
ABSTRACT
Ovarian cancer accounts for most deaths from gynecologic malignancies. Although more than 80% of patients respond to first-line standard of care, most of these responders present with recurrence and eventually succumb to carcinomatosis and chemotherapy-resistant disease. To improve patient survival, new modalities must, therefore, target or prevent recurrent disease. Here we describe for the first time a novel syngeneic mouse model of recurrent high-grade serous ovarian cancer (HGSOC), which allows immunotherapeutic interventions in a time course relevant to human carcinomatosis and disease course. Using this model, we demonstrate the efficacy of Transimmunization (TI), a dendritic cell (DC) vaccination strategy that uses autologous and physiologically derived DC loaded with autologous whole tumor antigens. TI has been proven successful in the treatment of human cutaneous T cell lymphoma and we report for the first time its in vivo efficacy against an intra-peritoneal solid tumor. Given as a single therapy, TI is able to elicit an effective anti-tumor immune response and inhibit immune-suppressive crosstalks with sufficient power to curtail tumor progression and establishment of carcinomatosis and recurrent disease. Specifically, TI is able to inhibit the expansion of tumor-associated macrophages as well as myeloid-derived suppressive cells consequently restoring T cell immune-surveillance. These results demonstrate the possible value of TI in the management of ovarian cancer and other intra-peritoneal tumors.
Introduction
Ovarian cancer accounts for most deaths from gynecologic malignancies with a dismal 5-year survival rate of 45.9%.Citation1,Citation2 In the US alone, in 2019 the American Cancer Society has estimated that 13,980 women will die of this disease. With these grim statistics, it is notable that ovarian cancer has a relatively high initial response rate to first-line standard of care. Even though a majority of patients are diagnosed at an advanced stage (Stage III–IV), 80% of these newly diagnosed patients will respond to first-line standard of care comprised of debulking surgery and combination chemotherapy.Citation3-Citation5 It is when these patients recur, however, that challenges arise as co-presentation of carcinomatosis and cross-resistance to various chemotherapy agents limit the value of surgery and chemotherapy, thus leading to treatment failure and death. Recurrence in ovarian cancer occurs in more than 90% of patients and usually presents within 2 to 5 years after the conclusion of first-line therapies.Citation6-Citation8
The reliable development of recurrent ovarian cancer, especially in patients that initially demonstrated complete clinical response, suggests that after the conclusion of first-line therapies, micro-metastatic residual malignancy remains, which eventually reemerge as a resistant variant of the initial disease.Citation6,Citation9 To prevent recurrence and improve patient survival, it is, therefore, necessary to eliminate residual disease. To achieve this, it is imperative to identify novel therapeutic modalities that can complement the current standard first-line treatment.
The quality of immune infiltrates in the ovarian tumor microenvironment greatly impacts the course of the disease and thus the prognosis of patientsCitation10-Citation12. Immunosuppressive cells such as tumor-associated macrophages (TAMs) and myeloid-derived suppressor cells (MDSC) have been shown to be major promoters of ovarian cancer progression and their presence has been demonstrated to inversely correlate with patient prognosisCitation13-Citation15 . TAMs and MDSC do not only suppress cytotoxic T cell responses but also secrete factors that promote angiogenesis and tumor cell invasiveness.Citation16 Overcoming these immunosuppressive components of the ovarian tumor microenvironment is, therefore, critical in controlling tumor progression and recurrence. Unfortunately, clinical trials employing immune-checkpoint inhibitors such as those that target PD-1 or CTLA-4 did not show robust activity in ovarian cancer patients.Citation17
A promising immunotherapy approach is extracorporeal photochemotherapy (ECP) or it is more recent iteration, transimmunization (TI).Citation18 ECP is an immunotherapy widely deployed across university medical centers in the USA and Europe for the treatment of cutaneous T cell lymphoma (CTCL) as well for the control of pathogenic T cells in the transplant setting.Citation19,Citation20 Since its FDA approval in 1988 for CTCL, ECP has been evaluated in more than 1,000 CTCL patients in >350 university centers around the world with a response rate of ~60%. It has also been evaluated in >1300 acute and chronic graft vs host disease patients with response rates between 40-80%.Citation21,Citation22 Recent findings have attributed the therapy’s bidirectional generation of immunity and tolerance to the large-scale physiologic production of functional antigen-loaded dendritic cells (DC), which are produced in the absence of added cytokines. These DC are produced through activation of blood monocytes via dynamic interactions with plasma proteins and platelets.Citation22-Citation25 The ECP process involves four sequential steps: ex vivo conversion of circulating blood monocytes to CD83+/MHC II++/CD14- dendritic antigen-presenting cells; reinfusion of DC back to patients; presumed loading of antigen from apoptotic malignant T cells (or pathogenic T cells in the case of GVHD) into the newly re-infused DC; and expansion of anti-tumor/tolerogenic T cells. A fifth step has been subsequently added to ECP with the objective of increasing the efficiency of antigen loading into the newly formed DC. The modified treatment, which we refer to as TI,Citation18,Citation26,Citation27 involves an added incubation step wherein apoptotic malignant cells are co-cultured ex vivo with the newly formed DC to allow for optimal tumor antigen internalization prior to re-infusion of the vaccinating DC. Recently, a miniaturized scalable device that reproduces the cellular effects of the human ECP device has been developed. This tabletop “TI chamber” allows further research on this modality using animal models,Citation28 and confirmed anti-cancer efficacy against murine melanoma.Citation29 Findings from utilizing the TI chamber defined key cellular and molecular requirements for an anti-tumor immune response and the mechanisms at play, with circulating monocytes acting as the key cellular player after platelet-induced DC conversion. We mechanistically dissected this effect and recently reported that platelet/monocyte interaction through the P-Selectin/PSGL-1 stimulatory junction leads to the creation of “physiological DC” (Han et al., Science Advances, 2020 in press). In addition to the rapid creation of vaccinating DC, the importance of the type of cell death induced in the tumor cells, which act as efficient DC internalization substrates in the TI device, has also been recently elucidated. The photoactivatable chemotherapeutic agent 8-methoxypsoralen (8-MOP), which is the apoptosis-inducing drug at the heart of the clinical ECP device, has been shown to be an extremely potent activator of immunogenic cell death (ICD), providing superior protection in in vivo vaccination assays and in vitro T cell stimulation assays when compared to other apoptosis-inducing agents.Citation30
We hypothesized that because of the history of clinical success associated with ECP/TI and the increased knowledge of the underlying mechanism driving its anti-tumor effects, TI may provide an effective approach to modify the suppressive ovarian tumor microenvironment, stimulate potent anti-tumor T cell responses, and prevent ovarian cancer recurrence in our human-relevant model. Here we report significant vaccination effects of TI treatment in the prevention of ovarian cancer recurrence in a syngeneic, immunocompetent mouse model. We first describe the optimization of the orthotopic mouse model of high-grade serous ovarian cancer (HGSOC) in C57BL/6 J mice, which was developed with the goal of establishing a platform for the timely evaluation of novel therapeutic modalities including immunotherapies. Using this model, we demonstrate the efficacy of TI in delaying the progression of established tumors and, critically, in the prevention of recurrence and formation of carcinomatosis. TI inhibits the expansion of immune-suppressive TAMs and MDSC and restores immune surveillance in the tumor microenvironment and relevant lymphoid organs. Our data suggest that TI is able to prevent recurrence through a bi-directional/two-pronged enhancement of antigen-specific anti-tumor responses through DC, in combination with downregulation of tumor microenvironment-borne immunosuppression, evidence that TI can overcome barriers that had previously stifled existing immunotherapeutic strategies in ovarian cancer and possibly other solid tumors.
Materials and methods
Cancer cell lines
Triple knock-out (TKO) mouse ovarian cancer cells were kindly provided by Dr. Martin Matzuk.Citation31 These cells were isolated from i.p. tumors that spontaneously formed in conditional Dicer-PTEN knock-out mice harboring p53 mutation ((p53LSL-R172H/+Dicerflox/floxPtenflox/flox Amhr2cre/+)..RCitation2,Citation17,Citation31 mCherry fluorescence was stably expressed in the isolated TKO cells using a lentivirusCitation32 to allow monitoring of i.p. tumors in real time. YUMM1.7 mouse melanoma cells were kindly provided by Dr. Marcus Bosenberg.Citation33 Both cell lines were cultured in DMEM-F12 (Thermo Fischer Scientific) supplemented with 10% heat-inactivated FBS and 1% penicillin-streptomycin under standard cell culture conditions. Cells were authenticated by detailed and accurate record-keeping and labeling, routinely tested for Mycoplasma by PCR methods, and injected in animals within 4 passages of thawing a frozen aliquot.
Establishment of TKO intra-peritoneal tumors and monitoring of disease progression
The Yale Institutional Animal Care and Use Committee approved all animal protocols that guided the in vivo experiments described in this study. All animals bear i.p. tumors from TKO ovarian cancer cells. 1 × 10Citation7 mCherry-positive TKO cells were injected in 4-week old female C57BL/6 J mice (Jackson Laboratories). Imaging was performed twice a week under isoflurane anesthesia using the Invivo FX PRO imaging system (Bruker Corp.). mCherry fluorescence was acquired at 550 excitation and 635 emission. Images were set at 0.5 min and 5000 max and mCherry region of interest (ROI) area was quantified as previously described using Bruker MI software.Citation34,Citation35 Body area, which was used as a surrogate for abdominal girth, was calculated using height and width measures from x-ray images. Body area measurement that is equal to or greater than 800 mm2 was considered to be positive for ascites.
Induction of apoptosis in tumor cell lines
Apoptosis was induced in 2.5 × 106 TKO cells or YUMM1.7 cells by treatment with 400 ng/ml or 200 ng/ml, respectively, of 8-methoxypsoralen (8-MOP; UVADEX, Therakos) in FBS followed by exposure to 16 J/cmCitation2 or 4 J/cm,Citation2 respectively, of UVA radiation. These doses were pre-determined as the minimum amount required to induce death in these cells. Apoptotic cells were mixed with peripheral blood mononuclear cells (PBMC) as described below.
Isolation of PBMC
200 μl of blood was collected from each mouse in both control and treated groups via cheek bleed. Blood was collected twice a week but the animals were rotated such as that each individual animal was bled only once a week. Platelet-containing PBMC were isolated from heparinized whole blood using Lymphocyte Separation Media (Lonza, Inc.). Red blood cells were lysed using a hypotonic solution. Isolated PBMC were resuspended in FBS. Plasma was collected and reserved for subsequent steps.
Transimmunization (TI) protocol
The TI protocol is illustrated in Supplementary Figures 1 and 2 and performed as previously described with minor modification.Citation29 Apoptotic TKO or apoptotic YUMM1.7 were combined 1:1 with the isolated PBMC and incubated in a sterile polystyrene TI chamber (Transimmune AG and Fraunhofer Institute for Biomedical Engineering) for 1 h at 37oC. The TI chamber is a miniaturized ECP device suitable for in vivo studies and has been previously described.Citation28 Following the 1 hr incubation, the cells were passed through the chamber using a syringe pump at a flow rate of 0.09 ml/min. The collected cells (PBMC and apoptotic cells) were then washed and cultured overnight in a 35 mm non-tissue-culture-treated sterile dish in standard cell culture conditions in phenol-free RPMI 1640 media (Thermo Fisher Scientific) supplemented with 15% autologous plasma or C57/BL6 mouse serum isolated previously and 1% penicillin/streptomycin.
Figure 1. Establishment of a novel mouse syngeneic model of HGSOC. 1 x 10Citation7 mCherry+ TKO ovarian cancer cells (detailed in Material and methods section) were injected i.p, in C57BL/6 mice. a. Live animal imaging of mCherry fluorescence showing i.p. tumor burden. Three representative animals are shown through time to demonstrate disease progression; b. Typical tumor kinetics observed in this model as quantified by measuring mCherry fluorescence ROI area as described in the Materials and methods Section. The three phases of disease progression described in the text are shown; ** p < .0001 comparing day 3 with days 11, 13, and 17; c. Abdominal area measurements from X-ray images as described in the Materials and methods Section showing the development of ascites; d. Necropsy image of a representative animal sacrificed on day 30 showing omental cake (yellow arrow) and hemorrhagic ascites (blue arrow).
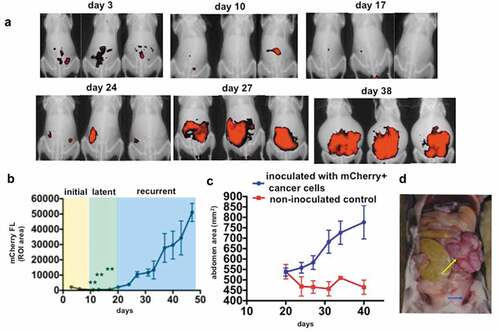
Figure 2. Transimmunization prevents recurrence. Mice bearing i.p. tumors were randomized into three groups 4 days after inoculation of cancer cells: group 1) PBS control (n = 20), group 2) TI TKO (n = 10), and group 3) TI YUMM (n = 10). Treatment was administered as described in the Materials and methods section. a. Tumor growth curves showing mCherry FL ROI area. Data are presented as mean ± SEM. Arrow indicates administration of treatment; **** p < .0001 PBS control vs TI TKO; ns, p = .1079 PBS control vs TI YUMM by Two-Way ANOVA. b. Live imaging of animals on day 27 showing mCherry fluorescence signal as a surrogate for i.p. tumor burden. Ten animals are shown per group; c. Graphical representation of tumor incidence by live animal imaging.
The cells were cultured overnight, and the following day were scraped, washed, resuspended in autologous plasma and administered i.p. in 100ul total volume containing between 5 × 105–1 × 106 cells/dose.
Treatment schedule
For the treatment of recurrent disease, bleeding commenced 25 days post i.p. injection of TKO cells and first i.p. administration of treatment was done 26 days post-injection of TKO cells. For the prevention of recurrence, bleeding commenced 4 days post-injection of TKO cells and first treatment given 5 days post-injection of TKO cells. Treatment was given twice a week for a total of six doses (Supp. Fig. 1,2).
Immunophenotyping by FACS analysis
Peritoneal cells were collected from ascites-free mice by intraperitoneal injection of sterile PBS and then fluid aspiration into heparin-containing tubes. Peritoneal fluid from ascites-containing mice was collected by peritoneal tap into heparin-containing tubes. Peritoneal cells were combined, if necessary, within each group to obtain sufficient cells for analysis. Only tumor-bearing animals were included in the peritoneal analysis. Splenocytes from tumor-bearing as well as tumor-free mice were prepared individually by homogenizing the tissue by passage through a nylon mesh (70 μm). Red blood cells were removed from all cell suspensions using ACK Lysis Buffer (Lonza) and washed in DPBS (Gibco Life Technologies). Splenocytes were analyzed individually for each animal. Obtained cell populations were stained following standard cell-surface FACS protocol. The following antibodies were used: FITC conjugated CD4 (GK1.5), Ly6C (HK1.4); APC conjugated CD62L (MEL-14), F4/80 (BM8), Gr-1 (RB6-8 C5); PE/Cy7 conjugated CD44 (IM7), Ly6G (1A8); BV421 conjugated CD8a (53–6.7), CD11b (M1/70). Fluorochrome-matched isotype control antibodies were obtained from the same vendors. Flow cytometry was carried out on a Stratedigm flow cytometer with electronic gates set on live cells by a combination of forward and side light scatter and EMA (Invitrogen) and mCherry (TKO cells) exclusion. A minimum of 5 × 104 events were collected per sample, and data were analyzed with FlowJo software (FlowJo LLC).
Immunohistochemistry staining and quantitation
Mouse tumors were excised and immediately fixed in buffered formalin for 48 h and then transferred to 70% ethanol for storage or immediate paraffin embedding. Embedded tissue was sectioned into 5-µm-thick sections and mounted onto slides, which were deparaffinized and rehydrated to distilled water. The CD4 and CD8 slides were then placed in EDTA epitope retrieval buffer and the FoxP3 slides were placed in citrate buffer, both at 95 deg C for 30 min, then cooled and rinsed and placed in TBS with tween. An identical solution was used in subsequent washing steps. Endogenous peroxidase is blocked using 3% hydrogen peroxide then rinsed. The primary antibodies were diluted as follows: CD4 (eBioscience 14–9766) 1:100, CD8 (Diagnostic Biosystems RPMD012) 1:100 and FoxP3 (eBioscience 14–5773) 1:80. The slides were then rinsed and the antibodies were detected with HRP conjugated anti-rat secondary antibody then rinsed. DAB is used to identify the reaction then the slides were washed and then counterstained in hematoxylin, dehydrated, cleared, and mounted with resinous mounting media.
Statistical analysis
Data are presented as mean ± SEM. Data were analyzed using Ordinary One-way ANOVA, Two-way ANOVA or unpaired t test as appropriate and as designated in the Figure legends. p < .05 was considered as significant and reported as * p < .05, ** p < .01, *** p < .001, **** p < .0001, ns p > .05 not significant.
Results
Establishment and characterization of a mouse HGSOC syngeneic model for testing novel therapies
In order to evaluate the potential efficacy of immunotherapy in the initiation, progression, and recurrence of ovarian cancer, we developed an intraperitoneal (i.p.) mouse model of ovarian cancer using mouse TKO ovarian cancer cells injected into C57/BL6 immune-competent mice. TKO ovarian cancer cells were isolated from TKO mice, a conditional Dicer-PTEN knock-out model with a p53 mutation.Citation2,Citation17,Citation31 On average, tumors in these mice begin at 8 weeks and mice reach an average survival of 32 weeks. To obtain a more rapid model that can be evaluated in real-time, TKO mouse ovarian cancer cells were isolated from spontaneously formed tumors and transduced to stably express the mCherry fluorescence. Fluorescent TKO ovarian cancer cells (mCherry+ TKO cancer cells) were then inoculated i.p. for real-time monitoring and evaluation of disease progression. Typical live animal imaging and tumor growth curves are presented in using mCherry fluorescence intensity as a readout for tumor burden. Three days after inoculation, mCherry+ TKO cancer cells can be observed within the peritoneal cavity. This initial mCherry signal gradually dissipates within 5–20 days post-injection without any treatment intervention (). Nevertheless, at approximately 2 weeks post-inoculation, mCherry signal reemerges and continues to progress, with animals developing substantial i.p. carcinomatosis and ascites by day 27–38 (). Upon necropsy, animals present with hemorrhagic ascites, ovarian tumors, and metastatic implants on the omentum, pelvic fat, and mesentery (), which mimic localization, distribution, and histology described for human disease.Citation32,Citation34,Citation35 Thus, in this animal model, we were able to define three distinct phases of i.p. tumor progression: 1) initial phase (days 1–9; yellow box); 2) latent phase (days 10–19; green box); and 3) recurrent phase (days 20 onwards, blue box) (). Distinguishing these phases would allow us to evaluate the effect of interventional therapy in the different stages of ovarian cancer progression.
TI inhibits progression of recurrent disease
With this pattern of disease progression, we initially tested whether TI vaccination could delay progression of recurrent disease. Thus, mCherry+ TKO cancer cells were inoculated i.p. and mice were randomized into PBS control and TI group on day 26 when tumors had recurred and reached its logarithmic growth phase (). The TI protocol and treatment schedule are illustrated in Supplementary Figure 1. TI was administered i.p. twice a week for 3 weeks for a total six doses. On day 43, after the 6th vaccination dose, we observed notable differences in tumor progression. While mice in the Control group had entered the log phase of growth, TI-treated mice exhibited suppression of tumor growth although statistical significance was not achieved. Nevertheless, tumor suppression was maintained for up to 3 weeks after the last DC vaccination dose (Supp. Figure 3a). Concurrently, abdominal girth measurements associated with active ascites formation were stabilized in the TI-treated mice in contrast to mice in the Control group (Supp. Figure 3b, c). Similarly however, statistical significance was not achieved. Mice in the Control group were sacrificed around day 43 due to extensive disease progression whereas median survival in TI-treated mice was extended to day 57 (Supp. Figure 3d, p = .0339).
TI promotes changes in the immune infiltrates
To determine whether TI-induced anti-tumor immune responses could be correlated with the observed tumor inhibition we performed a pilot study to characterize the intra-tumoral immunophenotype. Thus, we collected tumor explants from control (n = 2) and treatment groups (n = 2) and characterized the immune infiltrates by flow cytometry. Our results show that TI-treated mice have higher percentages of CD3+ tumor-infiltrating lymphocytes (TILs) compared to controls (Supp. Figure 4a) which has previously been associated with better prognosis in human HGSOC.Citation36,Citation37 In addition, TI-treated mice presented lower levels of tumor-promoting CD11b+/Gr1+ myeloid-derived suppressor cells (MDSC) (Sup. Figure 4b), also previously associated with favorable prognosis in HGSOC.Citation38 This difference was confirmed in the tumor-associated ascites fluid following ascites/peritoneal lavage as a shift in the ratio of TKO cancer cells to immune cells, where TI-treated mice exhibited reduced ratios of mCherry+ TKO cancer cells (38% compared to 68% in the Control) to CD45+ mCherry-negative leukocytes in the peritoneum (40% compared to 7% in the control) (Supp. Figure 4c). Although statistical significance is not reached, probably due to the low sample size tested, our findings suggest that TI treatment can stimulate anti-tumor immune responses through alterations of the tumor microenvironment both in the tumor interstitium and tumor-associated ascites.
TI prevents establishment of recurrent disease
Following our initial observation of TI-induced tumor growth inhibition in mice with recurrent disease and heavy peritoneal tumor burden, we sought to determine whether TI could prevent the establishment of recurrent disease. To this end, our treatment schedule was modified to begin 4 days post-tumor implantation, prior to logarithmic tumor growth. Frequency of TI administration was maintained at twice a week for six doses (Supp. Figure 2). To demonstrate the antigen specificity of TI treatment, a second control group was included where animals were treated with TI, however, with an unrelated apoptotic melanoma tumor as an antigen source. TKO ovarian cancer-bearing mice were randomized into three groups: (1) PBS control; (2) TI TKO, utilizing TKO-loaded TI cells (as previously generated in Supp. Figs. 1 and 3); and (3) TI YUMM, where apoptotic YUMM1.7 murine melanoma cells were substituted as the tumor source. We have previously demonstrated YUMM1.7 as an immunogenic murine melanoma suitable for modeling TI treatment against established subcutaneous tumors and have previously reported that 8-MOP-UVA treatment induces immunogenic cell death in these cells.Citation30 The addition of this third group would allow us to confirm the presence of antigen-specific T cell generation following TI treatment and rule out nonspecific vaccination effects as drivers of the observed tumor growth inhibition. As shown in , the initial and latent phases of tumor progression described in are observed in all three groups. By day 20, we observed uniformly logarithmic tumor growth commencing in the PBS control and TI YUMM groups, with some limited growth in the TI TKO group. By day 27, tumor growth in TI TKO-treated mice diverged dramatically from those in PBS control (p = .0001) (). Control and TI YUMM groups did not show significant differences in tumor growth by day 20 (p = .1079), with both groups exhibiting log phase of growth until day 27. In contrast, TI TKO group exhibited suppression in tumor growth by day 24 and continued to decrease until day 27, 7 days post the final administration of TI. Visualized graphically, by day 27, only 20% of the TI TKO-treated mice exhibited measurable disease in contrast to 65% and 60% in PBS control and TI YUMM-treated groups, respectively (. TI TKO vaccination led to both a significant reduction in the percentage of mice with recurrent disease and extension of the overall time to recurrence, metrics useful in evaluating any model treatment of human HGSOC. Our results in the parallel TI YUMM treatment show that the effect exhibited in TI TKO was predominantly specific to the ovarian tumors. Taken together, these results indicate that TI generated vaccinating DCs loaded with a relevant “personalized” antigen source can effectively delay or prevent the establishment of recurrent disease.
TI induces intra-tumoral influx of CD8 and CD4 T cells
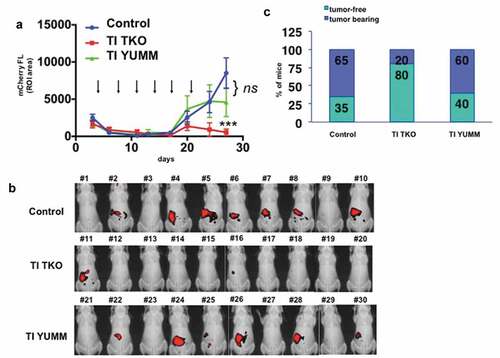
To validate our findings shown in Supp. Figure 4 that TI treatment is associated with an enhanced influx of TILs and reduced intra-tumoral immune-suppressive cells, we performed IHC on representative tumors from the Control and TI TKO groups. Intra-tumoral staining for CD8 and CD4 showed significantly higher levels of both T cell subpopulations in tumors from TI TK-treated mice compared to Control (p = .0037 and p = .0079, respectively; ). We also observed lower levels of FOXP3+ cells in tumors from TI TKO-treated mice compared to Control although statistical significance was not achieved (p = .0904).
Figure 3. Transimmunization enhances intra-tumoral T cell influx.Tumors from animals in Figure 3 were collected on day 30 and immunostained for a. CD8; b. CD4; and c. FOXP3. Top panel. For each tumor sample, quantification was performed on three randomly selected tumor areas and shown as mean ± SEM. ** p < .01; ns, p = .0904; bottom panel shows representative images for each immunostain.
TI upregulates intra-tumoral Rantes and MIP1β
To determine the effect of TI on intra-tumoral cytokine levels, we quantified 23 cytokines and chemokines from homogenized tumor lysates. We saw the most significant effect on the chemokine Rantes. Compared to Control (mean = 28.4 ± 8.4 pg/ml) and TI YUMM (mean = 34.7 ± 1.7 pg/ml) groups, mice in the TI TKO group (mean = 59.6 ± 10.8 pg/ml) showed significantly higher levels of Rantes (p = .0037 and p = .0051, respectively) (Supp. Figure 5). Rantes levels were not statistically different between Control and TI YUMM (p = .8373). We also observed changes in the levels of MIP1β. Significance was observed between the TI TKO group (mean = 50.67 ± 6 pg/ml) and TI YUMM group (mean = 37.2 ± 1.7 pg/ml) (p = .0058) but not when Control (mean = 42.16 ± 4 pg.ml) was compared to TI TKO (p = .0766) and to TI YUMM (p = .3414) (Supp. Figure 5). No significant differences in the levels of IL1α, IL1β, IL2, IL6, IL9, IL10, IL12p40, IL12p70, IL13, G-CSF, GM-CSF, IFNɣ, Eotaxin, KC, MIP1α, TNF α, MCP1 were observed (data not shown). IL3, IL4, IL5, and IL17 were below the detectable range.
TI reprograms the intra-peritoneal immune phenotype
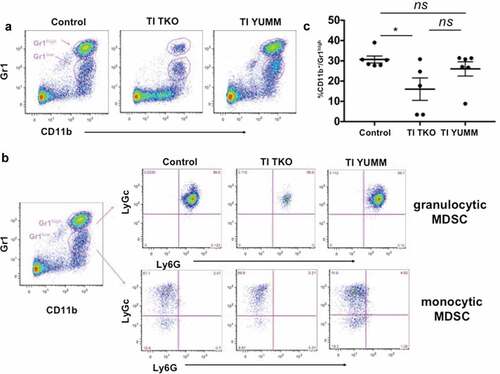
We next sought to further determine the effects of TI on the immune-profile of cells present in the local i.p. tumor environment. Thus, collections from the ascites fluid or when little or no ascites was available, peritoneal lavage from individual tumor-bearing mice were analyzed using three phenotyping panels designed for: 1) tumor/malignant ascites-associated macrophages (TAMs) utilizing antibodies against CD11b and F4/80; panel 2) MDSCs: CD11b, Gr1, Ly6 G, and Ly6 c; and 3) T cell memory and activation: CD4, CD8, CD44, and CD62 L.
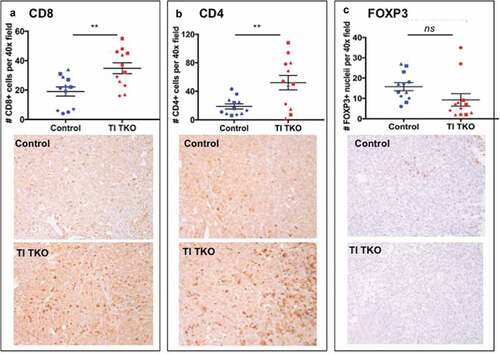
Percentages of i.p. TAMs are shown in . We observed a lower percentage of CD11b+/F4/80+ TAMs in TI TKO-treated animals (mean = 29.41%±17.83) compared to Control (mean = 49.42%±6.3) and TI YUMM groups (mean = 39.68%±11.05) suggesting that TI was able to prevent the full maturation of monocytes into pro-tumor TAMs. Interestingly, statistical analysis demonstrated significant differences in TI TKO group when compared to Control (p = .0422) but not when compared to TI YUMM (p = .37) . Further analysis of CD11b+ myeloid cells confirmed lower intensities of F4/80 staining in TI TKO-treated mice (mean = 86.02 ± 14.91 MFI) compared to Control (mean = 138.2 ± 37.18 MFI) and TI YUMM (mean = 101.9 ± 33.7 MFI). Similarly, statistical significance was observed only when TI TKO was compared to Control (p = .037) and not when compared to TI YUMM (p = .68). The significant suppression of the acquisition or maintenance of F4/80 positivity within the CD11b+ cells in the TI group suggests that vaccination was able to prevent or alter macrophage differentiation and inhibit macrophage-associated pro-tumor signals. The lack of statistical significance between TI TKO and TI YUMM suggests a non-antigen specific effect of TI on this immune cell population.
Figure 4. Transimmunization inhibits the formation of tumor/malignant ascites-associated macrophages (TAMs). Ascites or peritoneal lavage were collected at the end of the study and analyzed by flow cytometry for CD11b and F4/80. a. Left panel, representative dot plot images showing quadrant analysis; right panel, representative F4/80 MFI histogram plots for CD11b+ cells; b. Graphical representation of the percentage of CD11b+/F480+ cells in each group. Data are presented as mean ± SEM; * p = .042; ns, p > .05; c. Graphical representation of F4/80 MFI in CD11b+ cells in each group. Data are presented as mean ± SEM; * p = .037; ns, p > .05. Statistical analysis was performed using Ordinary One-way ANOVA with Tukey post hoc analysis.
We then characterized the effect of TI on MDSC. We co-stained for established MDSC markers, CD11b and Gr1, as well as the myeloid differentiation markers Ly6 C and Ly6 G. We confirmed the presence of CD11b+/Gr1+ MDSC in the peritoneum of animals in all groups and observed two distinct MDSC populations, which were distinguished by the intensity of Gr1 expression (i.e. CD11b+/Gr1high and CD11b+/Gr1low) (). Further analysis of these two populations indicated that CD11b+/Gr1high shows the characteristics of granulocytic MDSC (Ly6 G+/Ly6 Clow) whereas CD11b+/Gr1low MDSC appear to be monocytic MDSC (Ly6 G−/Ly6 Chigh) (). Quantification showed that TI TKO treatment (mean = 16.01 ± 12.38%) significantly lowered the levels of granulocytic MDSC when compared to Control (mean = 30.67 ± 4.2%; p = .0388) (). Statistical significance was however not observed when TI TKO was compared to TI YUMM (mean = 26.01 ± 8.67%; p = .18). Similarly, granulocytic MDSC levels were not significantly different between Control and TI YUMM (p = .63). In addition, analysis of the monocytic MDSC component demonstrated no statistical difference between groups (data not shown). These results are in line with previous studies, which indicate that MDSC in the human metastatic site typically demonstrate a granulocytic phenotype.Citation39 More importantly, these results demonstrate that similar to the effect on TAMs, TI is able to decrease the levels of immunosuppressive granulocytic MDSC in an antigen-independent manner. Thus taken together, the observed changes in the TAMs and MDSC makeup in both TI YUMM and TI TKO may suggest a general TI-induced effect on inhibition of suppressive myeloid populations regardless of antigen identity.
Figure 5. Transimmunization significantly decreases the intra-peritoneal levels of granulocytic myeloid-derived suppressor cells. Ascites or peritoneal lavage were collected at the end of the study and analyzed by flow cytometry for CD11b, Gr1, Ly6C, and Ly6 G. a. Representative dot plot images showing distinct cell populations (CD11b+/Gr1high and CD11b+/Gr1low). b. Representative dot plot images showing subanalysis of CD11b+/Gr1high and CD11b+/Gr1low populations for Ly6C and Ly6 G; c. Graphical representation of percentage of CD11b+/Gr1high cells in each group. Data are presented as mean ± SEM; * p = .0388; ns, p > .05 by Ordinary One-way ANOVA with Tukey post hoc analysis.
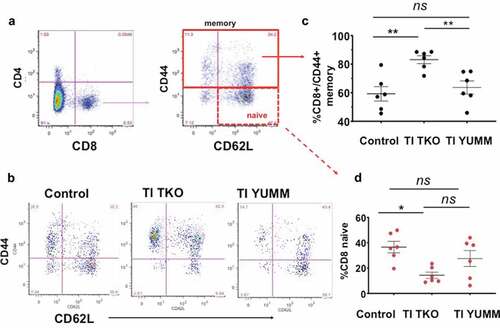
Finally, evaluation of effector cells engaging in antigen-specific immune responses displayed no superficial difference among treatment groups in the CD4 and CD8 makeup of T cells in the peritoneum (Supp. Figure 6). However, a more detailed analysis of the specific phenotype of CD8 T cells showed a significant increase in the percentage of CD44+ memory CD8 T cells in the TI TKO group (mean = 82.92 ± 7.52%) compared to Control (mean = 59.61 ± 12.34%; p = .0069) and TI YUMM (mean = 63.74 ± 15.14%; p = 00.88) ().In addition, we observed a significant decrease in the percentage of CD8+/CD44-/CD62 L+ naive T cells in the TI TKO-treated mice (mean = 14.53 ± 6.67%) compared to Control (mean = 36.11 ± 11.18%; p = .0125) () No statistically significant difference was observed in the percentage of CD8 naïve T cells between TI TKO and TI YUMM (p = .0591) and between Control and TI YUMM (p = .7508) ().
Figure 6. Transimmunization significantly improves memory T cell responses in the peritoneum. Ascites or peritoneal lavage were collected at the end of the study and analyzed by flow cytometry for CD4, CD8, CD44, and CD62 L. a. Dot plots showing gating strategy for analysis of different phenotypes of CD8 T cells; b. Representative dot plot images for each group showing sub-analysis for CD44 and CD62 L in CD8+ cells; c. Graphical representation of the percentage of CD8+/CD44+ memory T cells in each group. Data are presented as mean ± SEM; ** p < .01; ns, p > .05; d. Percentage of CD8 naive T cells in each group. Data are presented as mean ± SEM; * p = .0125; ns, p > .05. Statistical analysis is performed using Ordinary One-way ANOVA with Tukey post hoc analysis.
Taken together, these results suggest that the ability of TI TKO to prevent recurrence may be due to the inhibition of recruitment/differentiation of immunosuppressive cells. Results from our secondary control, TI YUMM, further suggest that TI may be targeting TAMs and MDSCs for reprogramming in an antigen-independent manner. In combination with effector and memory T cell populations exclusively arising from TI TKO treatment, TI is shown to employ both antigen-independent and antigen-specific mechanisms of myeloid reprogramming and effector stimulation to confer anti-tumor immunotherapy.
TI reprograms the systemic immune milieu
Our final objective was to determine not only the effect of treatment but also to characterize the changing systemic immune-profiles in an animal through successful tumor establishment in this novel, clinically relevant recurrence model of ovarian cancer. We tested both tumor-bearing and non-tumor bearing animals for deconvoluting changes to the immune phenotypes brought on by tumor formation and those caused by TI treatment, as we hypothesized that these changes will illuminate key events required for initial native immune suppression of the tumor as well as vaccine-induced therapeutic contributions. Thus, we analyzed dissociated splenocytes from animals in all the three groups, regardless of tumor incidence, as most animals in the TI TKO group did not develop tumors. We utilized a similar panel of myeloid and lymphoid markers for the characterization of MDSC and T cells as described above.
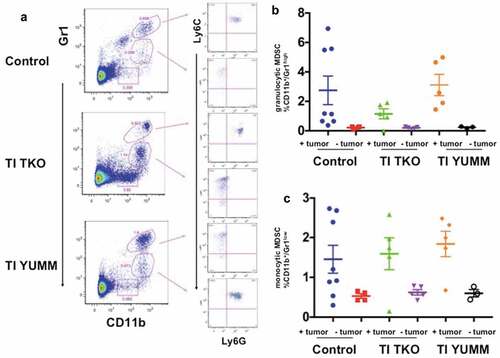
Analysis of MDSC populations in the spleen showed that independent of treatment, tumor-forming animals consistently exhibited higher percentages of both granulocytic and monocytic MDSC compared to non-tumor bearing animals, demonstrating a potential role for MDSCs in immune escape and progression of tumors (). Interestingly, we observed decreased percentages of granulocytic MDSCs in the spleen of the tumor-bearing TI TKO-treated group (mean = 1.15 ± 0.7%) compared to tumor-bearing Control (mean = 2.75 ± 2.7%) and tumor-bearing TI YUMM (mean = 3.11 ± 1.6%), though statistical significance was not achieved (). No significant difference was observed in the percentage of monocytic MDSCs in tumor-bearing animals between treatment groups ().
Figure 7. Transimmunization decreases differentiation/recruitment of granulocytic MDSC. Splenocytes were collected at the end of the study and analyzed by flow cytometry for CD11b, Gr1, Ly6C, and Ly6 G. a. Representative dot plot images showing distinct CD11b+/Gr1high and CD11b+/Gr1low cell populations and sub-analysis of these populations for Ly6C and Ly6 G; Graphical representation of the percentage of b. CD11b+/Gr1high cells and c. CD11b+/Gr1low cells in each treatment group with tumor-bearing and non-tumor-bearing animals shown; p < .05 for both graphs; ns, not significant by One-Way ANOVA.
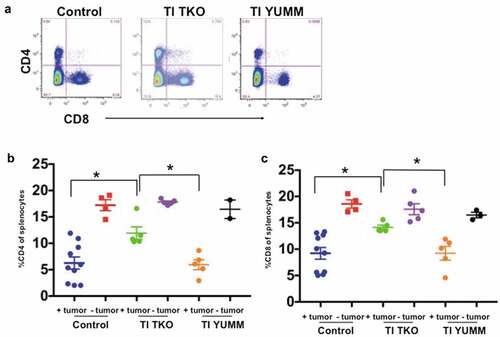
Finally, analysis of the T cell compartment showed that independent of treatment, tumor-bearing animals exhibited significantly lower percentages of CD4 and CD8 T cells compared to non-tumor bearing animals () suggesting that the presence of tumor can suppress effector activity. Evaluated together in the context of different treatment regimens, we found that tumor-bearing animals in the TI TKO group presented a significantly restored T cell effector compartment, and a profile most comparable to non-tumor bearing animals (). Compared to tumor-bearing Control (blue data points; mean = 9.2 ± 3.5%) and tumor-bearing TI YUMM-treated animals (orange data points; mean = 9.22 ± 2.9%), tumor-bearing TI TKO-treated animals (green data points; mean = 14.14 ± 0.9%) had significantly increased levels of CD4 T cells (p = .0196 compared to Control and p = .0451 compared to TI YUMM) (). Similarly, compared to tumor-bearing Control animals (blue data points; mean = 6.26 ± 3.64%) and tumor-bearing TI YUMM-treated animals (orange data points; mean = 5.93 ± 2.1%) tumor-bearing TI TKO-treated animals (green data points; mean = 11.92 ± 2.65%) also had significantly higher levels of CD8 T cells (p = .0108 compared to Control and p = .0195 compared to TI YUMM) (). These results suggest that one of the specific effects of TI may be counteracting the systemic effects of tumor progression by restoring/maintaining CD4 and CD8 T cell levels, while limiting recruitment of tumor-promoting suppressive myeloid cells.
Figure 8. Transimmunization enhances systemic CD4 and CD8 T cell proliferation. Splenocytes were collected at the end of the study and analyzed by flow cytometry for CD4 and CD8. a. Representative dot plot images showing quadrant analysis for CD4 and CD8 in the different groups. b. Graphical representation of the percentage of CD4 cells. Data are presented as mean ± SEM; * p < .05. c. Graphical representation of the percentage of CD8 cells. Data are presented as mean ± SEM; * p < .05. Statistical analysis is performed using Ordinary One-way ANOVA with Tukey post hoc analysis.
Taken together, our results demonstrate that TI treatment is associated with reprogramming of the suppressive myeloid environment and, simultaneously, generating DC-based vaccination for antigen-specific anti-tumor responses, delaying tumor progression, and preventing recurrence. These effects are observable not only in the tumor itself but also in the tumor-associated ascites fluid and surrounding lymphoid tissue. As both TI TKO and Control TI YUMM have helped demonstrate, TI treatment can be attributable for decreasing TAM and MDSC infiltration and, when utilized with the disease-specific antigen, can amplify its therapeutic effect through promotion of an anti-tumor adaptive immune response characterized by increased CD4+ and CD8 + T cells.
Discussion
We describe for the first time the in vivo efficacy of TI vaccination in the treatment of i.p. solid tumors. Using a syngeneic mouse model of HGSOC we showed that TI, using autologous apoptotic cancer cells as the source of antigen, can prevent recurrence and the establishment of carcinomatosis. TI, independent of antigen source, is able to decrease the levels of immunosuppressive TAMs and MDSCs in the tumor microenvironment. In addition, TI can promote CD4 and CD8 T cell survival and expansion. The effect on the T cell population is interestingly antigen-dependent, in contrast to the observed effect on the immunosuppressive myeloid population. It is this antigen-specific effect on the T cells that reflects the anti-tumor activity of TI pointing to the critical necessity of effector T cell mobilization in the restoration of immune surveillance. As such, the TI TKO-induced therapeutic effect is shown to be antigen-specific as anti-tumor responses were conditional to the use of autologous tumor as antigen source, but not upon use of an unrelated tumor of differing tissue-type. TI and its parent therapy ECP have previously been associated clinically with treatment-induced increases in circulating anti-tumor CD8 T cells and reduced T-regsCitation18,Citation40 as well as T cell-mediated anti-tumor effects in solid tumor mouse models;Citation29 however, this work is the first to report TI-driven increases in TIL and ascites CD4/CD8 cells as well as reduction in suppressive myeloid cells, MDSCs and TAMs, and T-regs, indicating TI may be an effective treatment for virtually any immunogenic solid tumor. As it is essential to correlate the effect of TI-induced changes in immunophenotypes to the extension of survival, we are aware that a limitation of our study is the lack of demonstration on the treatment effect on progression-free survival and overall survival. This is currently an active endeavor in our lab in addition to the determination of how many doses of TI are required to optimally improve survival. Nevertheless, we clearly demonstrate the promise of TI in preventing recurrence and curtailing the formation of carcinomatosis, two outcomes key to the translation potential of the therapy in the treatment of ovarian cancer.
Historically, ovarian cancer has not been considered an immunogenic malignancy given its low mutational tumor burden compared to highly mutated tumors such as melanoma and renal cancers.Citation41,Citation42 However, the demonstration in patients that the presence of TILs correlates with both PFS and OSCitation37,Citation43-Citation45 suggested the possible value of immunotherapy in ovarian cancer. Since then there has been a substantial increase in research activities concerning the development of immunotherapies targeting ovarian cancer and this has continued in the past decade (reviewed comprehensively inCitation46-Citation48). The current understanding is that ovarian tumors do express tumor-associated antigens and that the activation of T cell effector function concomitant with the downregulation of immunosuppressive checkpoint pathways can improve patient outcomes.Citation48 Achieving an effective anti-tumor immune response however required multiple modalities or combined administration with other therapies as in the case with NY-ESO-1 immunotherapy wherein the combination with TGFβ inhibition or epigenetic regulators is being investigated.Citation49-Citation52 Indeed, it has been suggested that effective anti-tumor immune responses as a result of immunotherapies require sufficient generation and intra-tumoral trafficking of effector T cells, the concomitant inhibition of immune-modulatory checkpoints, the recognition of tumor antigens and the generation and persistence of an anti-tumor immune response.Citation48 It is particularly notable that TI, as a single modality, is able to induce generation of effector T cells capable of sufficient tumor infiltration to be potentially associated with tumor control, and additionally downregulate two immunosuppressive networks, the TAMs and MDSC to result in a significant reduction of tumor burden. Our data showing upregulation of memory CD8 + T cells suggests that TI may provide lasting protection, though further studies are needed to better characterize the longevity of TI effect, the number of TI doses required and whether combination of TI vaccination and checkpoint inhibition could synergistically affect T cell responsiveness and lead to increased tumor control or complete prevention of tumor recurrence.
The observed inverse correlation between MDSCs and tumor progression is important to note. Multiple immune cell types are known to infiltrate the tumor microenvironment. MDSCs represent one of the most important effectors of immunosuppression and the support of tumor escape.Citation53 These cells can strongly inhibit antitumor immune responses mediated by both T cells and NK cells. We observed a decrease in the percentage of T cells and an increase in the percentage of MDSCs in animals with progressing tumors. This ratio is reversed in animals responding to TI. MDSCs are immature myeloid cells that under chronic inflammatory conditions fail to complete their maturation to macrophages, granulocytes, or DCs. This type of microenvironment is typical in ovarian cancer and supports tumor progression.Citation54-Citation56 Mouse MDSCs consist of two subsets: granulocytic CD11b+Ly6 G+Ly6 Clo MDSCs and monocytic CD11b+Ly6G−Ly6Chi MDSCs.Citation54,Citation55 They exert multiple mechanisms that allow tumor immune escape. These include the expression of immunosuppressive cytokines such as TGFβ and IL-10; upregulation of checkpoint inhibitors such as PD-L1, and induction of activation-inducible NO synthase, which results in the synthesis of NO as well as the production of reactive oxygen species (ROS). Both NO and ROS can induce T cell apoptosis or block T cell function.Citation54,Citation56 Thus, it is clear that MDSCs play a critical role in the development of an immunosuppressive tumor microenvironment, and inhibition of their recruitment or differentiation is a critical step in restoring immune surveillance. Indeed, several studies have shown the presence of MDSCs in ovarian tumor samples and their direct correlation with poor prognosis.Citation57-Citation59 Consequently, an immunotherapeutic approach that can reduce the generation of MDSCs will likely overcome generalized immunosuppression as well as regulation of anti-tumor/infiltrating T cells. Our data support that TI is able to achieve this goal by simultaneous inhibition of suppressive MDSCs and TAMs and stimulation of both intra-tumoral and intraperitoneal T cell responses. Interestingly, this effect on both reduction of suppressive myeloid cells and increases in T cells was also observed in the spleen, which is not traditionally regarded as part of the ovarian tumor microenvironment but often used to monitor systemic effects. Our conclusion is that TI deploys a bilateral attack on the tumor microenvironment by reprogramming the suppressive myeloid cells that negatively regulate adaptive immune responses, and by promoting tumor antigen-specific DC vaccines that stimulate effector T cell generation and infiltration.
The demonstration that the immune response is dependent on the source of tumor antigen provides important information, especially in the context of non-chemonaive or recurrent ovarian cancer. This highlights the importance of using the appropriate tumor-antigen source. Our group has previously shown that treatment with Paclitaxel can change the molecular signature of ovarian cancer and enhance the mesenchymal phenotype.Citation32 Hence, the ability of tumors to evolve as a response to the stress of chemotherapy and with disease progression should be taken into account in the choice of tumor antigen source. Residual disease has been observed during second-look laparoscopic surgeries post 1st line treatment even in patients classified as responders.Citation9,Citation60 As such biopsy samples from these lesions may be considered as a possible source of tumor antigen if TI will be administered after 1st-line standard of care. It is also important to note that the TI protocol precludes the need to identify a specific tumor-associated antigen since it uses all the constituents of apoptotic cell cultures.
The mouse model described uses cancer cells injected i.p. and allowed to attach, establish, and progress to carcinomatosis. This mimics the tumor environment during the process of recurrence in ovarian cancer patients wherein tumor cells remaining after 1st-line treatment can shed into the peritoneal fluid, reach a secondary site, establish, and grow. Our results show that TI can prevent or significantly slow the establishment of i.p. tumors, suggesting its value in averting the formation of recurrent disease following 1st line therapy failure. This strategy might be particularly effective if utilized in a setting of minimal residual disease, with an immunocompetent patient being vaccinated against precisely the antigens expressed by the chemo-resistant cells responsible for recurrence, a strategy we can now model using the TI TKO system.
The data presented in this study confirm the efficacy of TI-based vaccination in ovarian cancer and, additionally, point to the possibility of immunotherapeutic intervention directly in the peritoneal cavity for a variety of intra-peritoneal cancers. The antigen-specific nature of the observed response indicates that TI could be modified to provide “personalized” anti-tumor responses against neoantigens in any malignancy already demonstrating responses to immune-based therapies.Citation61,Citation62 This suggests TI could be active as a monotherapy but also opening the door to potential combination approaches in which TI could synergize with promising agents such as immune checkpoint inhibitors, CAR T cells, and adoptively transferred T cells.
The ability to add an effective immunotherapy regimen in the management and care of ovarian cancer patients would have a significant impact on patient survival, especially since the current clinical practice has not improved survival in the past few decades. The demonstration that TI, as a single therapy, can activate an immune response and inhibit immune-modulatory crosstalk with sufficient power to prevent recurrent disease and limit the progression of carcinomatosis suggests the possible value of testing this modality in clinical trial of recurrent ovarian cancer, as well as other peritoneal cancers.
Disclosure of Potential Conflicts of Interest
O. Sobolev is VP Immunology at Transimmune AG; M. Bosenberg is a consultant for Eli Lilly and Company; R.L. Edelson has ownership interest (including patents) in, and is a consultant/advisory board member of Transimmune AG.
Supplemental Material
Download ()Supplementary Material
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
References
- Audiger C, Rahman MJ, Yun TJ, Tarbell KV, Lesage S. The importance of dendritic cells in maintaining immune tolerance. J Immunol. 2017;198(6):2223–14. doi:10.4049/jimmunol.1601629.
- Mutyambizi K, Berger CL, Edelson RL. The balance between immunity and tolerance: the role of Langerhans cells. Cell Mol Life Sci. 2009;66:831–840. doi:10.1007/s00018-008-8470-y.
- Markman M. Pharmaceutical management of ovarian cancer: current status. Drugs. 2019;79(11):1231–1239. doi:10.1007/s40265-019-01158-1.
- Markman M. The evolving arena of ovarian cancer maintenance therapy. Oncology. 2019;97:202–205.
- Pokhriyal R, Hariprasad R, Kumar L, Hariprasad G. Chemotherapy resistance in advanced ovarian cancer patients. Biomark Cancer. (2019);11:1179299X19860815. doi:10.1177/1179299X19860815.
- Armbruster S, Coleman RL, Rauh-Hain JA. Management and treatment of recurrent epithelial ovarian cancer. Hematol Oncol Clin North Am. (2018);32(6):965–982. doi:10.1016/j.hoc.2018.07.005.
- Armstrong DK. Relapsed ovarian cancer: challenges and management strategies for a chronic disease. Oncologist. (2002);7(Suppl 5):20–28. doi:10.1634/theoncologist.7-suppl_5-20.
- Onnis A. The management of ovarian cancer: an update. Eur J Gynaecol Oncol. 1997;18:157–160.
- Chu CS, Rubin SC. Second-look laparotomy for epithelial ovarian cancer: a reappraisal. Curr Oncol Rep. 2001;3(1):11–18. doi:10.1007/s11912-001-0037-0.
- Alvero AB, Montagna MK, Craveiro V, Liu L, Mor G. Distinct subpopulations of epithelial ovarian cancer cells can differentially induce macrophages and T regulatory cells toward a pro-tumor phenotype. Am J Reprod Immunol. 2012;67(3):256–265. doi:10.1111/j.1600-0897.2011.01068.x.
- Gimotty PA, Zhang L, Alagkiozidis I, Cadungog M, Adams S, Chu C, Katsaros D, Coukos G. Immune prognostic factors in ovarian cancer: lessons from translational research. Dis Markers. 2007;23(5–6):445–452. doi:10.1155/2007/508350.
- Zsiros E, Dangaj D, June CH, Kandalaft LE, Coukos G. Ovarian cancer chemokines may not be a significant barrier during whole tumor antigen dendritic-cell vaccine and adoptive T-cell immunotherapy. Oncoimmunology. 2016;5(5):e1062210. doi:10.1080/2162402X.2015.1062210.
- Baert T, Vankerckhoven A, Riva M, Van Hoylandt A, Thirion G, Holger G, Mathivet T, Vergote I, Coosemans A. Myeloid derived suppressor cells: key drivers of immunosuppression in ovarian cancer. Front Immunol. 2019;10:1273. doi:10.3389/fimmu.2019.01273.
- Walankiewicz M, Grywalska E, Polak G, Kotarski J, Siwicka-Gieroba DJ, Roliński J. Myeloid-derived suppressor cells in ovarian cancer: friend or foe? Cent Eur J Immunol. 2017;42(4):383–389. doi:10.5114/ceji.2017.72823.
- Zhang AW, McPherson A, Milne K, Kroeger DR, Hamilton PT, Miranda A, Funnell T, Little N, de Souza CPE, Laan S. Interfaces of malignant and immunologic clonal dynamics in ovarian cancer. Cell. 2018;173(7):1755–1769 e1722. doi:10.1016/j.cell.2018.03.073.
- Raghavan S, Mehta P, Xie Y, Lei YL, Mehta G. Ovarian cancer stem cells and macrophages reciprocally interact through the WNT pathway to promote pro-tumoral and malignant phenotypes in 3D engineered microenvironments. J Immunother Cancer. 2019;7(1):190. doi:10.1186/s40425-019-0666-1.
- Gaillard SL, Secord AA, Monk B. The role of immune checkpoint inhibition in the treatment of ovarian cancer. Gynecol Oncol Res Pract. 2016;3(1):11. doi:10.1186/s40661-016-0033-6.
- Girardi M, Berger CL, Wilson LD, Christensen IR, Thompson KR, Glusac EJ, Edelson RL. Transimmunization for cutaneous T cell lymphoma: a Phase I study. Leuk Lymphoma. 2006;47(8):1495–1503. doi:10.1080/10428190600581419.
- Edelson R, Wu Y, Schneiderman J. American council on ECP (ACE): why now? J Clin Apher. 2018;33(4):464–468. doi:10.1002/jca.21627.
- Raval JS, Ratcliffe NR. Extracorporeal photopheresis and personalized medicine in the 21st century: the future’s so bright! J Clin Apher. 2018;33(4):461–463. doi:10.1002/jca.21633.
- Alfred A, Taylor PC, Dignan F, El-Ghariani K, Griffin J, Gennery AR, Bonney D, Das-Gupta E, Lawson S, Malladi RK. The role of extracorporeal photopheresis in the management of cutaneous T-cell lymphoma, graft-versus-host disease and organ transplant rejection: a consensus statement update from the UK Photopheresis Society. Br J Haematol. 2017;177(2):287–310. doi:10.1111/bjh.14537.
- Dunbar NM, Raval JS, Johnson A, Abikoff CM, Adamski J, Cooling LL, Grossman B, Kim HC, Marques MB, Morgan S. Extracorporeal photopheresis practice patterns: an international survey by the ASFA ECP subcommittee. J Clin Apher. 2017;32(4):215–223. doi:10.1002/jca.21486.
- Durazzo TS, Tigelaar RE, Filler R, Hayday A, Girardi M, Edelson RL. Induction of monocyte-to-dendritic cell maturation by extracorporeal photochemotherapy: initiation via direct platelet signaling. Transfus Apher Sci. 2014;50:370–378. doi:10.1016/j.transci.2013.11.008.
- Edelson RL. Mechanistic insights into extracorporeal photochemotherapy: efficient induction of monocyte-to-dendritic cell maturation. Transfusion Apheresis Sci: Off J World Apheresis Assoc: Off J Eur Soc Haemapheresis. 2014;50(3):322–329. doi:10.1016/j.transci.2013.07.031.
- Kibbi N, Sobolev O, Girardi M, Edelson RL. Induction of anti-tumor CD8 T cell responses by experimental ECP-induced human dendritic antigen presenting cells. Transfusion Apheresis Sci: Off J World Apheresis Assoc: Off J Eur Soc Haemapheresis. 2016;55(1):146–152. doi:10.1016/j.transci.2016.06.001.
- Edelson RL. Transimmunization: the science catches up to the clinical success. Transfus Apher Sci. 2002;26(3):177–180. doi:10.1016/S1473-0502(02)00010-1.
- Girardi M, Berger C, Hanlon D, Edelson RL. Efficient tumor antigen loading of dendritic antigen presenting cells by transimmunization. Technol Cancer Res Treat. 2002;1(1):65–69. doi:10.1177/153303460200100109.
- Ventura A, Vassall A, Yurter A, Robinson E, Filler R, Hanlon D, Meeth K, Ezaldein H, Girardi M, Sobolev O. Novel protocol for generating physiologic immunogenic dendritic cells. J Visualized Exp: JoVE. 2019;147. 10.3791/59370
- Ventura A, Vassall A, Robinson E, Filler R, Hanlon D, Meeth K, Ezaldein H, Girardi M, Sobolev O, Bosenberg MW, et al. Extracorporeal photochemotherapy drives monocyte-to-dendritic cell maturation to induce anticancer immunity. Cancer Res. 2018;78(14):4045–4058. doi:10.1158/0008-5472.CAN-18-0171.
- Tatsuno K, Yamazaki T, Hanlon D, Han P, Robinson E, Sobolev O, Yurter A, Rivera-Molina F, Arshad N, Edelson RL. Extracorporeal photochemotherapy induces bona fide immunogenic cell death. Cell Death Dis. 2019;10(8):578. doi:10.1038/s41419-019-1819-3.
- Kim J, Coffey DM, Ma L, Matzuk MM. The ovary is an alternative site of origin for high-grade serous ovarian cancer in mice. Endocrinology. 2015;156(6):1975–1981. doi:10.1210/en.2014-1977.
- Craveiro V, Yang-Hartwich Y, Holmberg JC, Sumi NJ, Pizzonia J, Griffin B, Gill SK, Silasi D-A, Azodi M, Rutherford T. Phenotypic modifications in ovarian cancer stem cells following Paclitaxel treatment. Cancer Med. (2013);2(6):751–762. doi:10.1002/cam4.115.
- Meeth K, Wang JX, Micevic G, Damsky W, Bosenberg MW. The YUMM lines: a series of congenic mouse melanoma cell lines with defined genetic alterations. Pigment Cell Melanoma Res. (2016);29(5):590–597. doi:10.1111/pcmr.12498.
- Alvero AB, Heaton A, Lima E, Pitruzzello M, Sumi N, Yang-Hartwich Y, Cardenas C, Steinmacher S, Silasi D-A, Brown D. TRX-E-002-1 induces c-Jun–Dependent apoptosis in ovarian cancer stem cells and prevents recurrence in vivo. Mol Cancer Ther. 2016;15(6):1279–1290. doi:10.1158/1535-7163.MCT-16-0005.
- Alvero AB, Kim D, Lima E, Sumi NJ, Lee JS, Cardenas C, Pitruzzello M, Silasi D-A, Buza N, Fahmy T. Novel approach for the detection of intraperitoneal micrometastasis using an ovarian cancer mouse model. Sci Rep. 2017;7(1):40989. doi:10.1038/srep40989.
- Milne K, Köbel M, Kalloger SE, Barnes RO, Gao D, Gilks CB, Watson PH, Nelson BH. Systematic analysis of immune infiltrates in high-grade serous ovarian cancer reveals CD20, FoxP3 and TIA-1 as positive prognostic factors. PLoS One. 2009;4(7):e6412. doi:10.1371/journal.pone.0006412.
- Zhang L, Conejo-Garcia JR, Katsaros D, Gimotty PA, Massobrio M, Regnani G, Makrigiannakis A, Gray H, Schlienger K, Liebman MN. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med. 2003;348(3):203–213. doi:10.1056/NEJMoa020177.
- Radestad E, Klynning C, Stikvoort A, Mogensen O, Nava S, Magalhaes I, Uhlin M. Immune profiling and identification of prognostic immune-related risk factors in human ovarian cancer. Oncoimmunology. 2019;8(2):e1535730. doi:10.1080/2162402X.2018.1535730.
- Ouzounova M, Lee E, Piranlioglu R, El Andaloussi A, Kolhe R, Demirci MF, Marasco D, Asm I, Chadli A, Hassan KA. Monocytic and granulocytic myeloid derived suppressor cells differentially regulate spatiotemporal tumour plasticity during metastatic cascade. Nat Commun. 2017;8(1):14979. doi:10.1038/ncomms14979.
- Shiue LH, Couturier J, Lewis DE, Wei C, Ni X, Duvic M. The effect of extracorporeal photopheresis alone or in combination therapy on circulating CD4+ Foxp3 + CD25 T cells in patients with leukemic cutaneous T-cell lymphoma. Photodermatol Photoimmunol Photomed. (2015);31(4):184–194. doi:10.1111/phpp.12175.
- Goodman AM, Kato S, Bazhenova L, Patel SP, Frampton GM, Miller V, Stephens PJ, Daniels GA, Kurzrock R. Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol Cancer Ther. 2017;16(11):2598–2608. doi:10.1158/1535-7163.MCT-17-0386.
- Spranger S, Luke JJ, Bao R, Zha Y, Hernandez KM, Li Y, Gajewski AP, Andrade J, Gajewski TF. Density of immunogenic antigens does not explain the presence or absence of the T-cell-inflamed tumor microenvironment in melanoma. Proc Natl Acad Sci U S A. 2016;113(48):E7759–e7768. doi:10.1073/pnas.1609376113.
- Clarke B, Tinker AV, Lee C-H, Subramanian S, van de Rijn M, Turbin D, Kalloger S, Han G, Ceballos K, Cadungog MG. Intraepithelial T cells and prognosis in ovarian carcinoma: novel associations with stage, tumor type, and BRCA1 loss. Mod Pathol. 2009;22(3):393–402. doi:10.1038/modpathol.2008.191.
- Hwang WT, Adams SF, Tahirovic E, Hagemann IS, Coukos G. Prognostic significance of tumor-infiltrating T cells in ovarian cancer: a meta-analysis. Gynecol Oncol. 2012;124(2):192–198. doi:10.1016/j.ygyno.2011.09.039.
- Li J, Wang J, Chen R, Bai Y, Lu X. The prognostic value of tumor-infiltrating T lymphocytes in ovarian cancer. Oncotarget. 2017;8(9):15621–15631. doi:10.18632/oncotarget.14919.
- Fan CA, Reader J, Roque DM. Review of immune therapies targeting ovarian cancer. Curr Treat Options Oncol. 2018;19(12):74. doi:10.1007/s11864-018-0584-3.
- Marth C, Wieser V, Tsibulak I, Zeimet AG. Immunotherapy in ovarian cancer: fake news or the real deal? Int J Gynecol Cancer. 2019;29(1):201–211. doi:10.1136/ijgc-2018-000011.
- Odunsi K. Immunotherapy in ovarian cancer. Ann Oncol. 2017;28:viii1–viii7. doi:10.1093/annonc/mdx444.
- Griffiths EA, Srivastava P, Matsuzaki J, Brumberger Z, Wang ES, Kocent J, Miller A, Roloff GW, Wong HY, Paluch BE. NY-ESO-1 vaccination in combination with decitabine induces antigen-specific T-lymphocyte responses in patients with myelodysplastic syndrome. Clin Cancer Res. (2018);24(5):1019–1029. doi:10.1158/1078-0432.CCR-17-1792.
- Moufarrij S, Dandapani M, Arthofer E, Gomez S, Srivastava A, Lopez-Acevedo M, Villagra A, Chiappinelli KB. Epigenetic therapy for ovarian cancer: promise and progress. Clin Epigenetics. 2019;11(1):7. doi:10.1186/s13148-018-0602-0.
- Odunsi K, Matsuzaki J, James SR, Mhawech-Fauceglia P, Tsuji T,Miller A, Zhang W, Akers SN, Griffiths EA, Miliotto A, et al. Epigenetic potentiation of NY-ESO-1 vaccine therapy in human ovarian cancer. Cancer Immunol Res. 2014;2:37–49. doi:10.1158/2326-6066.CIR-13-0126.
- Thomas R, Al-Khadairi G, Roelands J, Hendrickx W, Dermime S, Bedognetti D, Decock J. NY-ESO-1 based immunotherapy of cancer: current perspectives. Front Immunol. 2018;9:947. doi:10.3389/fimmu.2018.00947.
- Umansky V, Blattner C, Fleming V, Hu X, Gebhardt C, Altevogt P, Utikal J. Myeloid-derived suppressor cells and tumor escape from immune surveillance. Semin Immunopathol. 2017;39(3):295–305. doi:10.1007/s00281-016-0597-6.
- Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12(4):253–268. doi:10.1038/nri3175.
- De Sanctis F, Solito S, Ugel S, Molon B, Bronte V, Marigo I. “MDSCs in cancer: conceiving new prognostic and therapeutic targets.” Biochim Biophys Acta. (2016);1865(1):35–48. doi:10.1016/j.bbcan.2015.08.001.
- Parker KH, Beury DW, Ostrand-Rosenberg S. Myeloid-derived suppressor cells: critical cells driving immune suppression in the tumor microenvironment. Adv Cancer Res. 2015;128:95–139.
- Okla K, Czerwonka A, Wawruszak A, Bobiński M, Bilska M, Tarkowski R, Bednarek W, Wertel I, Kotarski J. Clinical relevance and immunosuppressive pattern of circulating and infiltrating subsets of myeloid-derived suppressor cells (MDSCs) in epithelial ovarian cancer. Front Immunol. 2019;10:691. doi:10.3389/fimmu.2019.00691.
- Santegoets S, de Groot AF, Dijkgraaf EM, Simões AMC, van der Noord VE, van Ham JJ, Welters MJP, Kroep JR, van der Burg SH. The blood mMDSC to DC ratio is a sensitive and easy to assess independent predictive factor for epithelial ovarian cancer survival. Oncoimmunology. 2018;7:e1465166. doi:10.1080/2162402X.2018.1465166.
- Pastaki Khoshbin A, Eskian M, Keshavarz-Fathi M, Rezaei N. “Roles of myeloid-derived suppressor cells in cancer metastasis: immunosuppression and beyond. Arch Immunol Ther Exp (Warsz). 2019;67:89–102. doi:10.1007/s00005-018-0531-9.
- Sijmons EA, Heintz AP. Second-look and second surgery: second chance or second best? Semin Surg Oncol. 2000;19(1):54–61. doi:10.1002/1098-2388(200007/08)19:1<54::AID-SSU9>3.0.CO;2-9.
- Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature. 2017;541(7637):321–330. doi:10.1038/nature21349.
- Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348(6230):69–74. doi:10.1126/science.aaa4971.