.onc.1206775.
- D R R, Wu YM, Lonigro RJ, Vats P, Cobain E, Everett J, Cao X, Rabban E, Kumar-Sinha C, Raymond V, et al. Integrative clinical genomics of metastatic cancer. Nature. 2017;548:297–303. doi:10.1038/nature23306.
- D S C, Bashel B, Balasubramanya SAH, Creighton CJ, Ponce-Rodriguez I, Chakravarthi B, UALCAN: VS. A portal for facilitating tumor subgroup gene expression and survival analyses. Neoplasia (New York, NY). 2017;19:649–658. doi:10.1016/j.neo.2017.05.002.
- Liu J, Lichtenberg T, Hoadley KA, Poisson LM, Lazar AJ, Cherniack AD, Kovatich AJ, Benz CC, Levine DA, Lee AV, et al. An integrated TCGA Pan-cancer clinical data resource to drive high-quality survival outcome analytics. Cell. 2018;173:400–416.e411. doi:10.1016/j.cell.2018.02.052.
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi:10.1093/bioinformatics/bts635.
- Anders S, Pyl PT, Huber W. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi:10.1093/bioinformatics/btu638.
- M I L, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi:10.1186/s13059-014-0550-8.
- Kramer A, Green J, Pollard J Jr., Tugendreich S. Causal analysis approaches in ingenuity pathway analysis. Bioinformatics. 2014;30:523–530. doi:10.1093/bioinformatics/btt703.
- Dompe N, Rivers CS, Li L, Cordes S, Schwickart M, Punnoose EA, Amler L, Seshagiri S, Tang J, Modrusan Z, et al. A whole-genome RNAi screen identifies an 8q22 gene cluster that inhibits death receptor-mediated apoptosis. Proc Natl Acad Sci U S A. 2011;108:E943–951. doi:10.1073/pnas.1100132108.
- Stankic M, Pavlovic S, Chin Y, Brogi E, Padua D, Norton L, Massague J, Benezra R. TGF-beta-Id1 signaling opposes Twist1 and promotes metastatic colonization via a mesenchymal-to-epithelial transition. Cell Rep. 2013;5:1228–1242. doi:10.1016/j.celrep.2013.11.014.
- Kong Y, Cao W, Xi X, Ma C, Cui L, He W. The NKG2D ligand ULBP4 binds to TCRgamma9/delta2 and induces cytotoxicity to tumor cells through both TCRgammadelta and NKG2D. Blood. 2009;114:310–317. doi:10.1182/blood-2008-12-196287.
- Xu Y, Zhou L, Zong J, Ye Y, Chen G, Chen Y, Liao X, Guo Q, Qiu S, Lin S, et al. Decreased expression of the NKG2D ligand ULBP4 may be an indicator of poor prognosis in patients with nasopharyngeal carcinoma. Oncotarget. 2017;8:42007–42019. doi:10.18632/oncotarget.14917.
- Cho H, Chung JY, Kim S, Braunschweig T, Kang TH, Kim J, Chung EJ, Hewitt SM, Kim JH. MICA/B and ULBP1 NKG2D ligands are independent predictors of good prognosis in cervical cancer. BMC Cancer. 2014;14:957. doi:10.1186/1471-2407-14-957.
- M A M, Saunders DN, Henderson MJ, Clancy JL, Russell AJ, Lehrbach G, Musgrove EA, Watts CK, Sutherland RL. The E3 ubiquitin ligase EDD regulates S-phase and G(2)/M DNA damage checkpoints. Cell Cycle. 2007;6:3070–3077. doi:10.4161/cc.6.24.5021.
- Smits VA. EDD induces cell cycle arrest by increasing p53 levels. Cell Cycle. 2012;11:715–720. doi:10.4161/cc.11.4.19154.
- Ling S, Lin WC. EDD inhibits ATM-mediated phosphorylation of p53. J Biol Chem. 2011;286:14972–14982. doi:10.1074/jbc.M110.182527.
- Li J, Yang Z, Chen Z, Bao Y, Zhang H, Fang X, Yang W. ATF3 suppresses ESCC via downregulation of ID1. Oncol Lett. 2016;12:1642–1648. doi:10.3892/ol.2016.4832.
- San-Marina S, Han Y, Suarez Saiz F, Trus MR, Minden MD. Lyl1 interacts with CREB1 and alters expression of CREB1 target genes. Biochim Biophys Acta. 2008;1783:503–517. doi:10.1016/j.bbamcr.2007.11.015.