
ABSTRACT
Type I (IFNα/β) interferon signaling represents a critical transduction pathway involved in recognition and destruction of nascent tumor cells. Downregulation of this pathway to promote a more immunosuppressed microenvironment contributes to the ability of tumor cells to evade the immune system, a known Hallmark of Cancer. The present study investigates the progesterone receptor (PR), which is expressed in the vast majority of breast cancers, and its ability to inhibit efficient interferon signaling in tumor cells. We have shown that PR can block the interferon signaling cascade by promoting ubiquitination and degradation of STAT2. Targeting STAT2 is critical, as we show that it is an essential protein in inducing transcription of interferon-stimulated genes (ISG); shRNA-mediated knockdown of STAT2 severely abrogates the interferon response in vitro. Importantly, we were able to reverse this inhibition by treating with onapristone, an anti-progestin currently being investigated in breast cancer clinical trials. Additionally, we have found that an interferon-related gene signature (composed of ISGs) is inversely correlated with PR expression in human tumors. We speculate that PR inhibition of interferon signaling may contribute to creating an immunosuppressed microenvironment and reversal of this through anti-progestins may present a novel therapeutic target to promote immune activity within the tumor.
KEYWORDS:
Introduction
Hanahan and Weinberg revolutionized our understanding of cancer biology through the issuing of the “Hallmarks of Cancer” close to two decades ago, with a vital update occurring 10 y later.Citation1,Citation2 Among these updated hallmarks-i.e. characteristics shared by virtually all tumors-was ‘evasion of the immune system.’ As such, our understanding of how the immune system is involved in all steps of the tumor life cycle (initiation, progression, metastasis, etc.) has grown exponentially and allowed for the development of specific immune-targeting therapies.Citation3,Citation4
Multiple signaling pathways involved in the immune response are responsible for identification and elimination of malignantly transformed cells.Citation4 Among these key signaling mechanisms are a diverse group of cytokines called interferons. Type I interferons (IFNα/β) are cytokines canonically released by immune cells during viral infection and act through binding their cognate transmembrane receptor (IFNαR1/2). This binding leads to dimerization of the receptor, cross-phosphorylation of receptor associated Janus Activated Kinases (JAK1 and TYK2), and recruitment of Signal Transducer and Activator of Transcription family members (STAT1 and STAT2) that are then subsequently phosphorylated. Phosphorylated STAT1 and STAT2, in conjunction with Interferon Regulatory Factor 9 (IRF9), form a transcriptional complex that translocates to the nucleus, binds DNA at interferon-stimulated response elements (ISRE), and activates transcription of a diverse subset of Interferon Stimulated Genes (ISGs).Citation5 While canonically a pathway activated in response to viral infection, growing evidence has implicated type I interferons as crucial mediators in tumor detection and destruction.Citation6–Citation8 Immune “cold” tumors – those deficient in lymphocyte infiltration/activity – characteristically lack efficient interferon signaling, justifying the usage of Stimulator of Interferon Genes (STING) agonists to bolster activity within this pathway.Citation9 As such, induction of interferon signaling is a key step in bridging the innate and adaptive immune systems to mount an effective, anti-tumorigenic response.
Breast cancer represents the second leading cause of cancer-related death in women within the United States.Citation10 Of the over 250,000 new cases diagnosed annually in the United States, the vast majority of these express the estrogen (ER) and progesterone (PR) receptors.Citation11 PR is a ligand-activated nuclear receptor that functions as a transcription factor, regulating a variety of genes involved in proliferation and differentiation in the reproductive tract and mammary gland.Citation12 Clinical trial data emerging over the last few decades have suggested a role for PR, independent of ER, in the development of breast cancer.Citation13,Citation14 In light of these data, there is clinical impetus for identifying how PR is signaling in breast cancer in order to better inform treatments for patients with hormone receptor (ER/PR) positive cancer.
Our previous studies have concluded that PR can inhibit type I interferon signaling at multiple steps along the cascade, including prevention of STAT1 phosphorylation, DNA binding, and ISG transcription.Citation15,Citation16 Because of the biological importance of interferon signaling (prevention of infection and disease spread), compensatory mechanisms have been discovered in the absence of a functional STAT1 complex. In some instances, STAT2 is able to propagate the interferon response without STAT1, thus suggesting that multiple antagonistic mechanisms are necessary to fully hinder interferon signaling.Citation17 The present study investigates PR’s ability to inhibit STAT2 signaling through a mechanism distinct from PR’s ability to inhibit STAT1. Through inhibition of both STAT1 and STAT2, PR effectively shuts down interferon signaling, a vital pathway for the identification and destruction of nascent tumor cells.
Methods
Cell lines and treatments
T47D and BT474 cells were acquired from ATCC and cultured in DMEM (Cellgro) supplemented with 5% FBS and 1% penicillin/streptomycin. PR, STAT1, and STAT2 shRNA knockdown cells were created using viral particles (GE/Dharmacon) targeting three different regions of each respective gene. Viral transduction protocol was followed as per manufacturer’s instructions. Transduced, stable cell line pools expressing NS, PR, STAT1, or STAT2 shRNA were created in T47D cells following 14 d of selection in 2.5 ug/ml Puromycin (MP Biomedicals). Target shRNA sequences are listed in Supplementary Table 1. RFP and STAT1 overexpressing cells were created using lentiviral ORF particles (GE/Dharmacon). Transduced, stable cell line pools overexpressing RFP (control) or STAT1 were created in T47D cell lines stably expressing NS or STAT2 shRNA following 14 d of selection in 15 ug/mL Blasticidin S HCL (Corning). Sequences are shown in Supplementary Table 1. Cells were treated with the following reagents where indicated: R5020 (10 nM, Sigma), human rIFN-α (1000 IU/mL, IFN-α2A, SPR4594; Sigma-Aldrich, vehicle-H20), onapristone (10uM, provided through an MTA with Context Therapeutics), cycloheximide (200ug/mL, Calbiochem), and MG132 (5uM, Selleck Chemical).
Western blot analysis
Western blot analysis was performed as previously described.Citation15,Citation16 Primary antibodies for immunoblotting are as follows: PR (sc-7208; Santa Cruz Biotechnology), phospho-STAT2 (4441; Cell Signaling), Total STAT2 (sc-1668; Santa Cruz Biotechnology), IFIT3 (sc-393512; Santa Cruz Biotechnology), IFIT1 (14769; Cell Signaling), STAT1 (9172; Cell Signaling), Ubiquitin (3933; Cell Signaling), and beta-tubulin (2128; Cell Signaling). Densitometry to quantify immunoblots was performed using ImageJ.
Co-immunoprecipitation
Co-immunoprecipitation experiments were performed as previously described.Citation15 In brief, 1000ug of protein from isolated protein lysates were incubated with 2 mg of STAT2 (SC-1668, Santa Cruz Biotechnology) or control IgG antibody and rocked overnight at 4°C. Protein G agarose (Roche Diagnostics) was added for the final 2 h of incubation time. Beads were washed three times in supplemented RIPA buffer, bound protein eluted in Laemmili sample buffer, then subjected to immunoblotting as described above.
Quantitative RT-PCR
RNA isolation, cDNA generation, and qRT-PCR were performed as previously described.Citation15,Citation16 qPCR primer sequences are listed in Supplementary Table 1.
Chromatin immunoprecipitation
Chromatin immunoprecipitation (ChIP) experiments were performed using the ChIP-IT Express Kit (Active Motif) according to the manufacturer’s instructions. Isolated chromatin was sheared using sonication and sheared chromatin was IP’d overnight using the following primary antibodies (or species-specific IgG control): STAT1 (ab47425; abcam), STAT2 (sc-1668; Santa Cruz Biotechnology), and IRF9 (76684; Cell Signaling). IP’d chromatin was analyzed using qRT-PCR with primers designed to amplify specific ISG promoters. Data shown as percentage IP’d DNA over input DNA. ChIP-qPCR primer sequences are listed in Supplementary Table 1.
TCGA analysis
Gene expression data for the 50-gene IRDS signatureCitation18 (full list of genes in IRDS shown in Supplementary Table 2) were extracted from TCGA breastCitation19 ‘Provisional’ dataset and associated clinical metadata were accessed via the cBio portal.Citation20 The IRDS signature score was calculated by summing the normalized log2 Z-scores of the expression data from the 50 genes. Clinical metadata were used to stratify signature scores by ER/PR status, and the degree of PR expression by IHC. PGR gene expression was also extracted from the TCGA dataset via the cBio portal.Citation20
Statistical analyses
Statistical significance for all experiments was determined using an unpaired Student’s t-test unless otherwise specified. A p value ≤0.05 is considered statistically significant. The Delta method was used to calculate standard deviation for the ratio of two variables using their individual standard deviations, as seen when plotting fold relative RNA expression data between two treatment groups/cell lines.Citation21
Results
PR and STAT2 interact without affecting STAT2 phosphorylation
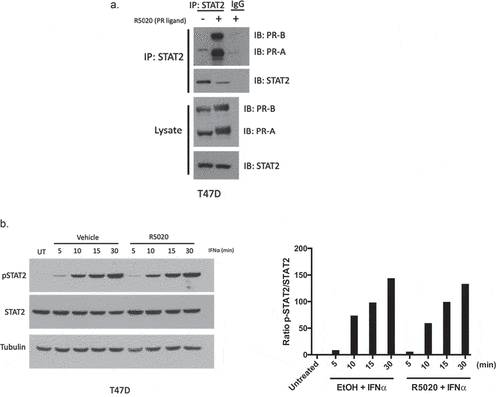
As we have previously shown that PR interacts with STAT1, we proposed that PR may be interacting with multiple proteins in the type I interferon signaling pathway to inhibit efficient signal transduction. To test whether PR was interacting with STAT2, we utilized co-immunoprecipitation in T47D cells (ER/PR-positive human breast cancer). Following treatment with the synthetic PR ligand, 10 nM R5020, we found an increase in the formation of a PR:STAT2 complex when compared to the vehicle control ()). Importantly, this was independent of STAT1, as STAT1 was not involved in PR:STAT2 complex formation (Supplementary Figure 1). Like other signal transduction pathways, type I interferon signaling is heavily regulated through the concerted addition and removal of post-translational modifications such as phosphorylation, acetylation, ubiquitination, etc.Citation22 To identify whether PR interacting with STAT2 impeded phosphorylation of STAT2, we treated with IFNα for 0–30 min in the presence or absence of PR ligand (R5020) and found no differences in STAT2 phosphorylation with PR activation ()). These data suggest that the interaction between PR and STAT2 does not affect interferon-induced STAT2 phosphorylation.
Figure 1. PR and STAT2 interact without affecting STAT2 phosphorylation.
PR activation promotes STAT2 ubiquitination and subsequent protein degradation
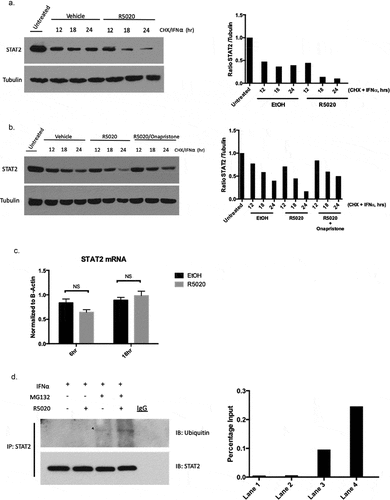
Multiple mechanisms exist to inhibit effective interferon signaling in the context of a viral infection – blocking the production of the interferons themselves, prevention of phosphorylation of JAK/STAT proteins, promotion of JAK/STAT degradation, etc.Citation23 We observed that phosphorylation of STAT2 was unaffected ()), so we investigated how PR activation affects STAT2 protein turnover. To investigate whether PR is affecting STAT2 expression in this manner, we utilized the inhibitor cycloheximide (CHX) which prevents translation and allows for the observation of protein turnover. We treated T47D cells with CHX and IFNα at varying time points (0–24 h) in the presence or absence of PR ligand and found that STAT2 protein was destabilized/degraded at a higher rate when PR was activated with ligand (), Supplementary Figure 2). We observed this phenotype in an additional PR+ breast cancer cell line, BT474 (Supplementary Figure 3) Onapristone, a potent PR antagonist, is currently being evaluated in a surgical window of opportunity trial in treatment naïve, PR-positive breast cancers (Onward 203, ONAWA trial). Additionally, onapristone is currently being used in a Phase I clinical trial for treatment of patients with late-stage breast cancer.Citation24 When treating with onapristone, we were able to effectively reverse the ability of PR to promote STAT2 turnover ()). This illustrates 1) this effect on STAT2 degradation is PR dependent, and 2) a drug already in clinical development for breast cancer prevents PR-mediated STAT2 degradation. Importantly, we did not see an effect on STAT2 mRNA in response to R5020 treatment indicating that this regulation is exclusively post-translational ()). Increased STAT2 turnover in the presence of PR ligand suggests that PR may be promoting ubiquitination of STAT2 protein, effectively targeting the protein for proteasomal degradation. To test this, we used the proteasome inhibitor MG132 to observe the accumulation of ubiquitinated protein. Following pre-treatment with MG132 and IFNα and subsequent treatment with R5020, we found that when PR is activated, we observed an increased population of ubiquitinated STAT2 ()). Taken together, these data suggest that PR interaction with STAT2 promotes ubiquitination, thereby targeting it for degradation.
Figure 2. PR activation promotes STAT2 ubiquitination and subsequent protein degradation.
STAT2 is essential for type I interferon signaling
Much of the current knowledge regarding interferon signaling has focused on the role of STAT1, as it is a major component of multiple interferon-regulated transcriptional complexes. Emerging data, however, has illuminated the essential role for STAT2 in promotion of type I interferon signaling during viral infection.Citation25,Citation26 The dependence on STAT2 to propagate the immune response in breast cancer, however, has been understudied. To tease apart how loss of STAT proteins affect ISGF3 component DNA binding, we first utilized T47D cells stably expressing either non-silencing (NS) or STAT1 shRNA (mRNA to confirm STAT1 knockdown with shRNA shown in ), protein in )). Using chromatin immunoprecipitation (ChIP) assays, we found that in cells lacking STAT1, STAT2, and IRF9 were still able to bind DNA at Interferon Stimulated Gene (ISG) promoters (ISREs) in response to IFNα (), Supplementary Table 3); recruitment at select representative ISGs is shown. To then test the dependence upon STAT2, we used shRNA to stably knockdown STAT2 (mRNA shown in ), protein in )). Contrary to what occurred when STAT1 was knocked down, we observed abrogated recruitment of STAT1 and IRF9 to ISG promoters, suggesting STAT2 is necessary to promote efficient DNA binding of other ISGF3 components (), Supplementary Table 4). These data suggest that STAT2, not STAT1, is essential in mediating the interferon response.
Figure 3. Interferon-induced DNA binding is maintained in the absence of STAT1.
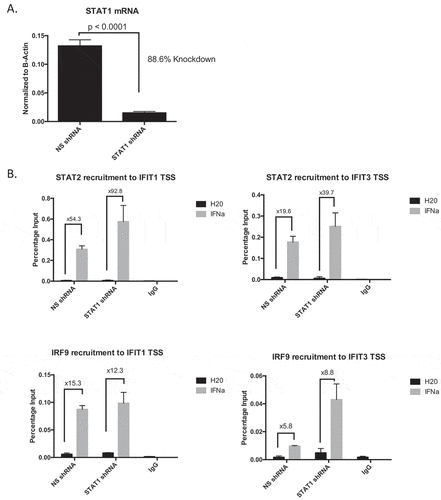
Figure 4. ISGF3 binding is severely hindered in the absence of STAT2.
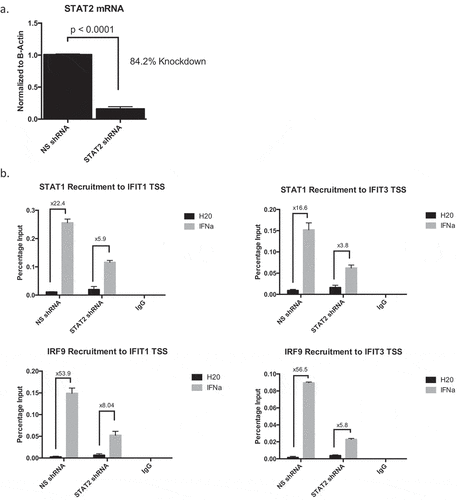
Figure 5. STAT2 is essential for efficient ISG transcription.
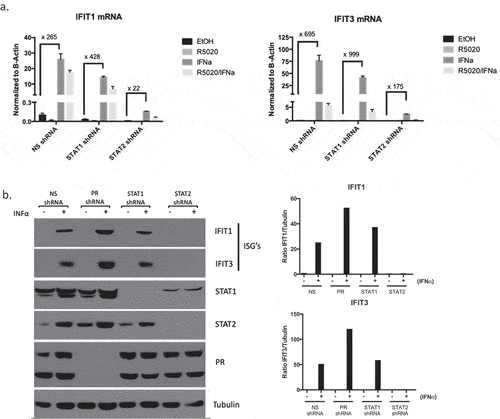
We next measured ISG transcription as a readout of activated interferon signaling in both shRNA knock down (STAT1 and STAT2) cell lines. We found that knocking down STAT1 did not affect ISG transcription ()), indicating compensatory mechanisms involving STAT2 can effectively overcome the lack of STAT1 in breast cancer cells. Studies in viral models have shown that STAT2 can direct prolonged, constitutive interferon activation in the absence of a functional STAT1 complex.Citation25,Citation27 We observe a similar phenotype in our breast cancer cells as exemplified by increased recruitment of STAT2 and IRF9 to ISREs and increased ISG transcription when STAT1 is knocked down. Conversely, knocking down STAT2 severely hindered the ability of these cells to respond to IFNα, further demonstrating the indispensability of STAT2 in the propagating the interferon signaling cascade ()). Finally, observing ISG protein output, we compared the interferon response between cells expressing NS, PR, STAT1, or STAT2 shRNA. We found that knocking down PR increased the response to IFNα (consistent with previous data from our laboratoryCitation15,Citation16). Furthermore, IFNα response in STAT1 shRNA cells was comparable to the NS control, and STAT2 knockdown completely eliminated ISG protein production ()). As STAT1 itself is an ISG whose expression is dampened in STAT2 shRNA cells ()), we performed a STAT1 re-expression experiment in these cells. Cells without STAT2, but with overexpression of STAT1 via lentiviral transduction, exhibited a similar phenotype in both STAT1 DNA binding and ISG protein production when compared to STAT2 shRNA alone (Supplementary Figure 4). These data indicate that the hinderance of the interferon response is solely a result of loss of STAT2. These data taken together establish the essential role of STAT2 in the type I interferon response in breast cancer, and therefore underscore the consequences of PR-mediated degradation of STAT2 protein.
PR status in human tumors is correlated with decreased interferon gene signature
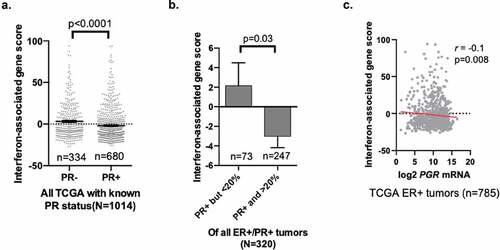
To investigate how PR status affects interferon signaling in human breast cancers, we analyzed data acquired from The Cancer Genome Atlas (TCGA). Our data suggest PR promotes STAT2 degradation and thereby inhibits proper interferon signaling. Because this regulation occurs post-translationally and not at the gene/transcript level, we found that STAT2 gene expression was expectedly not significantly different between PR-positive and PR-negative tumors (data not shown). While protein expression data is limited in TCGA, we can instead identify changes in gene signatures among PR-positive and PR-negative breast cancers as a readout for active interferon signaling. We utilized TCGA breast cancer gene expression datasets (TCGA Breast, provisionalCitation19) and assessed differences in expression of an established interferon gene signature (that includes the ISGs shown herein, Citation18 full list of genes shown in Supplementary Table 2) to identify any differences between PR-positive and PR-negative breast cancers ()). We found that PR expression was significantly associated with a decrease in this interferon gene signature when analyzing all tumors in this TCGA dataset. Moreover, when stratifying PR-positive breast cancers based upon level of expression (< or >20% PR-positive cellular expression), we found that expression of the interferon-related gene signature is inversely correlated with level (% positivity) of PR in ER/PR-positive breast tumors; tumors with higher percentage PR-positivity exhibited lower expression of the interferon gene signature ()). Moreover, we see a negative correlation (spearman correlation coefficient [r = −0.1; p = .008]) between PR (PGR) mRNA levels and expression of the interferon gene signature in the TCGA dataset containing all ER-positive tumors. Cumulatively, these data show that PR expression in human breast tumors correlates with decreased interferon signaling. These data suggest that PR positivity promotes an immunosuppressive phenotype as measured by decreased interferon signaling; a potential mechanism by which early malignancies progress to clinically relevant tumors.
Figure 6. PR status in human tumors is correlated with decreased interferon gene signature.
Discussion
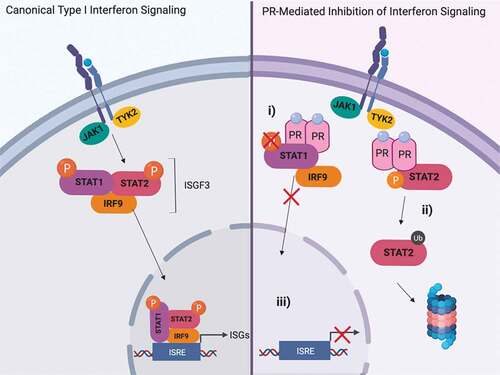
In the present study, we have shown that PR interacts with STAT2. While this interaction does not affect STAT2 phosphorylation, we do observe an increase in STAT2 ubiquitination and degradation when PR is activated by ligand. Previous studies in virally infected cells have discovered that in the absence of a functional STAT1 complex, compensatory STAT2-dependent signaling mechanisms are employed to maintain active interferon signaling.Citation17,Citation25-Citation29 A recent study established the indispensability of STAT2 in interferon signaling in Hela cells and our work has shown a similar vital role of STAT2 in breast cancer.Citation30 As our previous study examined PR’s ability to inhibit STAT1 functionality in breast cancer, we have exhibited a mechanism by which breast cancer cells attempt to overcome this inhibition. By inhibiting both STAT1 and STAT2 functionalities, PR is able to fully abrogate the interferon response, as exemplified through significantly decreased ISG transcription (). Data from our previous studies, as well as analyzed TCGA data in the present study, have shown that this PR-dependent downregulation of ISGs is seen in human tumors as well.
Figure 7. PR inhibits type I interferon signaling by targeting both STAT1 and STAT2.
For patients with ER/PR-positive breast cancer, the current standard of care primarily involves ER-targeted therapies like aromatase inhibitors or selective estrogen modulators/down regulators (i.e. fulvestrant, tamoxifen).Citation31 While such therapies have proven to be effective in these patients, they are accompanied by undesirable side effects which can hinder compliance. Furthermore, many patients, even when compliant, will still relapse.Citation32,Citation33 Our increased understanding of immune signaling in tumorigenesis has led the development of multiple immunotherapies – checkpoint inhibitors (CTLA-4, PD-L1), CAR-T therapy, vaccination, etc.Citation34 These innovative approaches to cancer treatment have proven to be effective in several tumor types but have been largely disappointing in luminal breast cancer.Citation35 Tumors more likely to respond to these therapies are considered immune “hot” – higher mutational burden, cytotoxic lymphocyte infiltration, increased antigen presentation, etc.Citation36 Breast cancer, particularly hormone receptor-positive breast cancer, lacks immunogenicity and therefore is immune “cold”.Citation37 Identifying a mechanism by which immune signaling is hindered in PR-positive breast cancer, and finding a way to target and convert to a “hot” environment could open up the possible therapies these patients will respond to and vastly improve clinical outcomes.Citation38 The data shown herein illustrates that PR is repressing the interferon response, which we speculate may contribute to the promotion of an immunosuppressed microenvironment. Additionally, data from this study and previous studies from our lab have illustrated a ligand-independent effect of PR downregulating interferon response, further reinforcing a potent effort in inhibiting this particular immune response.Citation15,Citation16 It is essential to further define how PR suppresses immune signaling, as it may be a promising therapeutic target to prime tumors for immune-directed therapies and current experiments are underway to further characterize this connection.
Pre-clinical and clinical data have shown an essential role for type I interferons in bridging the innate and adaptive immune responses to promote dendritic cell (DC) priming and cytotoxic T cell recruitment/activation.Citation8 In particular, mice lacking the interferon receptor (IFNαR) show defects in CD8α+ DC ability to cross-present to cytotoxic CD8 + T cells; lymphocytes established to be vital in tumor rejection.Citation39 Interferon stimulated genes (ISGs), are a diverse group of proteins produced in response to active interferon signaling. Previous studies have shown that many of these proteins are involved in lymphocyte trafficking, inhibition of proliferation and cell motility, antigen presentation, and apoptosis.Citation40
Additionally, PD-L1 expression is induced on immune cells in response to interferons.Citation41 As PD-L1 expression is necessary in determining whether a patient will respond to anti-PD-1/PD-L1 directed checkpoint inhibitors, the implications of inhibiting interferon signaling in the tumor microenvironment cannot be overstated.Citation42 The upregulation of PD-L1 on immune cells is not unique to type I interferons, as crosstalk exists between type I and type II (i.e. INFgamma) signaling via STAT1-dependent mechanisms.Citation43 Current studies are underway to identify the impact of PR and progesterone on type II interferon signaling. Finally, a study recently published by Anurag et al., showed that endocrine therapy-resistant tumors exhibited an inverse correlation between PR expression and immune checkpoint expression, further illustrating the role of PR in promoting an immunosuppressive microenvironment.Citation44 The initial data herein and previous studies from our lab have demonstrated a concerted effort of PR to decrease interferon signaling in breast cancer cells which warrants further investigation into hormone mediation of the immune response in tumor development.
There is precedence for utilizing ubiquitination of STAT2 to inhibit type I interferon signaling. Multiple viruses employ viral proteins to function as E3 ubiquitin ligases to promote degradation of STAT2, thereby effectively inhibiting interferon signaling and allowing for the progression of infection.Citation45–Citation47 Much like viruses that need to overcome type I interferon signaling to replicate within their hosts, transformed cells need to downregulate or inhibit this pathway to avoid immune detection. Immune evasion allows for nascent tumor cells to avoid destruction by cytotoxic immune cells and progress to palpable tumors. Interestingly, mutations in STAT1 and STAT2 are not common in breast cancer.Citation19 As the majority of breast cancers are PR-positive and our data have shown PR inhibits these pathways, genomic mutations of these proteins would not be necessary to establish an immunosuppressive, tumorigenic microenvironment; PR inhibition of STAT1 and STAT2 activity circumvents the need for loss/mutation of these proteins.
We have identified multiple ways in which PR attacks the interferon signaling pathway, which we speculate may offer a potential mechanism by which nascent breast cancer cells (the majority of which are PR-positive) evade immune detection and effectively escape immune-mediated destruction. We have also demonstrated that treatment with onapristone, a potent anti-progestin can reverse the ability of PR to promote STAT2 degradation. Because of this, it is imperative to investigate further whether targeting PR signaling could offer an advantage as a chemopreventative agent to prime immunosuppressed tumors to respond to other types of therapy. Current studies are underway to further characterize PR’s role in immune escape and how this affects tumor formation and progression in vivo.
Declaration of interest statement
In accordance with Taylor & Francis policy and my ethical obligation as a researcher, I am reporting that Justin Balko, a co-author on this manuscript, receives research support from Genentech/Roche, Bristol Myers Squibb, and Incyte Corporation, has received consulting/expert witness fees from Novartis, and is an inventor on provisional patents regarding immunotherapy targets and biomarkers in cancer. I have disclosed these interests fully to Taylor & Francis, and I have in place an approved plan for managing any potential conflicts arising from this involvement.
Supplemental Material
Download ()Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
References
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–11. doi:10.1016/j.cell.2011.02.013.
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57–70. doi:10.1016/S0092-8674(00)81683-9.
- Helmy KY, Patel SA, Nahas GR, Rameshwar P. Cancer immunotherapy: accomplishments to date and future promise. Ther Deliv. 2013;4(10):1307–1320. doi:10.4155/tde.13.88.
- Vinay DS, Ryan EP, Pawelec G, Talib WH, Stagg J, Elkord E, Lichtor T, Decker WK, Whelan RL, Kumara HMCS, et al. Immune evasion in cancer: mechanistic basis and therapeutic strategies. Semin Cancer Biol. 2015;35(Suppl):S185–s198. doi:10.1016/j.semcancer.2015.03.004.
- Snell LM, McGaha TL, Brooks DG. Type I interferon in chronic virus infection and cancer. Trends Immunol. 2017;38(8):542–557. doi:10.1016/j.it.2017.05.005.
- Stone ML, Chiappinelli KB, Li H, Murphy LM, Travers ME, Topper MJ, Mathios D, Lim M, Shih I-M, Wang T-L, et al. Epigenetic therapy activates type I interferon signaling in murine ovarian cancer to reduce immunosuppression and tumor burden. Proc Natl Acad Sci U S A. 2017;114(51):E10981–e10990 doi: 10.1073/pnas.1712514114.
- Fuertes MB, Kacha AK, Kline J, Woo S-R, Kranz DM, Murphy KM, Gajewski TF. Host type I IFN signals are required for antitumor CD8(+) T cell responses through CD8α(+) dendritic cells. J Exp Med. 2011;208(10):2005–2016. doi:10.1084/jem.20101159.
- Fuertes MB, Woo S-R, Burnett B, Fu Y-X, Gajewski TF. Type I interferon response and innate immune sensing of cancer. Trends Immunol. 2013;34(2):67–73. doi:10.1016/j.it.2012.10.004.
- Woo S-R, Corrales L, Gajewski TF. The STING pathway and the T cell-inflamed tumor microenvironment. Trends Immunol. 2015;36(4):250–256. doi:10.1016/j.it.2015.02.003.
- Breast cancer facts & figures 2017–2018. 2016. [accessed 2019 Nov 1] . www.cancer.org/statistics.
- Knutson TP, Lange CA. Tracking progesterone receptor-mediated actions in breast cancer. Pharmacol Ther. 2014;142(1):114–125. doi:10.1016/j.pharmthera.2013.11.010.
- Grimm SL, Hartig SM, Edwards DP. Progesterone receptor signaling mechanisms. J Mol Biol. 2016;428(19):3831–3849. doi:10.1016/j.jmb.2016.06.020.
- Narod SA. Hormone replacement therapy and the risk of breast cancer. Nat Rev Clin Oncol. 2011;8(11):669–676. doi:10.1038/nrclinonc.2011.110.
- Fournier A, Berrino F, Riboli E, Avenel V, Clavel-Chapelon F. Breast cancer risk in relation to different types of hormone replacement therapy in the E3N-EPIC cohort. Int J Cancer. 2005;114(3):448–454. doi:10.1002/ijc.20710.
- Goodman ML, Trinca GM, Walter KR, Papachristou EK, D’Santos CS, Li T, Liu Q, Lai Z, Chalise P, Madan R, et al. Progesterone receptor attenuates STAT1-mediated IFN signaling in breast cancer. J Immunol. 2019;202(10):3076–3086. doi:10.4049/jimmunol.1801152.
- Walter KR, Goodman ML, Singhal H, Hall JA, Li T, Holloran SM, Trinca GM, Gibson KA, Jin VX, Greene GL, et al. Interferon-stimulated genes are transcriptionally repressed by PR in breast cancer. Mol Cancer Res. 2017;15(10):1331–1340. doi:10.1158/1541-7786.MCR-17-0180.
- Fink K, Grandvaux N. STAT2 and IRF9: beyond ISGF3. JAKSTAT. 2013;2:e27521.
- Khodarev NN, Beckett M, Labay E, Darga T, Roizman B, Weichselbaum RR. STAT1 is overexpressed in tumors selected for radioresistance and confers protection from radiation in transduced sensitive cells. Proc Natl Acad Sci U S A. 2004;101(6):1714–1719. doi:10.1073/pnas.0308102100.
- The Cancer Genome Access Network.Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. doi:10.1038/nature11412.
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–404. doi:10.1158/2159-8290.CD-12-0095.
- Casella G, Berger RL. Statistical inference. Pacific Grove: CA Duxbury Press; 2001.
- Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol. 2014;14(1):36–49. doi:10.1038/nri3581.
- Devasthanam AS. Mechanisms underlying the inhibition of interferon signaling by viruses. Virulence. 2014;5(2):270–277. doi:10.4161/viru.27902.
- Cottu PH, Bonneterre J, Varga A, Campone M, Leary A, Floquet A, Berton-Rigaud D, Sablin M-P, Lesoin A, Rezai K, et al. Phase I study of onapristone, a type I antiprogestin, in female patients with previously treated recurrent or metastatic progesterone receptor-expressing cancers. PLoS One. 2018;13(10):e0204973. doi:10.1371/journal.pone.0204973.
- Blaszczyk K, Nowicka H, Kostyrko K, Antonczyk A, Wesoly J, Bluyssen HAR. The unique role of STAT2 in constitutive and IFN-induced transcription and antiviral responses. Cytokine Growth Factor Rev. 2016;29:71–81. doi:10.1016/j.cytogfr.2016.02.010.
- Le-Trilling VTK, Wohlgemuth K, Rückborn MU, Jagnjic A, Maaßen F, Timmer L, Katschinski B, Trilling M, et al. STAT2-dependent immune responses ensure host survival despite the presence of a potent viral antagonist. J Virol. 2018;92(14):e00296-18.
- Blaszczyk K, Olejnik A, Nowicka H, Ozgyin L, Chen Y-L, Chmielewski S, Kostyrko K, Wesoly J, Balint B, Lee C-K, et al. STAT2/IRF9 directs a prolonged ISGF3-like transcriptional response and antiviral activity in the absence of STAT1. Biochem J. 2015;466(3):511–524. doi:10.1042/BJ20140644.
- Abdul-Sater AA, Majoros A, Plumlee CR, Perry S, Gu A-D, Lee C, Shresta S, Decker T, Schindler C. Different STAT transcription complexes drive early and delayed responses to type I IFNs. J Immunol. 2015;195(1):210–216. doi:10.4049/jimmunol.1401139.
- Platanitis E, Demiroz D, Schneller A, Fischer K, Capelle C, Hartl M, Gossenreiter T, Müller M, Novatchkova M, Decker T, et al. A molecular switch from STAT2-IRF9 to ISGF3 underlies interferon-induced gene transcription. Nat Commun. 2019;10(1):2921. doi:10.1038/s41467-019-10970-y.
- Urin V, Shemesh M, Schreiber G. CRISPR/Cas9-based knockout strategy elucidates components essential for type 1 interferon signaling in human HeLa cells. J Mol Biol. 2019;431(17):3324–3338. doi:10.1016/j.jmb.2019.06.007.
- Clarke R, Tyson JJ, Dixon JM. Endocrine resistance in breast cancer–An overview and update. Mol Cell Endocrinol. 2015;418(Pt 3(0 3)):220–234. doi:10.1016/j.mce.2015.09.035.
- Condorelli R, Vaz-Luis I. Managing side effects in adjuvant endocrine therapy for breast cancer. Expert Rev Anticancer Ther. 2018;18(11):1101–1112. doi:10.1080/14737140.2018.1520096.
- D’Souza A, Spicer D, Lu J. Overcoming endocrine resistance in metastatic hormone receptor-positive breast cancer. J Hematol Oncol. 2018;11(1):80. doi:10.1186/s13045-018-0620-6.
- Criscitiello C, Curigliano G. Immunotherapy of breast cancer. Prog Tumor Res. 2015;42:30–43.
- Yu LY, Tang J, Zhang CM, Zeng WJ, Yan H, Li MP, Chen XP, et al. New immunotherapy strategies in breast cancer. Int J Environ Res Public Health. 2017;14(1).
- Maleki Vareki S. High and low mutational burden tumors versus immunologically hot and cold tumors and response to immune checkpoint inhibitors. J Immunother Cancer. 2018;6(1):157. doi:10.1186/s40425-018-0479-7.
- Thomas A, Routh ED, Pullikuth A, Jin G, Su J, Chou JW, Hoadley KA, Print C, Knowlton N, Black MA, et al. Tumor mutational burden is a determinant of immune-mediated survival in breast cancer. Oncoimmunology. 2018;7(10):e1490854. doi:10.1080/2162402X.2018.1490854.
- Tolba MF, Omar HA. Immunotherapy, an evolving approach for the management of triple negative breast cancer: converting non-responders to responders. Crit Rev Oncol Hematol. 2018;122:202–207. doi:10.1016/j.critrevonc.2018.01.005.
- Diamond MS, Kinder M, Matsushita H, Mashayekhi M, Dunn GP, Archambault JM, Lee H, Arthur CD, White JM, Kalinke U, et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med. 2011;208(10):1989–2003. doi:10.1084/jem.20101158.
- Cheon H, Borden EC, Stark GR. Interferons and their stimulated genes in the tumor microenvironment. Semin Oncol. 2014;41(2):156–173. doi:10.1053/j.seminoncol.2014.02.002.
- Xiao W, Klement JD, Lu C, Ibrahim ML, Liu K. IFNAR1 controls autocrine type I IFN regulation of PD-L1 expression in myeloid-derived suppressor cells. J Immunol. 2018;201(1):264–277. doi:10.4049/jimmunol.1800129.
- Shen X, Xu Z, Zhang L, Li J, Wang Y, Li Y, et al. Recent findings in the regulation of programmed death ligand 1 expression. Front Immunol. 2019;10:1337.
- Garcia-Diaz A, Shin DS, Moreno BH, Saco J, Escuin-Ordinas H, Rodriguez GA, Zaretsky JM, Sun L, Hugo W, Wang X, et al. Interferon receptor signaling pathways regulating PD-L1 and PD-L2 expression. Cell Rep. 2017;19(6):1189–1201. doi:10.1016/j.celrep.2017.04.031.
- Anurag M, Zhu M, Huang C, Vasaikar S, Wang J, Hoog J, Burugu S, Gao D, Suman V, Zhang XH, et al. Immune checkpoint profiles in luminal B breast cancer (Alliance). J Natl Cancer Inst. 2019. doi:10.1093/jnci/djz213.
- Ashour J, Laurent-Rolle M, Shi PY, García-Sastre A, et al. NS5 of dengue virus mediates STAT2 binding and degradation. J Virol. 2009;83(11):5408–5418. doi:10.1128/JVI.02188-08.
- Elliott J, Lynch OT, Suessmuth Y, Qian P, Boyd CR, Burrows JF, Buick R, Stevenson NJ, Touzelet O, Gadina M, et al. Respiratory syncytial virus NS1 protein degrades STAT2 by using the Elongin-Cullin E3 ligase. J Virol. 2007;81(7):3428–3436. doi:10.1128/JVI.02303-06.
- Grant A, Ponia S, Tripathi S, Balasubramaniam V, Miorin L, Sourisseau M, Schwarz M, Sánchez-Seco M, Evans M, Best S, et al. Zika virus targets human STAT2 to inhibit type I interferon signaling. Cell Host Microbe. 2016;19(6):882–890. doi:10.1016/j.chom.2016.05.009.