
ABSTRACT
In our previous studies, using a B cell vaccine (scFv-Her2), the targeting of tumor-associated antigen Her2 (human epidermal growth factor receptor-2) to B cells via the anti-CD19 single chain variable fragment (scFv) was shown to augment tumor-specific immunity, which enhanced tumor control in the prophylactic and therapeutic setting. However, the fusion protein displayed limited activity against established tumors, and local relapses often occurred following scFv-Her2 treatment, indicating that scFv-Her2-induced responses are inadequate to maintain anti-tumor immunity. In this study, targeting the IV region (D4) of the extracellular region of Her2 to B cells via CD19 molecules (scFv-Her2D4) was found to enhance IFN-γ-producing-CD8+ T cell infiltration in tumor tissues and reduced the number of tumor-infiltrating myeloid-derived suppressor cells (MDSCs). However, negative co-stimulatory molecules such as programmed cell death protein-1 (PD-1), CD160, and LAG-3 on T cells and programmed death protein ligand-1 (PD-L1) on tumor cells were upregulated in the tumor microenvironment after scFv-Her2D4 treatment. Further, anti-PD1 administration enhanced the efficacy of scFv-Her2D4 and anti-tumor immunity, as evidenced by the reversal of tumor-infiltrating CD8+ T cell exhaustion and the reduction of MDSCs and Treg cells, which suppress T cells and alter the tumor immune microenvironment. Moreover, combining this with anti-PD1 antibodies promoted complete tumor rejection. Our data provide evidence of a close interaction among tumor vaccines, T cells, and the PD-L1/PD-1 axis and establish a basis for the rational design of combination therapy with immune modulators and tumor vaccine therapy.
Introduction
Breast cancer is the second most common cancer globally, which threatens women’s health.Citation1 Common therapies for breast cancer management include surgery, radiotherapy, chemotherapy, and immunotherapy.Citation2,Citation3 Immunotherapy is a new treatment for breast cancer, inducing the body’s immune system to fight cancer.Citation4,Citation5 The Her2/neu (human epidermal growth factor receptor 2) gene encodes an epidermal growth factor receptor-(EGFR)-related tyrosine kinase that is overexpressed in 20–25% of invasive breast cancers. As such, Her2 has become an important therapeutic target in breast cancer.Citation6 Herceptin, a recombinant humanized monoclonal antibody directed against the extracellular domain (ECD) of the Her2 protein is widely used in oncology for Her2+ patient care.Citation7 However, the objective response rates to Herceptin monotherapy are low, with a median duration of 9 months. Therefore, overcoming antibody tolerance is critical to improve the survival of patients with Her2-overexpressing tumors.Citation3,Citation8 CD8+ T cell responses were found to be effective against these tumors. Thus, generating sustained and active immune responses to the Her2 protein is essential for this existing approach.Citation9,Citation10
B cells are capable of eliciting antitumor responses through antibody (Ab) production and by serving as antigen-presenting cells (APCs) to induce T cell responses.Citation11,Citation12,Citation13 CD19 is a B cell-specific member of the Ig superfamily expressed at almost every stage of B cell development, except after differentiation into plasma cells.Citation13 CD19 is also considered a co-receptor for BCR (B cell receptor) and is essential for B cell activation by promoting B cell receptor–antigen microcluster formation in response to membrane-bound ligands.Citation14 Our previous studies also demonstrated the utility of targeting B cells through CD19 molecules (scFv-Her2) for cancer therapy.Citation15 Nonetheless, the targeting of tumor-associated antigens to B cells has displayed limited activity against established tumors, and local relapses have occurred following scFv-Her2 treatment, indicating that scFv-Her2-induced responses are inadequate to maintain anti-tumor immunity.Citation15,Citation16 Therefore, elucidating the molecular mechanism through which tumor cells escape immune surveillance is needed to improve the efficacy of B cell vaccines.
Many co-inhibitory molecules play major roles in tumor evasion from immunosurveillance, such as PD1, CD160, and LAG-3, which are linked to the intratumoural over-expression of some cognate ligands, such as PD-L1 on APCs, thereby favoring a tolerogenic environment.Citation17 Programmed cell death protein 1 (PD1)–PD-L1 (a PD-1 ligand) interaction plays a very important role in tumor immune escape.Citation18–Citation20 PD-1, predominantly expressed on activated T cells, is an important immune checkpoint receptor. PD-1 transmits inhibitory signals to T cells after binding to PD-Ls in the tumor microenvironment.Citation21,Citation22 Tumor cells promote T cell dysfunction through the expression of ligands binding to inhibitory receptors, including PD-L1 (as known as CD274).Citation23 Currently, checkpoint blockade therapies such as anti-PD1 immunotherapy have been noticeably effective in reactivating T cell responses and providing long-term protection to patients.Citation24 However, no objective responses were found when large patient populations were treated with checkpoint blockade monotherapies.Citation25 Thus, combinations with other drugs are needed to promote synergistic action on these two major oncogenic pathways, which might result in better response rates and potential benefit from these therapies.
In this study, we fused the IV region (D4) of the extracellular region of Her2 with scFv by constructing a CD19 molecule single-chain antibody (scFv). Targeting the tumor-associated antigen Her2D4 to B cells combined with a PD1 antibody not only effectively induced the production of Herceptin-like antibodies, but also enhanced the killing effect of antigen-specific T cells in vivo. Moreover, it reversed the tumor-induced immunosuppressed microenvironment, significantly prolonging the survival time of tumor-bearing mice. Thus, this approach offers a new avenue for cancer immunotherapy.
Materials and methods
Mice and cell lines
Six-to-eight-week-old female BALB/c were purchased from the Animal Experimentation Center of Xi’an Jiaotong University, China. All mice used in the experiments were housed under specific pathogen-free conditions in the animal facility at Xi’an Jiaotong University and were treated in accordance with the guidelines of the Institutional Animal Care and Use Committee (xjtu2014136). The mouse colon cancer cell line CT26 and mouse breast cancer cell line 4T1, obtained from the American Type Culture Collection, were maintained in RPMI1640 medium (Hyclone, Thermo Scientific) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Sigma), 2 mM L-glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin. Further, 293 T cells were stored in our laboratory and cultured in Dulbecco’s modified Eagle’s medium (Hyclone) supplemented with 10% FBS, non-essential amino acids, and penicillin-streptomycin (Hyclone) at 37°C with 5% CO2. OrigamiB (DE3) pLysS competent cells were purchased from Novagen.
Reagents
Purified anti-mouse PD1 mAb (clone J43) and control Ig (2A3) were purchased from BioXCell and used with the schedule and dose as indicated in vivo. The fluorescently labeled anti-mouse PD1 mAb (clone RMP1-30) from BioLegend was used in vitro.The blocking Ab anti-mouse CD16/32 (553141) was purchased from BD Pharmingen. For flow cytometric analysis, fluorescently-labeled anti-mouse CD3e (4341614) and anti-mouse Granzyme B (1919536) antibodies were purchased from eBioscience. Fluorescently-labeled anti-mouse CD4 antibody (563933), anti-mouse IFN-γ antibody (554412), anti-mouse PD-L1 antibody (558091), and anti-mouse CD8a antibody (563068) were purchased from BD Pharmingen. Fluorescently-labeled streptavidin (405203), anti-mouse B220 (103212), anti-mouse CD80 antibody (104706), anti-mouse CD86 antibody (105007), anti-mouse I-A/I-E antibody (107614), anti-mouse CD45 antibody (103126), anti-mouse ly-6 G/ly-6 C antibody (108412), anti-mouse PD1 antibody (109111), anti-mouse CD127 (121111), anti-mouse/human CD44 (103006), anti-mouse CD160(143011), anti-mouse CD223 (LAG-3; 125207), and anti-mouse TNF-α (506305) were purchased from BioLegend. CD19+ microbeads (130-052-201) were purchased from Miltenyi Biotec.
Generation of CD19 scFv and fusion proteins
The pET-20b(+)–anti-CD19–scFv–c-ErbB-2cDNA constructs that encode the Herceptin-binding domain (designated P3–4) were generated.Citation15 The IV region (D4) of the extracellular region of Her2 (designated D4) was isolated from the plasmid pET-20b(+)–anti-CD19–scFv–c-ErbB-2cDNA using primers summarized in Supplemental Table I. The pET-43.1a-scFv, pET-43.1a-D4, and pET43.1a-scFv-D4 vectors were generated. These plasmids were transfected into OrigamiB(DE3)pLysS cells. Next, the cells were induced with 0.1 mM isopropyl β-D-thiogalactoside. Subsequently, the proteins were purified using His-Select Nickel Affinity Gel (Sigma-Aldrich), and LPS contamination was removed by Detoxi-Gel Endotoxin Removing Columns (GenScript Corp.). After this, the proteins were dialyzed in PBS and analyzed by SDS-PAGE and western blotting. The endotoxin level was 0.5 EU/mg, as measured by the LAL assay (GenScript Corp.).
Western blotting
Recombinant anti-CD19 scFv-D4 (scFv–D4), Her2/Neu D4 fusion proteins, and the anti-CD19 scFv miniAb proteins were produced and purified. Protein samples were loaded on a 10% (w/v) tris-HCl SDS-PAGE gel for electrophoresis, transferred to PVDF membranes (Millipore), and blotted. Subsequently, the membrane was probed with anti-His (1:3000, ab5000, Abcam) and anti-Her2 (1:0000, AF1129, RD) antibodies at 4°C overnight. The signal was further detected using the following secondary antibodies: goat anti-mouse (1:3000, ab6789, Abcam) and rat anti-goat IgG (1:5000,ZB-2306, Beijing Zhongshang Jinqiao Biotechnology Co., Ltd.) conjugated with horseradish peroxidase at room temperature for 1 h. Band signals were visualized using the Western Chemiluminescent substrate (Millipore).
Fusion protein binding assay
For the binding assay in vitro, splenocytes were incubated with biotin-labeled proteins, scFv-biotin, D4-biotin, or scFv-D4-biotin, and then stained with PE-streptavidin and APC–anti-mouse B220. Cells were washed and assessed by flow cytometry. For the binding assay in vivo, mice were injected i.v. with biotinylated fusion proteins scFv, D4, or scFv-D4. Peripheral blood was drawn 10 min after injection. Cells were stained with APC-anti-mouse B220 and PE streptavidin and then assessed by flow cytometry.
B cell activation assay
Purified B cells were stimulated with scFv, D4, or scFv-D4 (5 μg/mL) for 24 h, followed by harvesting to detect CD80, CD86, and MHCII surface marker expression by flow cytometry.
Mouse immunization and B cell presentation assay in vitro
Six-to-eight-week-old BALB/c mice were immunized i.v. with scFv, D4, or scFv–D4 at 50 μg per mouse per injection on days 0, 7, and 14. A group of mice immunized with PBS was used as a control. On days 21, the sera were collected for Her-2/Neu Ab measurements. Further, the splenocytes were harvested for T cell activation measurements. For the B cell presentation assay in vitro, splenocyte B cells were depleted using CD19+ microbeads. After that, splenocytes with or without B cells were stimulated with scFv-D4 for 3 days and then re-stimulated with PMA and ionomycin in the presence of brefeldin A (BD) for 4 h. Finally, IFN-γ production by CD4+ T cells was assayed by flow cytometry.
Detection of Her-2D4 Abs by ELISA
At first, 96-well plates were coated with recombinant Her-2/Neu D4 protein (1 μg per well) overnight at 4°C and blocked with 1% BSA/PBST (200 μL per well). Subsequently, the pre- or post-immune sera from mice were diluted (1:200) and further reacted with goat anti-mouse IgG HRP conjugates (Abcam, ab97023). Finally, the assays were developed by the addition of the Soluble TMB Substrate Solution (TIANGEN BIOTECHCO, LTD), and the OD450 nm was determined.
Ab competitive inhibition assay
Briefly, 96-well plates were coated with recombinant Her-2 D4 (1 μg/well) overnight at 4°C and then blocked with 1% BSA/PBST (200 μL per well) for 2 h at room temperature. The diluted immune sera (1:10) were added into wells for 1 h at room temperature, followed by Herceptin (Roche Pharma (Schweiz) Ltd) for 1 h at room temperature. Pre-immune sera were used as controls. Subsequently, the wells were incubated with anti-human IgG HRP conjugates (Abcam, ab6858) and the TMB Microwell Substrate. The OD450 nm was measured. The following formula was used to calculate the percent inhibition: (ODpre − ODpost)/ODpre × 100%.
Lentivirus production and stable cell line generation
To generate CT26 and 4T1 stable cell lines with the integrated human HER2 gene (CT26/E2, 4T1/E2), the pWPI-Her2 vectors were generated using primers summarized in Supplemental Table I. Further, the plasmids pWPI-Her2 were transfected into 293 T cells seeded at a density of 1 × 10Citation6 cells per 5-cm plate, and the cells were co-transfected with the ΔR9 and pVSVG helper plasmids using the X-tremeGENE HP DNA transfection reagent (Roche, Mannheim). Supernatants were collected after 72 h of transfection. CT26 and 4T1 cells (1 × 10Citation6) were infected with a freshly prepared Her2 lentivirus at room temperature in the presence of 8 μg/mL polybrene. Her2+CT26 and Her2+4T1 tumor cells were sorted by flow cytometry. Human Her2 expression was confirmed by western blotting.
Tumor challenge
Six-to-eight-week-old female BALB/c mice were injected s.c. with 4T1/E2 (1 × 10Citation6) or CT26/E2 (1 × 10Citation6) cells into the lower right flank. ScFv-D4 and anti-PD1 treatment commenced when the average tumor diameter reached 3–5 mm. ScFv, D4, and scFv-D4 were injected i.v. (50 μg per mouse) on days 5, 12, and 19. Anti-PD1 was injected i.v. at a dose of 100 μg per mouse every 3 days. Tumor size (microcalipers) and mouse weight were measured weekly. Mice were sacrificed when the tumor size reached 12 mm in diameter. Tumor volume was calculated using the formula: 0.5 × length × widthCitation2, where the length was the longer dimension. In some experiments, the mice were sacrificed on day 26 or 34 after tumor cell inoculation, and surface and intracellular cytokine-staining patterns of MDSCs and T cells were analyzed by flow cytometry.
Flow cytometry
Single-cell suspensions from tumors, tumor draining lymph nodes, tumor non-draining lymph nodes, and spleens were generated by macerating the tissues through a 70-μM nylon mesh. All flow cytometric analysis were performed on a Beckman CytoFLEX and analyzed using Kaluza software2.1.
Surface and intracellular molecular staining
For surface and intracellular staining of splenocytes, cells from tumor tissues and tumor draining and tumor non-draining lymph nodes were stimulated with PMA plus ionomycin for 4 h in the presence of brefeldin A, which was followed by staining with APC- or FITC-conjugated mAbs against mouse CD8 or CD4, PD1, and PE-conjugated anti-mouse IFN-γ or anti-mouse gzm B. For surface staining of tumors, isolated tumors were cut into small pieces and incubated in dissociation solution with 2 mg/mL collagenase type I (Sigma), 2 mg/mL collagenase type IV (Sigma), and 25 μg/mL DNase (Sigma). The solution was mixed up and down while incubating for 45 min at 37°C, and the suspension was filtered through 70-μm cell strainers to generate single-cell suspensions. After counting viable cells, the samples were incubated with an anti-CD16/CD32-blocking Ab and then stained with fluorochrome-labeled Abs against CD45, CD3, CD4, CD8, CD44, CD127, LAG3, CD160, CD11b, Gr1, PD1, and PD-L1. For intracellular staining of tumors, TILs were isolated from dissociated tumors and stimulated with PMA and ionomycin in the presence of brefeldin A (BD) for 4 h. Subsequently, the cells were fixed, permeabilised, and stained with Abs against IFN-γ, TNF-α, and gzmB. Fluorescence data were acquired on a Beckman CytoFLEX and analyzed using Kaluza software.
Statistical analysis
One-way ANOVA followed by Tukey’s multiple comparison test was applied to assess the statistical significance of differences between multiple treatment groups. The tumor volumes on indicated days were evaluated by one-way ANOVA. Kaplan–Meier survival analysis was used to determine the significance of the observed differences in results for in vivo tumor therapy. p < .05 (*), p < .01 (**), and p < .001(***) were considered statistically significant.
Results
Generation and characterization of an anti-CD19 scFv fusion protein
Our previous studies have suggested that targeting of antigens via CD19 can lead to enhanced Ag-specific T cell responses, which has demonstrated significant efficacy for some cancers.Citation15 Based on a previous report that the Herceptin-binding domain is located in the Her-2/neu ECD D4 domain,Citation26 we generated her-2/neu ECD D4 cDNA, which was ligated with anti-CD19 scFv plasmids. Hereafter, these recombinant proteins were produced, purified, and characterized. As indicated in , recombinant anti-CD19 scFv-D4 (scFv–D4), her-2/neu D4 fusion proteins were detected by both His-Tag and her-2/neu Abs. In contrast, the anti-CD19 scFv miniAb protein was detected with the His-Tag Ab but not with the her-2/neu Ab.
Figure 1. Generation and characterization of anti-CD19 scFv fusion protein
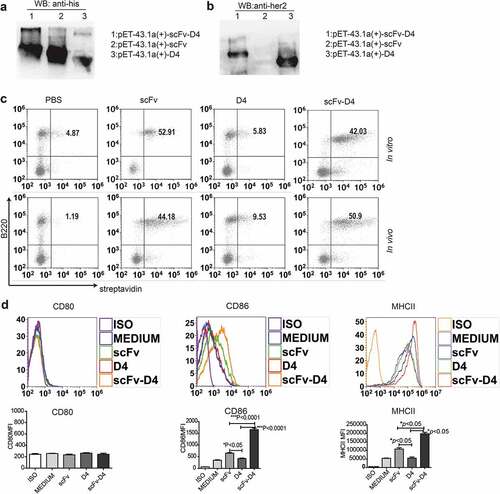
To verify that the scFv, scFv–D4 proteins retained the Ag-binding activity of the parental Ab, we tested specific binding to B cells in vitro and in vivo by flow cytometry. For the binding assay in vitro, splenocytes were incubated with protein biotin-scFv, biotin-D4, and biotin-scFv-D4. For the binding assay in vivo, biotin-labeled proteins were intravenously (i.v.) injected into mice. Peripheral blood was taken 10 min after the injection. Successful targeting of B cells was observed by identifying double positive cells ()). These results suggested that the anti-CD19 scFv miniAb with or without labeled Ag was capable of specifically binding B cells. Next, we measured whether the fusion protein scFv–D4 could activate B cells. Purified B cells were incubated with fusion proteins scFv, D4, or scFv-D4 for 24 h. Subsequently, we examined the expression of surface activation markers on B cells by flow cytometry. We found that the fusion protein scFv-D4, but not D4, significantly upregulated CD86 and MHCII on B cells but not CD80 ()). ScFv alone also stimulated moderate expression of CD86 and MHC class II, but the levels were significantly lower than those stimulated by scFv–D4 ()).
Targeting Ag to B cells elicits antibody generation and T cell responses
We next examined whether targeting TAA her-2/neu Ag to B cells could elicit anti-tumor Abs. As shown in ), mice immunized with scFv–D4 secreted large amounts of her-2/neu Abs. ScFv or D4 protein immunization did not induce secretion of any significant level of her-2/neu Ab. Because D4 contains the Herceptin-binding domain, we examined whether Abs from mice immunized with scFv–D4 possessed Herceptin-like activity. A competitive inhibition assay was performed using a solid-phase immunoassay with recombinant Her-2 D4 as the target Ag. The results showed that the post-immune sera from mice immunized with scFv–D4, but not scFv or D4, were capable of competing with Herceptin binding ()).
Figure 2. CD19-mediated Ag targeting of B cells induces antibody and T cell responses
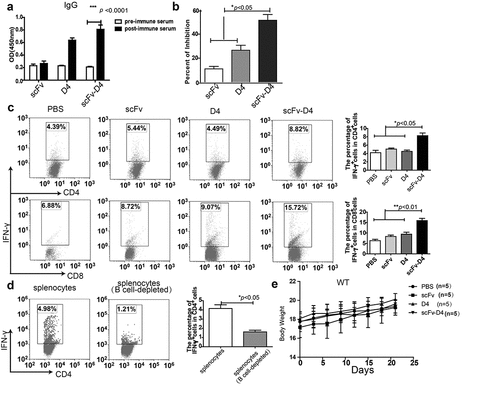
Next, we determined whether anti–her-2/neu T cell responses were generated by this vaccination strategy. The splenocytes from mice vaccinated with different fusion proteins were harvested and stimulated with scFv–D4 protein. We discovered that both IFN-γ-producing CD8+T cells and IFN-γ-producing CD4+T cells were significantly increased in mice immunized with the scFv-D4 fusion protein, compared to those from unimmunized, scFv-immunized, or D4-immunized mice ()). To further confirm that targeting TAA Her2/neu Ag to B cells could elicit T cell responses through B cell antigen presentation, the splenocytes from mice vaccinated with the scFv-D4 fusion protein were harvested, and then B cells were depleted. Subsequently, splenocytes with or without B cells were stimulated with scFv–D4 protein. The results showed that IFN-γ-producing CD4+T cells from splenocytes without B cells were significantly decreased compared to those from splenocytes with B cells ()). Additionally, no signs of toxicity or weight loss were observed in any of the treatment groups ()).
Targeting Ag to B cells shows remarkable anti-tumor activity in a 4T1/E2 breast cancer model
To determine whether scFv-D4 would induce greater inhibition of tumor growth than that in any control group, murine breast cancer cells (4T1/E2) and colon cancer cells (CT26/E2) that express human her-2/neu were prepared; the her-2/neu+ tumor cells were sorted by flow cytometry, and her-2/neu expression in tumor cells was confirmed (suppl. Figure 1(a,b)). Next, we treated BALB/c mice with established 4T1/E2 tumors with the scFv-D4 fusion protein. The 4T1/E2 cells were inoculated into BALB/c mice subcutaneously (s.c.). When tumors reached a size of approximately 3–5 mm in any direction, mice were either treated with PBS, scFv, D4, or scFv-D4 (50 μg per mouse i.v. once per week, for 3 weeks; )). Tumor measurements over the treatment period showed that the tumor size was significantly decreased in the scFv-D4 group, compared to that in the PBS, scFv, or D4 groups ()). Comparisons of the average tumor volume at days 24, 27, and 30 revealed statistically significantly differences in average tumor volumes between scFv-D4 and D4 groups (p < .05). Additionally, differences for each, in comparison with values in vehicle control or scFv groups, were statistically significant (p < .05; )). When mice were treated with PBS, scFv, D4, or scFv-D4 (50 μg per mouse, once per week, three times, i.v.), the survival rates at day 60 were 0%, 0%, 0%, and 40%, respectively. The scFv-D4 fusion protein further increased the overall survival time compared to that with any therapy (p < .05 vs. D4 and scFv; )).
Figure 3. Targeting Ags to B cells induces anti-tumor activity in a 4T1/E2 breast cancer model
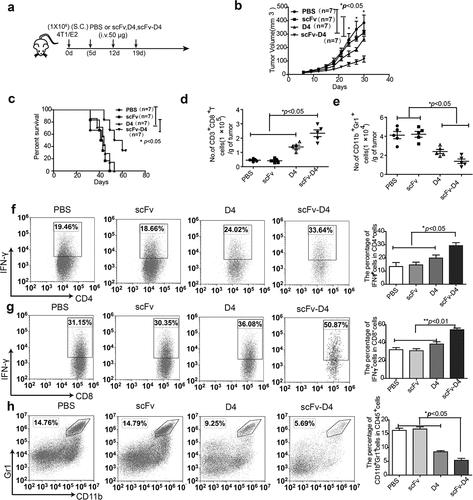
To determine whether the fusion protein scFv–D4 boosts or inhibits anti-her-2/neu T cell responses, BALB/c mice with established 4T1/E2 tumors were treated with PBS, scFv, D4, or scFv-D4, as previously described. On day 26 after tumor cell inoculation, tumors of different groups were harvested, weighted, and analyzed by flow cytometry. CD45+CD3+CD4+T cells, CD45+CD3+CD8+ T cells, and CD45+CD11b+GR1+cells were gated (suppl. Figure 2(a, b)), and IFN-γ production and the MDSC ratio in splenocytes, lymph nodes and tumor tissues from tumor-bearing mice were evaluated. Compared to those with PBS, scFv, and D4 therapies, we found that scFv-D4 treatment increased the number of tumor-infiltrating CD8+T cells and decreased the number of MDSCs in tumor tissues (). Furthermore, we observed that scFv-D4 treatment resulted in an increase in the percentage of IFN-γ-producing CD4+T cells in the tumor ()), indicating a shift toward a Th1 response. We also found that IFN-γ-expressing CD8+T cells were significantly increased in mice treated with the scFv–D4 fusion protein, compared to those in scFv-, D4-, or PBS-treated mice ()). Additionally, a lower proportion of tumor-infiltrating MDSCs in the scFv-D4 fusion protein treatment group was found compared to that in controls ()). These effects appeared to be limited to the tumor and tumor draining lymph node (suppl. Figure 3), as treatment had no impact on T cell function in the tumor non-draining lymph node (suppl. Figure 4) or the number of MDSCs in the spleen (suppl. Figure 5). Collectively, these data demonstrated that the scFv-D4 fusion protein augmented the number of functionally-active T cells within tumors and decreased the proportion of MDSCs.
Targeting Ag to B cells enhances PD1 and PD-L1 expression in the tumor microenvironment
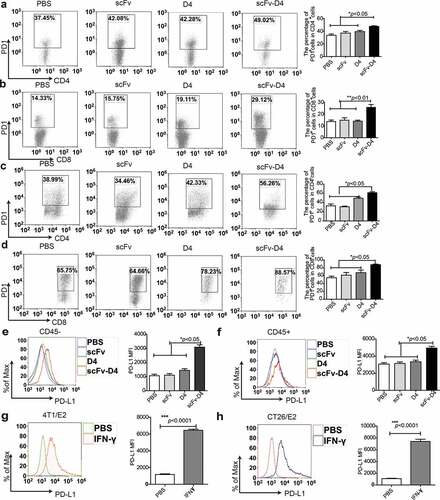
Targeting antigens to B cells generated Herceptin-like antibodies and promoted the T cell response in tumor-bearing mice, but only ∼40% of mice achieved long-term, tumor-free survival in the therapeutic setting, which suggested that a large number of tumor cells still evaded immune surveillance. We next sought to determine whether immune escape occurred after immunization with scFv-D4 fusion protein. The inhibitory molecule PD-L1 has been shown to exhaust cytotoxic T lymphocyte (CTL) by binding to PD1.Citation23 First, we examined whether PD1 expression in T lymphocytes was increased by this vaccination method. The splenocytes from mice vaccinated with different kinds of fusion proteins were harvested, and PD1 expression was examined by flow cytometry. We found that the PD1 expression levels in both CD4+T cells and CD8+T cells were significantly increased in mice immunized with the scFv–D4 fusion protein, compared to those in the control groups (). Moreover, we evaluated whether PD1 expression on tumor-infiltrating T lymphocytes was increased in scFv-D4-treated mice bearing 4T1/E2 tumors. We observed increased PD1 expression on tumor-infiltrating T lymphocytes after scFv-D4 treatment (). PD1 is not only a marker of T cell activation but also eventually leads to subsequent T cell exhaustion by binding to PD-L1, which is expressed on tumors and many immune cells.Citation27 Therefore, we evaluated whether scFv-D4 treatment would affect PD-L1 expression on tumor cells and tumor-infiltrating lymphocytes (TILs) in the tumor microenvironment. Results showed increased PD-L1 expression on tumor cells (CD45− cells; )) and TILs ()) following scFv-D4 treatment. As our data confirmed elevated IFN-γ production in tumor tissues following scFv-D4 treatment (), we next assessed whether IFN-γ promoted PD-L1 expression in both 4T1/E2 and CT26/E2 cells (). These data suggested that increased inflammatory cytokines might promote PD-L1 expression on tumor cells. Therefore, the high expression of PD1 and PD-L1 in tumor tissues after immunization with scFv-D4 fusion protein could be one of the important mechanisms underlying the immune escape of tumor cells.
Figure 4. Targeting Ags to B cells promotes PD1 and PDL1 expression in the tumor microenvironment
Combined treatment with scFv-D4 and anti-PD1 reverses tumor-induced immunosuppressive microenvironment and induces significant anti-tumor effects in a 4T1/E2 breast cancer model
The high PD1 and PD-L1 expression in the tumor microenvironment from scFv-D4-treated tumor-bearing mice prompted us to further investigate whether combining scFv-D4 and anti-PD1 would bring about greater inhibition of tumor growth than that with any agent alone. The 4T1/E2 cells were inoculated into BALB/c mice s.c. Once the tumors reached a size of approximately 3–5 mm in any direction, mice were treated with scFv-D4 (50 μg per mouse i.v. once per week). Subsequently, anti-PD1 (100 μg per mouse, i.v., and twice per week) was administered after the cycle of scFv-D4 treatment was complete ()). Tumor measurements over the treatment period showed that the tumor sizes were significantly decreased in the scFv-D4 and combination treatment groups compared to those in isotype control antibody or anti-PD1 groups. However, the smallest tumors were found in the combination group ()). When mice were treated with isotype control antibody, anti-PD1 antibody (100 μg per mouse, i.v., twice per week), scFv-D4 (50 μg per mouse, once per week, i.v.), or a combination of the two, the survival rates at day 66 were 0%, 28.6%, 42.9%, and 85.7%, respectively. The combination of scFv-D4 and anti-PD1 antibody further increased the overall survival time compared to that with any monotherapy (p < .05 vs. scFv-D4 and anti-PD1; )). These data suggest that the PD1/PD-L1 pathway inhibits activation of the immune system. Thus, anti-PD1 can block the PD1/PD-L1 inhibitory signal and boost the efficacy of scFv-D4.
Figure 5. Combined treatment with scFv-D4 and anti-PD-1 induces immune cell infiltration and shows remarkable anti-tumor activity in a 4T1/E2 breast cancer model
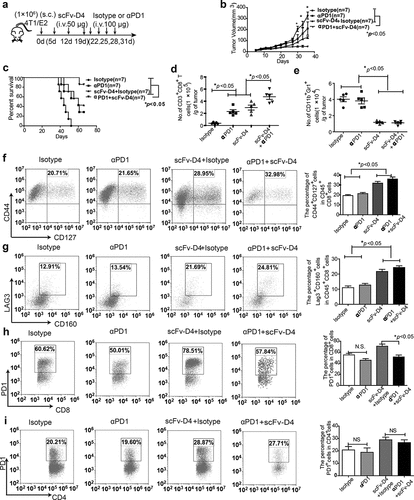
Since the data have confirmed that the combined therapy could enhance anti-tumor effects, we next investigated whether the beneficial anti-tumor effects of the combination therapy could be related to the reversion of a CD8+ T cell exhaustion phenotype. The 4T1/E2 tumor-bearing BALB/c mice were treated with scFv-D4 and anti-PD1. On day 34 after tumor cell inoculation, tumors of different groups were harvested, weighted, and analyzed by flow cytometry. Compared to that with scFv-D4 and anti-PD1 antibody monotherapies, we found that combination treatment increased the number of tumor-infiltrating CD8+T cells ()). The number of tumor-infiltrating MDSCs was also reduced by the combined therapy ()). Further, 4T1/E2 tumor-infiltrating CD8+ T cells that were subjected to the scFv-D4 treatment in combination with anti-PD-1 blockade had a higher proportion of memory T cells (CD44+CD127+CD8+) ()) and lower surface expression of PD-1 ()). PD1 expression in CD4+T cells was not affected in the groups treated with combined scFv-D4 and anti-PD1 ()). We also examined surface LAG3 and CD160 expression on tumor-infiltrating CD8+T cells ()). Interestingly, the expression of these checkpoint molecules on CD8+T cells was induced by scFv-D4 treatment by not PD1 antibody treatment. This conclusion is consistent with a result of a previous report in which PD1 blockade did not affect T cell expression of other inhibitory receptors co-expressed with PD-1, including CD160 and LAG-3.Citation28 Taken together, these results indicated that reduced tumor growth mediated by combination treatment was associated with a change in the tumor-induced immunosuppressive microenvironment state.
Combined treatment with scFv-D4 and anti-PD1 enhances the anti-tumor activity of T cells in the tumor microenvironment
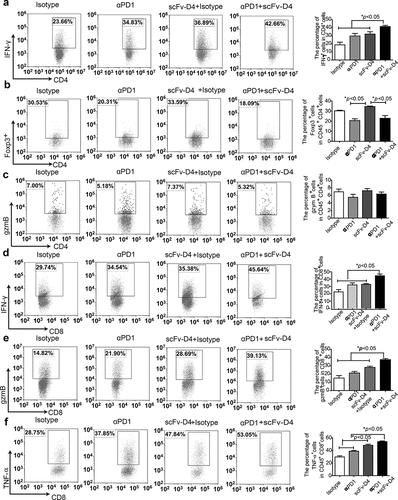
To further explore the mechanism underlying the reduction in tumor growth mediated by an increase in T cells within tumors, the 4T1/E2 tumor-bearing BALB/c mice were treated as performed to obtain for the results of ). We then analyzed effector CD4+ and CD8+T cells within tumors. Compared to that with scFv-D4 and anti-PD1 antibody monotherapies, we found that combination treatment increased the proportion of IFN-γ-expressing cells among tumor-infiltrating CD4+T cells ()) and decreased the percentage of CD4+Foxp3+ T cells within tumors ()). We also showed Granzyme B (gzmB) production by tumor-infiltrating CD4+T cells, but there was no significant difference among the groups ()). Furthermore, we found significantly increased IFN-γ, gzmB, and TNF-α production by tumor-infiltrating CD8+T cells in the combination group (). These data suggested that the reduction in Treg cells and increase in effector CD8+T cells and CD4+T cells contributed to the anti-tumor immune responses induced by the combined therapy. These results further confirmed that combined treatment can reverse the tumor-induced immunosuppressive microenvironment.
Figure 6. scFv-D4 and anti-PD1 combination therapy enhances T cell responses in a 4T1/E2 tumor model
Treatment with fusion protein scFv–D4 and anti-PD1 induces significant antitumor effects in a CT26/E2 tumor model
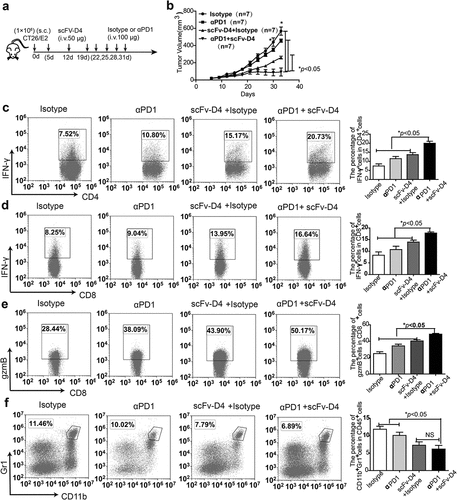
To further confirm the combined therapeutic effect, BALB/c mice implanted with human her-2/neu-expressing murine colon carcinoma CT26 cells were treated with different regimens. Similar to that with the 4T1/E2 BALB/c model, the tumor-bearing mice were treated with αPD1 and scFv-D4 alone or combination ()). Combination treatment groups had a significantly lower tumor burden than anti-PD1- or scFv-D4-treated mice or those of the isotype–control group ()). Further, we analyzed whether the effect of combined scFv-D4 and anti-PD1-mediated enhanced T cell responses in CT26/E2 tumors was the same as that in 4T1/E2 tumors. We found that combination treatment increased the proportion of IFN-γ-expressing cells among tumor-infiltrating CD4+ and CD8+ T cells, compared to that with scFv-D4 and anti-PD1 monotherapies (). Furthermore, combination treatment significantly increased gzmB production by tumor-infiltrating CD8+T cells upon assessing primary tumors of CT26/E2 mice ()). Of note, there were fewer tumor-infiltrating MDSCs in the combination treatment groups than in controls ()).
Figure 7. Treatment with fusion protein scFv–D4 and anti-PD1 induces significant anti-tumor effects in a CT26/E2 tumor model
Discussion
This study aimed to produce a sustained anti-tumor T cell response. Although targeting antigens to B cells via CD19miniAb has been shown to generate humoral and potent T cell responses in our previous study,Citation15 sustained T cell responses are required to eradicate tumor cells. Therefore, we established a combined treatment protocol comprising a novel CD19-targeting fusion protein and a PD1 antibody. We showed that the combined treatment generated both humoral and sustained T cell responses. The efficacy of this combined treatment was demonstrated in murine breast and colorectal cancer models. It appears that the combined treatment reversed tumor-induced immunosuppression in cancer tissues and inhibited the progression of HER2-overexpressing breast and colon cancer in mice. Moreover, this strategy can be further extended to other solid tumors.
The CD19-targeted CAR-T cell therapy in B-cell acute lymphoblastic leukemia has proven to be effective;Citation29,Citation30 however, targeting antigens to CD19 also activates B cells to become potent APCs,Citation15,Citation31 during which the activated B cells present antigens to prime T cell activation.Citation15,Citation31,Citation32 Additionally, there are still some challenges to be addressed. First, targeting antigens to B cells inhibits the growth of breast cancer cells. However, the effect of the fusion protein on immune cells in the tumor microenvironment is unclear. Second, targeting antigens to CD19 could prolong the survival time of tumor-bearing mice, but only ∼20% of treated mice achieved long-term, tumor-free survival in the therapeutic setting in terms of tumor progression, which indicated that a large number of tumor cells evaded immune surveillance. To resolve these issues, we fused the IV region of the extracellular region (D4) of the human epidermal growth factor receptor 2 (HER2) tumor antigen with the CD19 molecule single chain antibody (scFv) and obtained a soluble, functional fusion protein by reducing the size of the protein and using new expression strains. The uniqueness of the B cell-based vaccine approach is that antigens targeted to B cells elicit exaggerated Ag-specific Ab responses, although targeting antigens to dendritic cells (DCs) via lectins such as DEC205 has been shown to indirectly promote B cell humoral responses.Citation33 When DCs capture antigens to induce Ab responses, antigens are digested and dominant epitopes are presented on the surface as MHC class I or class II molecules.Citation34,Citation35 The generation of blocking antibodies requires the presentation of intact Ag to B cells.Citation36 Data from the present and previous studies have confirmed that targeting the Her2 antigen to B cells elicits the generation of a Herceptin-like antibody. DCs are considered more potent APCs to induce T cell responses;Citation35 however, activated B cells also serve as APCs to induce T cell responses and play an important role in anti-tumor immune responses.Citation12,Citation37 Our study suggested that the new B cell vaccine activates B cells to up-regulate the surface molecules CD86 and MHC class II molecules, which are critical for T cell characteristics. Moreover, the data showed that the fusion protein scFv-D4 could not activate CD4+T cells in the absence of B cells, which indicated that the targeting of B cells could stimulate B cells to present antigens to CD4+T cells. In addition, targeting antigens to B cells in vivo did not result in weight loss and the appearance of autoimmunity phenomena in mice, which indicated that the fusion protein was not toxic to the mice.
The ability of CTLs to provide effective anti-tumor immunity in vivo is clearly seen in animal experiments using carcinogen-induced and DNA virus-induced tumors. CTLs perform a surveillance function by recognizing and killing potentially malignant cells that express peptides derived from tumor antigens and presented in association with class I MHC molecules.Citation38 In particular, the number and activity of CD8+T cells in tumor tissues correlate with better prognosis.Citation39 Data have confirmed that the number of CD8+T cells was increased in scFv-D4-treated tumors compared to that in control-treated tumors. However, ~60% of tumor-bearing mice from the scFv-D4 group ultimately died, indicating that many tumors successfully evaded the immune system. The most important mechanism of immune evasion by tumors is the inhibition of anti-tumor immune responses.Citation40 Tumor-induced immunosuppression inhibits T cell infiltration. However, some checkpoint molecules on tumor cells, APCs, or T cells might be involved in inhibiting the activation of tumor-specific T cells.Citation41,Citation42 In this study, we found that the percentage of IFN-γ-producing-CD4+T cells, INF-γ-producing CD8+T cells, and CD44+CD127+CD8+T cells was increased in the scFv-D4 treatment group. However, PD-L1 expression on tumor cells and expression of the checkpoint molecules PD1, LAG3, and CD160 on CD8+T cells were also up-regulated ( and ). Meanwhile, scFv-D4 treatment also enhanced the infiltration of CD4+Foxp3+Treg cells in tumor tissues (). CD4+Foxp3+ Treg cells and checkpoint molecules are essential for maintaining self-tolerance and preventing excessive immune responses,Citation43 but immune suppression can also be induced by an increase in Treg cells and the expression of PD-1 and LAG3Citation43,Citation44on T cells. The checkpoint molecules PD1, LAG3, and TIM3 have been shown to be hijacked by tumor cells to evade the host immune system in both mice and humans, and immunogenic tumor cells might induce the anergy of tumor-specific T cells by expressing these molecules on their surfaces,Citation22 which explains why some tumor cells could escape immune surveillance in this study. It is not surprising that the expression of PD-L1, PD1, and LAG3 was increased in the tumor tissues of the scFv-D4-treated group, because inflammatory cytokines such as IFN-γ stimulate T cells to express PD1 and LAG3, stimulate tumor cells or myeloid cells to express PD-L1, and inhibit the activity of tumor-specific T cells.Citation23 Results also confirmed that more IFN-γ was produced in tumor tissues from the scFv-D4 treatment group than from those of control groups. Thus, targeting of PD1or LAG3 might overcome the shortcoming of the fusion protein and inhibit tumor cell growth.
Herceptin, an anti-HER2 monoclonal antibody is a widely-used therapy for patients with metastatic breast cancer.Citation45 Herceptin can slow or stop the growth of the breast cancer by blocking HER2/neu receptor from receiving growth signals or the cytotoxicity of Fc-driven innate immune effector functions. However, some breast cancer patients are refractory to Ab therapy despite high levels of her-2/neu expression on tumor cells. Furthermore, many patients who initially respond to Ab therapy ultimately develop resistance, leading to disease progression.Citation46 CD8+T cells are essential for anti-tumor associated antigen (TAA) monoclonal antibody efficacy, Her-2-specific CD8+T cell responses could eradicate drug-refractory tumors, but the cytotoxicity of antigen-specific CD8+T cells is blocked by tumor-induced immunosuppression in the tumor microenvironment.Citation9,Citation45,Citation47 Thus, it would be ideal if the treatment against her-2/neu TAA could generate both antibodies and lasting T cell responses.
In breast cancer, the blockade of PD1 or its ligand PD-L1 by specific monoclonal antibodies has been shown to reverse this effect and to potentiate cancer therapeutic immunity.Citation48,Citation49 However, single blockade of immune checkpoint molecules (such as PD1 and PD-L1) cannot effectively eliminate cancer cells.Citation42,Citation50 A combination with cancer immunotherapy might enhance the anti-tumor effect.Citation42 The results also suggested that the combination therapy could reverse tumor-induced immunosuppression and enhance anti-tumor immunity, as evidenced by the increase in tumor-infiltrating effector CD4+T cells and CD8+ T cells, decrease in Treg cells and MDSCs, and the longer survival time of the tumor-bearing mice. Interestingly, we also found that the combination therapy could not completely change the status of exhausted CD8+T cells; specifically, the expression of LAG3 on CD8+T cells was still increased, which would probably result in the escape of tumor cells from immunosurveillance. These effects on T cells reflect the complexity of tumor cell escape from immunosurveillance in vivo. The therapeutic efficacy of this combined treatment comprising a novel B cell vaccine and PD1 antibody was further tested on BALB/c mice using a CT26 colon carcinoma model. Similarly, tumor progression was significantly decreased in the tumor-bearing mice of the combination therapy group, even though the CT26 model exhibits a lower percentage of IFN-γ-producing-CD4+T cells and CD8+T cells. This is because there are fewer TILs in CT26 tumor tissues.Citation51 Thus, our study established that combined treatment could significantly inhibit the growth of tumor cells. This combined treatment strategy could be further extended to other fields including chronic infectious disease, particularly for the control of viral infection.
This study and our previous research suggest that the targeting of antigens to B cells combined with a PD1 antibody is more effective than either monotherapy. This combined therapy induced both the production of specific antibodies against tumor antigens and antigen-specific T cells. Moreover, the combined therapy maintained the cytotoxicity of CD8+T cells in tissues, although the mechanism of reversing the immunosuppressive state of the tumor microenvironment needs to be further addressed.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Authors’ contributions
Lv Z carried out the immunoassays and drafted the manuscript. Zhang P and Li D carried out the immunoassays. Qin M, Nie L, Wang X, and Ai L participated in the data analysis. Feng Z performed the statistical analysis. Woodvine O helped to revise the manuscript. Ma Y carried out the design of the study and helped to revise the manuscript. Ji Y carried out the design of the study. All authors read and approved the final manuscript.
Ethics approval and consent to participate
All animals were treated in this study in accordance with the guidelines of the Institutional Animal Care and Use Committee and were approved by the Institutional Animal Care and Use Committee of Xi’an Jiaotong University (xjtu2014136).
Availability of data and materials
Additional data are available as Supplementary information
Supplemental Material
Download ()Acknowledgments
We would like to thank Editage [http://online.editage.cn/] for English language editing.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
References
- Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–15. doi:10.3322/caac.21492.
- Force J, Leal J, McArthur HL. Checkpoint blockade strategies in the treatment of breast cancer: where we are and where we are heading. Curr Treat Options Oncol. 2019;20:35. doi:10.1007/s11864-019-0634-5.
- Takegawa N, Yonesaka K. HER2 as an emerging oncotarget for colorectal cancer treatment after failure of anti-epidermal growth factor receptor therapy. Clin Colorectal Cancer. 2017;16:247–251. doi:10.1016/j.clcc.2017.03.001.
- Rabinovich GA, Gabrilovich D, Sotomayor EM. Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol. 2007;25:267–296. doi:10.1146/annurev.immunol.25.022106.141609.
- Jenne CN, Kubes P. Immune surveillance by the liver. Nat Immunol. 2013;14:996–1006. doi:10.1038/ni.2691.
- Nahta R, Esteva FJ. Herceptin: mechanisms of action and resistance. Cancer Lett. 2006;232:123–138. doi:10.1016/j.canlet.2005.01.041.
- Valabrega G, Montemurro F, Aglietta M. Trastuzumab: mechanism of action, resistance and future perspectives in HER2-overexpressing breast cancer. Ann Oncol. 2007;18:977–984. doi:10.1093/annonc/mdl475.
- Meric-Bernstam F, Johnson AM, Dumbrava E, Raghav K, Balaji K, Bhatt M, Murthy RK, Rodon J, Piha-Paul SA. Advances in HER2-targeted therapy: novel agents and opportunities beyond breast and gastric cancer. Clin Cancer Res. 2019;25:2033–2041. doi:10.1158/1078-0432.CCR-18-2275.
- Ritter CA, Bianco R, Dugger T, Forbes J, Qu S, Rinehart C, King W, Arteaga CL. Mechanisms of resistance development against trastuzumab (Herceptin) in an in vivo breast cancer model. Int J Clin Pharmacol Ther. 2004;42:642–643. doi:10.5414/CPP42642.
- Vu T, Claret FX. Trastuzumab: updated mechanisms of action and resistance in breast cancer. Front Oncol. 2012;2:62. doi:10.3389/fonc.2012.00062.
- Constant SL. B lymphocytes as antigen-presenting cells for CD4+ T cell priming in vivo. J Immunol. 1999;162:5695–5703.
- Dubois Cauwelaert N, Baldwin SL, Orr MT, Desbien AL, Gage E, Hofmeyer KA, Coler RN. Antigen presentation by B cells guides programing of memory CD4+ T-cell responses to a TLR4-agonist containing vaccine in mice. Eur J Immunol. 2016;46:2719–2729. doi:10.1002/eji.201646399.
- Fujimoto M, Fujimoto Y, Poe JC, Jansen PJ, Lowell CA, DeFranco AL, Tedder TF. CD19 regulates Src family protein tyrosine kinase activation in B lymphocytes through processive amplification. Immunity. 2000;13:47–57. doi:10.1016/S1074-7613(00)00007-8.
- Depoil D, Fleire S, Treanor BL, Weber M, Harwood NE, Marchbank KL, Tybulewicz VL, Batista FD. CD19 is essential for B cell activation by promoting B cell receptor-antigen microcluster formation in response to membrane-bound ligand. Nat Immunol. 2008;9:63–72. doi:10.1038/ni1547.
- Ma Y, Xiang D, Sun J, Ding C, Liu M, Hu X, Li G, Kloecker G, Zhang HG, Yan J. Targeting of antigens to B lymphocytes via CD19 as a means for tumor vaccine development. J Immunol. 2013;190:5588–5599. doi:10.4049/jimmunol.1203216.
- Lorenzo-Herrero S, Sordo-Bahamonde C, González S, López-Soto A. Immunosurveillance of cancer cell stress. Cell Stress. 2019;3:295–309. doi:10.15698/cst2019.09.198.
- Burugu S, Dancsok AR, Nielsen TO. Emerging targets in cancer immunotherapy. Semin Cancer Biol. 2018;52:39–52. doi:10.1016/j.semcancer.2017.10.001.
- Perica K, Varela JC, Oelke M, Schneck J. Adoptive T cell immunotherapy for cancer. Rambam Maimonides Med J. 2015;6:e0004. doi:10.5041/RMMJ.10179.
- Tey SK, Bollard CM, Heslop HE. Adoptive T-cell transfer in cancer immunotherapy. Immunol Cell Biol. 2006;84:281–289. doi:10.1111/j.1440-1711.2006.01441.x.
- Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity. 2014;41:49–61. doi:10.1016/j.immuni.2014.06.010.
- Xu-Monette ZY, Zhou J, Young KH. PD-1 expression and clinical PD-1 blockade in B-cell lymphomas. Blood. 2018;131:68–83. doi:10.1182/blood-2017-07-740993.
- Jin HT, Ahmed R, Okazaki T. Role of PD-1 in regulating T-cell immunity. Curr Top Microbiol Immunol. 2011;350:17–37. doi:10.1007/82_2010_116.
- Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–264. doi:10.1038/nrc3239.
- Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. 2015;27:450–461. doi:10.1016/j.ccell.2015.03.001.
- Swart M, Verbrugge I, Beltman JB. Combination approaches with immune-checkpoint blockade in cancer therapy. Front Oncol. 2016;6:233. doi:10.3389/fonc.2016.00233.
- Cho HS, Mason K, Ramyar KX, Stanley AM, Gabelli SB, Denney DW, Leahy DJ. Structure of the extracellular region of HER2 alone and in complex with the Herceptin fab. Nature. 2003;421:756–760. doi:10.1038/nature01392.
- Sharpe AH, Wherry EJ, Ahmed R, Freeman GJ. The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat Immunol. 2007;8:239–245. doi:10.1038/ni1443.
- Seung E, Dudek TE, Allen TM, Freeman GJ, Luster AD, Tager AM. PD-1 blockade in chronically HIV-1-infected humanized mice suppresses viral loads. PLoS One. 2013;8:e77780. doi:10.1371/journal.pone.0077780.
- Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, Chung SS, Stefanski J, Borquez-Ojeda O, Olszewska M, et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014;6:224ra25. doi:10.1126/scitranslmed.3008226.
- Klebanoff CA, Rosenberg SA, Restifo NP. Prospects for gene-engineered T cell immunotherapy for solid cancers. Nat Med. 2016;22:26–36. doi:10.1038/nm.4015.
- Ding C, Wang L, Marroquin J, Yan J. Targeting of antigens to B cells augments antigen-specific T-cell responses and breaks immune tolerance to tumor-associated antigen MUC1. Blood. 2008;112:2817–2825. doi:10.1182/blood-2008-05-157396.
- Roe K, Shu GL, Draves KE, Giordano D, Pepper M, Clark EA. Targeting antigens to CD180 but not CD40 programs immature and mature B cell subsets to become efficient APCs. J Immunol. 2019;203:1715–1729. doi:10.4049/jimmunol.1900549.
- Zaneti AB, Yamamoto MM, Sulczewski FB, Almeida B, Souza H, Ferreira NS, Maeda D, Sales NS, Rosa DS, Ferreira L, et al. Dendritic cell targeting using a DNA vaccine induces specific antibodies and CD4+ T cells to the dengue virus envelope protein domain III. Front Immunol. 2019;10:59. doi:10.3389/fimmu.2019.00059.
- Kastenmüller W, Kastenmüller K, Kurts C, Seder RA. Dendritic cell-targeted vaccines–hope or hype. Nat Rev Immunol. 2014;14:705–711. doi:10.1038/nri3727.
- Cohn L, Delamarre L. Dendritic cell-targeted vaccines. Front Immunol. 2014;5:255. doi:10.3389/fimmu.2014.00255.
- Melchers M, Bontjer I, Tong T, Chung NP, Klasse PJ, Eggink D, Montefiori DC, Gentile M, Cerutti A, Olson WC, et al. Targeting HIV-1 envelope glycoprotein trimers to B cells by using APRIL improves antibody responses. J Virol. 2012;86:2488–2500. doi:10.1128/JVI.06259-11.
- Heesters BA, van der Poel CE, Das A, Carroll MC. Antigen presentation to B cells. Trends Immunol. 2016;37:844–854. doi:10.1016/j.it.2016.10.003.
- Fu C, Jiang A. Dendritic cells and CD8 T cell immunity in tumor microenvironment. Front Immunol. 2018;9:3059. doi:10.3389/fimmu.2018.03059.
- Baxevanis CN, Sofopoulos M, Fortis SP, Perez SA. The role of immune infiltrates as prognostic biomarkers in patients with breast cancer. Cancer Immunol Immunother. 2019;68:1671–1680. doi:10.1007/s00262-019-02327-7.
- Friedrich M, Jasinski-Bergner S, Lazaridou MF, Subbarayan K, Massa C, Tretbar S, Mueller A, Handke D, Biehl K, Bukur J, et al. Tumor-induced escape mechanisms and their association with resistance to checkpoint inhibitor therapy. Cancer Immunol Immunother. 2019;68:1689–1700. doi:10.1007/s00262-019-02373-1.
- McDermott DF, Atkins MB. PD-1 as a potential target in cancer therapy. Cancer Med. 2013;2:662–673. doi:10.1002/cam4.106.
- Mahoney KM, Rennert PD, Freeman GJ. Combination cancer immunotherapy and new immunomodulatory targets. Nat Rev Drug Discov. 2015;14:561–584. doi:10.1038/nrd4591.
- Littringer K, Moresi C, Rakebrandt N, Zhou X, Schorer M, Dolowschiak T, Kirchner F, Rost F, Keller CW, McHugh D, et al. Common features of regulatory T cell specialization during Th1 responses. Front Immunol. 2018;9:1344. doi:10.3389/fimmu.2018.01344.
- Oda K, Hamanishi J, Matsuo K, Hasegawa K. Genomics to immunotherapy of ovarian clear cell carcinoma: unique opportunities for management. Gynecol Oncol. 2018;151:381–389. doi:10.1016/j.ygyno.2018.09.001.
- Dieras V, Vincent-Salomon A, Degeorges A, Beuzeboc P, Mignot L, de Cremoux P. [Trastuzumab (Herceptin) and breast cancer: mechanisms of resistance]. Bull Cancer. 2007;94:259–266.
- Escrivá-de-Romaní S, Arumí M, Bellet M, Saura C. HER2-positive breast cancer: current and new therapeutic strategies. Breast. 2018;39:80–88. doi:10.1016/j.breast.2018.03.006.
- Wittrup KD. Antitumor antibodies can drive therapeutic T cell responses. Trends Cancer. 2017;3:615–620. doi:10.1016/j.trecan.2017.07.001.
- Hasan A, Ghebeh H, Lehe C, Ahmad R, Dermime S. Therapeutic targeting of B7-H1 in breast cancer. Expert Opin Ther Targets. 2011;15:1211–1225. doi:10.1517/14728222.2011.613826.
- Sabatier R, Finetti P, Mamessier E, Adelaide J, Chaffanet M, Ali HR, Viens P, Caldas C, Birnbaum D, Bertucci F. Prognostic and predictive value of PDL1 expression in breast cancer. Oncotarget. 2015;6:5449–5464. doi:10.18632/oncotarget.3216.
- Filippova N, Yang X, An Z, Nabors LB, Pereboeva L. Blocking PD1/PDL1 interactions together with MLN4924 therapy is a potential strategy for glioma treatment. J Cancer Sci Ther. 2018;10:190–197. doi:10.4172/1948-5956.1000543.
- Kapp K, Volz B, Oswald D, Wittig B, Baumann M, Schmidt M. Beneficial modulation of the tumor microenvironment and generation of anti-tumor responses by TLR9 agonist lefitolimod alone and in combination with checkpoint inhibitors. Oncoimmunology. 2019;8:e1659096. doi:10.1080/2162402X.2019.1659096.