
ABSTRACT
Triple-negative breast cancer (TNBC) is a highly metastatic and aggressive disease with limited treatment options. Recently, the combination of the immune checkpoint inhibitor (ICI) atezolizumab (anti-PD-L1) with nab-paclitaxel was approved following a clinical trial that showed response rates in at least 43% of patients. While this approval marks a major advance in the treatment of TNBC it may be possible to improve the efficacy of ICI therapies through further modulation of the suppressive tumor immune microenvironment (TIME). Several factors may limit immune response in TNBC including aberrant growth factor signaling, such as VEGFR2 and cMet signaling, inefficient vascularization, poor delivery of drugs and immune cells, and the skewing of immune cell populations toward immunosuppressive phenotypes. Here we investigate the immune-modulating properties of AXT201, a novel 20 amino-acid integrin-binding peptide in two syngeneic mouse TNBC models: 4T1-BALB/c and NT4-FVB. AXT201 treatment improved survival in the NT4 model by 20% and inhibited the growth of 4T1 tumors by 47% over 22 days post-inoculation. Subsequent immunohistochemical analyses of 4T1 tumors also showed a 53% reduction in vascular density and a 184% increase in pericyte coverage following peptide treatment. Flow cytometry analyses demonstrated evidence of a more favorable anti-tumor immune microenvironment following treatment with AXT201, including significant decreases in the populations of T regulatory cells, monocytic myeloid-derived suppressor cells, and PD-L1 expressing cells and increased expression of T cell functional markers. Together, these findings demonstrate immune-activating properties of AXT201 that could be developed in combination with other immunomodulatory agents in the treatment of TNBC.
Introduction
Breast cancer has the second highest incidence rate of all cancers and is the second leading cause of cancer death for women in the United States.Citation1 A subtype of breast cancer, known as triple-negative breast cancer (TNBC) for its lack of the estrogen, HER2, and progesterone receptors commonly used for targeted therapy, is often more aggressive and has a poorer prognosis for patient survival than other subtypes.Citation2,Citation3 However, the recent surge of cancer immunotherapy research, notably immune checkpoint inhibitors (ICIs), may provide new therapeutic opportunities. ICIs block immune suppressive receptors (e.g. programmed cell death protein 1 (PD-1), programmed death-ligand 1 (PD-L1), and cytotoxic T-lymphocyte associated protein 4 (CTLA4)) on T cells, cancer cells, and other cell types (e.g. macrophages, dendritic cells, or endothelial cells) to enhance anti-tumor immune responses.Citation4–Citation6 Importantly, TNBC is the most responsive form of breast cancer to immunotherapy and the FDA approval of the ICI atezolizumab (Tecentriq™), which binds PD-L1 to inhibit its interaction with PD-1, for its treatment marks a major advance.Citation7–Citation9 Nonetheless, ICI-based treatments fail to benefit most TNBC patients, and provide small progression-free survival advantage, suggesting that a better understanding of how to predict and improve responses to these therapies is an urgent need.
Variability in patient responses to immunotherapy is often attributed to the interplay between tumor and immune cells known as the tumor immune microenvironment (TIME). In comparison to other breast cancers, the TIME in TNBC is associated with greater numbers of tumor-infiltrating lymphocytes (TILs) and an abundance of tumor-associated macrophages (TAMs).Citation10,Citation11 High numbers of CD8+ T cells, a high ratio of CD8+ effector T cells to the immunosuppressive FoxP3+ regulatory T cells (Tregs), and low levels of TAMs are all generally associated with positive prognostic values.Citation12–Citation14 Myeloid-derived suppressor cells (MDSCs), another immunosuppressive cell type, increase in number with breast cancer stage and, along with TAMs, facilitate tumor progression and decrease anti-tumor immune responses through the release of immunosuppressive cytokines and factors, stimulation of angiogenesis, and promotion of metastasis.Citation15–Citation19 Therefore, therapies that aim to reprogram the TIME to be more permissive to immune responses are of great interest for immunotherapy research.
In addition to the more canonical immune cells, the tumor vasculature plays a critical and dynamic role in the tumor immune responses. TNBC has among the greatest microvascular densities (MVD) of all breast cancer subtypes, in part due to the high expression of angiogenic factors from immune and stromal cells.Citation16,Citation17 Notably, vascular endothelial growth factor (VEGF) expression levels are significantly higher in TNBC relative to other breast cancers and are associated with poorer prognosis.Citation20–Citation22 Like in most solid tumors, the vasculature in TNBC is often tortuous, hyperpermeable, and with incomplete perivascular coverage, leading to reduced delivery of therapeutic agents and cells, including ICIs and T cells, into poorly perfused regions of the tumor.Citation23–Citation27 Additionally, VEGF inhibits dendritic cell maturation, antigen presentation, and T cell infiltration into the tumor while stimulating pro-apoptotic signaling of T cells by the endothelial cell expression of FasL.Citation28,Citation29 Moreover, T cell infiltration and normalization of tumor vasculature have demonstrated reciprocating effects in mouse models of breast cancer.Citation30 Therefore, investigations that block or modify the activities of growth factors, like VEGF, highlight the importance of these targets through their induction of tumor vascular normalization with enhanced blood perfusion and deeper penetration of chemotherapeutics, reduced tumor growth and metastasis, and altered immune cell infiltration.Citation25,Citation27,Citation31–Citation33
Our work focuses on AXT201, a novel 20-mer anti-angiogenic peptide that belongs to a class of peptides for which we have demonstrated potent influences on tumor vasculature through the inhibition of VEGF signaling.Citation34–Citation37 Moreover, peptides from this class inhibit several other tumor-promoting signaling pathways including HGF, IGF1R and PDGF and have been previously demonstrated to inhibit the growth and metastasis of mouse tumor xenografts, including TNBC models, and an autochthonous model of hepatocellular carcinoma by blocking the growth of blood and lymphatic vessels.Citation36,Citation38–Citation42 We have confirmed these properties for AXT201 in the present study. Given that many of these growth factor receptors also influence immune cell signaling, we hypothesize that AXT201 will modulate the TIME.
Here we investigate the effects of AXT201 treatment on tumor growth and the TIME in two immunocompetent models of TNBC. We find that peptide treatment decreases tumor growth, significantly increases overall survival, and normalizes the vasculature in these models. Subsequently, we demonstrate that AXT201 treatment can alter various aspects of the immune microenvironment, including increasing the activation of CD8+ T cells and their secretion of IFNγ and decreasing PD-L1 expression and the numbers of Tregs and MDSCs.
Materials and methods
Peptide handling
AXT201 (LRRFSTAPFAFININNVINF) was manufactured by solid-phase synthesis at New England Peptide. The lyophilized peptide was dissolved in sterile water to a concentration of 4 mg/ml (in vivo) or 2 mM (cell culture). Reconstituted peptide was stored at 4°C and used within 3 to 4 days.
Endothelial cell experiments
Human umbilical vein endothelial cells (HUVECs) were purchased from Lonza (CC-2519) and maintained in VascuLifeTM VEGF Endothelial Medium Complete kit (Lifeline Cell Technologies; LL-0003) at 37°C in 5% CO2 and used between passages 2 to 7. For all cell culture experiments, 6-well culture dishes were coated with 10 μg/ml fibronectin 1 (FN1) in PBS for 2 h at 37°C. The FN1 solution was then removed and the cells cultured normally. Cells were serum starved for 4 h before the addition of growth factors. For serum starvation, cells were washed twice with DPBS containing calcium and magnesium, once with VascuLifeTM Basal Media (Lifeline Cell Technologies; LL-0005), and maintained in 1.5 ml of endothelial base media. Peptide or water vehicle was added at the indicated concentrations for 1.5 h before growth factor treatment. Growth factors were reconstituted according to manufacturer’s instructions. Where applicable, cells were treated with 20 ng/ml VEGFA (R&D Systems; 293-VE) for 10 min, 50 ng/ml HGF (Gibco; PHG0324) 15 min, or 50 ng/ml IGF1 (Gibco; PHG0078) for 15 min. After treatment, cells were washed twice in cold dPBS with calcium and magnesium and lysed in 120 μl of 1x Blue Loading Buffer (Cell Signaling Technologies; 7722S), sonicated, boiled, and resolved by SDS-PAGE using 4–12% Bis-Tris NuPAGE (Life Technologies; NP0335BOX) gels in MOPS buffer (Life Technologies; NP0001). Samples were then transferred to nitrocellulose membranes and analyzed by Western blot using the following primary antibodies: phospho -VEGFR2 (Y1175) (Cell Signaling; 2478), pMet (Y1234/Y1235) (Cell Signaling; 3077), phospho-IG1FRβ (Y1135/1136)/insulin receptor β (Y1150/1151) (Cell Signaling; 3024), GAPDH (Cell Signaling; 2118). Bands were imaged using the HRP-conjugated anti-rabbit or anti-mouse secondary antibodies (Kindle Biosciences) and the KwikQuant imaging system (Kindle Biosciences) and quantified by densitometry using ImageJ.
Animal care and use
All protocols have been approved by the Johns Hopkins University Institutional Animal Care and Use Committee. FVB and BALB/c mice, 4 to 5 weeks old, were purchased from Jackson Laboratories or Charles River Laboratories respectively. Mice were allowed to acclimate at least one week before each experiment.
NT4 survival studies
The NT4 cell line was derived from a Neu-N mouse model that was negative for Her2 and confirmed to be negative for mycoplasma contamination. NT4 cells were cultured in RPMI media supplemented with 10% FBS, penicillin/streptomycin, and L-glutamine at 37°C and 5% CO2. For survival studies, female FVB mice were inoculated orthotopically with 5*103 NT4 cells in the first mammary fat pad. Animals were treated daily with 20 mg/kg AXT201 or equivalent volume of the water vehicle by intraperitoneal (IP) injection starting on day 3 post inoculation. Tumor volume measurements were taken using calipers once tumors were palpable and continued twice a week until the end of the study. Tumor volumes were calculated according to the following equation: volume = 0.52*length*width.2 Survival was considered the time until mice were required to be euthanized according to IACUC standards (ie. tumors exceeded 2 cm in any direction, moribund appearance, or according to veterinary request). The study was continued for an additional two measurements after the last control was euthanized, at which point all mice were euthanized.
4T1 tumor models
The 4T1 cell line was purchased from ATCC (Manassas, VA) and confirmed to be negative for mycoplasma contamination. The cells were cultured as described for the NT4 cell line above. For tumor experiments, 2.5*104 cells were inoculated into the first mammary fat pad of BALB/c mice. Peptide or water vehicle were administered daily by IP injection starting on day 3 post inoculation. Tumor volume measurements were made twice a week once tumors were palpable as described for the NT4 model above and continued until mice were euthanized. For tumor growth measurements the studies proceeded until mice were required to be euthanized according to IACUC protocol, while tumor dissociation studies were terminated after 2 weeks to avoid tumor necrosis. Mice were euthanized by CO2 exposure and the tumors excised carefully to avoid the accidental capture of axillary lymph nodes as well. The tumors were then saved for IHC or dissociated for flow cytometry or T cell purification as described below.
Immunohistochemistry and immunofluorescence of 4T1 tumors
For IHC, excised tumors were rinsed in PBS, halved, and fixed for 48 h in at least 15 times volume of 10% neutral buffered formalin with gentle shaking at room temperature. The formalin was then exchanged for 70% ethanol twice, each exchange lasting 24 h at 4°C. The tumors were paraffin embedded and sectioned into 4 μm thick slices. For IHC staining, the sections were deparaffinized by two 10 min washes in 100% xylene and rehydrated in a series of 2 min washes in 100%, 95%, 70% and 50% ethanol, water, 0.165% NaCl in water, and PBS. Antigen retrieval was then performed in Dako Target Retrieval Solution (Dako; S1699) using a vegetable steamer for 25 min followed by a 45 min cooldown. The sections were washed, blocked in 5% goat serum, 0.2% gelatin, 1% BSA, and 0.2% Triton X-100 in PBS for 1 h, and then treated for 10 min with BloxAll (Vector Labs; SP-6000) to inhibit endogenous peroxidases. Samples were then incubated with primary antibody diluted in blocking buffer overnight at 4°C, followed by HRP-conjugated secondary antibodies for 1 h. Antibodies were detected using the SignalStain® DAB Substrate Kit (Cell Signaling Technologies; 8059) and counterstained with hemoxylin for 30 sec. The slides were then dehydrated in two washes each of 95% ethanol, 100% ethanol, and 100% xylene, air-dried, and mounted with Organo/Limonene MountTM (Sigma; O-8015). Images were taken using the Olympus BX51TF with DP70 color camera. The staining density was quantified with ImageJ (NIH) as the percent area of DAB staining relative to the total area analyzed. The analysis regions included at least three separate tumors. Tumor edges as well as necrotic or damaged regions of the tissue were not included in these analyses. Primary antibodies include CD31 (77699; Cell Signaling).
For immunofluorescence, tumors were halved, embedded in OCT compound, and frozen rapidly on dry ice. The tissue was then sliced into 10 μm sections and attached to slides. Sections were then thawed at room temperature and fixed for 10 min in 100% ice-cold acetone. The samples were air dried, blocked with 5% goat serum, 0.2% gelatin, 1% BSA, and 0.2% Triton X-100 in PBS for 1 h at room temperature, and incubated overnight at 4°C in primary antibody diluted in blocking buffer. The samples were then incubated with fluorophore-conjugated secondary antibodies diluted in blocking buffer for 1 h at room temperature followed by DAPI staining for 10 min. Images were acquired using a Zeiss AxioObserver with LSM700 confocal module. Pericyte coverage was quantified using ImageJ as the area of NG2 staining within 3 μm of CD31 staining as a percentage of CD31 staining. Primary antibodies include: CD31 (BD Pharmingen; 553370) and NG2-chondroitin (Millipore; AB5320).
Tumor dissociation and flow cytometry
Excised tumors were minced into approximately 1 mm3 pieces and further digested using the mouse tumor dissociation kit (Miltenyi Biotech; 130-096-730) and GentleMACs octodissociator (Miltenyi Biotech) according to the manufacturers protocol using the adaption to the R enzyme recommended for tumor-infiltrating immune cells. The dissociation enzymes were then immediately quenched in RPMI media containing 10% FBS, washed in PBS, and the red blood cells lysed using RBC lysis buffer (Biolegend; 420301).
For flow analysis of total cell populations, aliquots of 1*106 cells were then transferred to flow tubes, washed with PBS, and stained with UV Live/Dead stain (Life Technologies) according to manufacturer’s instructions. The tubes were washed twice in stain buffer (4% FBS and 0.1% sodium azide in PBS) and then stained with a cocktail of fluorophore-conjugated antibodies against external targets and FcR block (Miltenyi Biotech; 130-092-575) for 45 min. The samples were next washed three times with stain buffer and fixed using the FoxP3 fixation/permeabilization kit (eBiosciences; 00-5523-00) according to the manufacturer’s instructions. The samples were washed twice in PBS and stored overnight at 4°C in the dark. The following morning, the samples were permeabilized and stained for internal targets according to the FoxP3 fixation/permeabilization kit and analyzed by flow cytometry using the LSRII systems (BD Biosciences). All antibodies used for flow cytometry are listed in .
Table 1. Flow cytometry antibodies.
For the analysis of CD8+ T cell secretions, CD8+ T cell were negatively selected from tumor samples following RBC lysis using the EasySep mouse CD8 isolation kit (StemCell; 19853). Samples of 1*106 cells were then transferred to wells of a 24-well plate and activated overnight at 37°C with anti-CD3/CD28 Dynabeads (Life Technologies; 11452D) in RPMI media supplemented with 10% FBS, penicillin/streptomycin, L-glutamine, and 50 μM β-mercaptoethanol. The following morning, the cells were treated with GolgiStop containing monensin (BD Biosciences; 554724) for 4 h. The beads were then removed from the cells using a magnet and the cells stained for Live/Dead, internal, and external markers as described for flow cytometry of whole tumor samples above.
Data analysis and statistics
Unless otherwise stated, error bars are presented as SEM. Tumor growth curves were analyzed by two-way ANOVA matched for days since inoculation followed by Bonferroni posttests to compare individual treatments. Vessel density and pericyte coverage were analyzed using an unpaired Student’s t-test (two-tailed). For flow cytometry data, sample distributions were assessed for normality using the D’Agostino and Pearson omnibus normality test. Samples that were identified as normally distributed were analyzed using an unpaired Student’s t-test (two-tailed). Samples that did not pass the normality test or had too-few points for assessment were analyzed using the non-parametric Mann-Whitney test. All statistical analyses were performed using GraphPad Prism® software v. 5.0. A p-value less than 0.05 was considered significant. All image analysis was performed with ImageJ.
Results
AXT201 inhibits VEGFR2, IGF1R, and cMet signaling
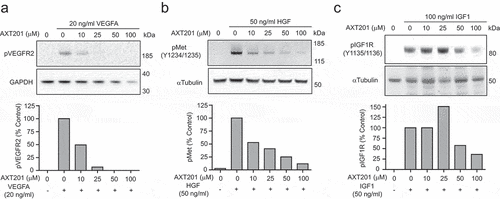
Similar to other peptides of the same class, AXT201 has been previously shown to potently inhibit angiogenesis.Citation35 However, the specific effects of AXT201 on growth factor signaling has not been investigated. Therefore, we investigated the effects of AXT201 on VEGFR2, cMet, and IGF1R phosphorylation in HUVECs. AXT201 treatment resulted in a dose-dependent inhibition of VEGFR2, cMet, and IGF1R, .
Figure 1. AXT201 inhibits VEGFR2, cMet, and IGF1 R signaling. (a-c) Top – Western blot images showing changes in the phosphorylation of (a) VEGFR2, (b) cMet, and (c) IGF1 R in HUVECs exposed to VEGFA, HGF, or IGF1 respectively and varying concentrations of AXT201. GAPDH and αTubulin are provided as loading controls. Bottom – quantification of band intensities normalized for loading and presented as percentages of growth factor only controls.
AXT201 inhibits tumor growth in two TNBC mouse models
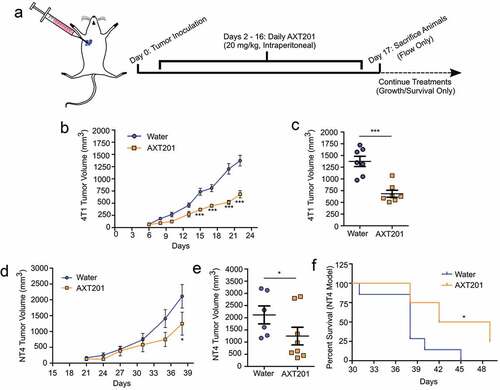
To investigate the effect of AXT201 on the TIME in TNBC, we utilized two mouse-derived tumor cells lines injected orthotopically into the right mammary fat pad of syngeneic mice. The 4T1 model is an aggressive and highly metastatic line derived from BALB/c mice.Citation43 The NT4 model is a basal-like TNBC cell line derived from a Neu-N model lacking HER2 expression and injected into FVB/N mice instead of Neu-N mice to avoid contamination with spontaneous tumors.Citation16,Citation44 To investigate the effect of AXT201 on 4T1 tumor growth, BALB/c mice were injected with 2.5*104 cells and treated with peptide daily by intraperitoneal injection starting on day 2 after inoculation (). As shown in , treatment with AXT201 significantly reduced tumor growth by day 15 (p < .001) with the greatest difference on day 22 (), in which peptide-treated animals exhibited a 47% reduction in tumor volume compared to vehicle controls. The effects of the peptide on survival were then investigated in FVB/N mice. Mice were injected with 5*103 NT4 cells and treated daily with peptide by intraperitoneal injection starting on day 3 after inoculation. As shown in ), mice treated with AXT201 showed a decreasing trend in tumor volume with a significant 41% reduction (p < .05) on day 38, the last day all mice were alive (). The AXT201-treated mice also survived significantly longer (), with a median survival rate of 45.5 days compared to 38 days for control animals (p = .0135) and 25% of AXT201-treated animals remaining after the death of the last control animal. Based on the greater response to treatment observed in our studies and the more extensive use in the literature, we decided to further investigate the effects of AXT201 on vessel normalization and immune cell infiltration in the 4T1 model.
Figure 2. AXT201 inhibits the growth of NT4 and 4T1 tumors in mice. (a) Schematic showing dosing schedule. The blue arrow indicates the start of daily peptide injections from day 2 on. The dashed lines indicate deviations in survival and growth experiments from flow experiments, which were terminated on day 17. (b, c) Tumor growth of water- (blue) and AXT201-treated (orange) 4T1 tumors showing both averages over time (b) and individual tumor measurements from Day 22 (c). *** indicates p < .001, two-way ANOVA with Bonferroni posttest. (d-f) Tumor growth of water- (blue) and AXT201-treated (orange) NT4 tumors showing both averages over time (d) and individual tumor measurements from Day 38 (e). * indicates p < .05, two-way ANOVA with Bonferroni posttest. (f) Kaplan-Meier curve showing differences in survival for water- and AXT201-treated mice. * indicates p < .05, Log-Rank Test.
AXT201 inhibits tumor angiogenesis and normalizes tumor vasculature in 4T1 tumors
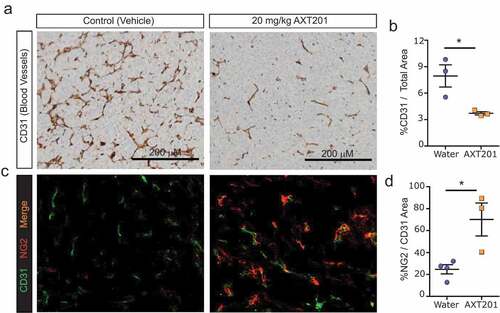
Previously, AXT201 or peptides from the same class were found to stabilize the vasculature and inhibit tumor vessel density.Citation36,Citation37,Citation40–Citation42 Based on these observations, we hypothesized that AXT201 treatment would reduce the vessel density of 4T1 tumors while increasing the perivascular coverage of the remaining vasculature. To investigate vessel density, tumors were isolated from mice after two weeks and stained by IHC for blood vessels using the CD31 marker (). Mice treated with AXT201 exhibited a significant 53% reduction in stained area compared to controls (). To investigate the effects of the peptide, frozen tumor sections were co-stained by immunofluorescence for CD31 and the pericyte marker NG2-chondroitin (). Consistent with vessel normalization, peptide treatment increased the pericyte coverage on blood vessels significantly by 184% ().
Figure 3. AXT201 alters tumor vasculature. (a) Immunohistochemical staining of water- and AXT201-treated tumors for CD31. (b) Quantification of CD31 staining area in water- and AXT201-treated tumors compared to the total area of the tumor slice. N = 3, *p < .05 Student’s t-test. (c) Immunofluorescence staining of water- and AXT201-treated tumors for CD31 (green) and NG2 (red). (d) Percentage of NG2 staining area overlapping with the CD31 staining area in water- and AXT201-treated tumors. N ≥ 3, *p < .05 Student’s t-test.
AXT201 treatment alters T lymphocyte cell populations and secretions within 4T1 tumors
The status of the tumor vasculature can profoundly impact the TIME by influencing the delivery of cells and treatment into the tumor tissue and altering the signaling pathways within the environment.Citation25,Citation31–Citation33 As such, the vascular normalization and growth factor inhibition effects of AXT201 suggested that the peptide be able to modulate the tumor immune response through regulation of the vasculature and signaling pathways.Citation29,Citation45–Citation48 To further investigate the effects of AXT201 treatment on the TIME, we isolated 4T1 tumors after two weeks of treatment with AXT201 and used flow cytometry to assess changes in various lymphoid cell populations within the tumors induced by peptide treatment (flow cytometry gating strategies can be found in Figures S1). A summary of the findings can be found in .
Table 2. Effects of AXT201 on TIME cell populations.
Exposure to AXT201 had no significant effects on the total number of CD4+ or CD8+ T lymphocytes, which averaged around 2% and 1% respectively relative to all living cells within the tumor regardless of treatment (). However, the number of Tregs decreased significantly (p = .0378) by 50% in the AXT201-treated mice ().
Figure 4. Effect of AXT201 treatment on lymphocyte populations in 4T1 tumors. (a-c) Percentage of CD4+ T cells (a), CD8+ T cells (b), and Tregs (c) out of all live cells from isolated whole 4T1 tumor samples. N ≥ 10, *p < .05, Mann-Whitney test. (d-k) Percentage of subpopulations out of total (d-j) or out of effector (k) CD8+ T-cells from whole CD8+ T-cells isolated from 4T1 tumors by negative selection and stimulated overnight. (d-f) Percentage of CD8+ T cells expressing the secreted factors IFNγ (d), Granzyme B (e), or TNFα (f). (g) Owing to high variability in baseline levels between experimental sets, the percentage of CD8+ T cells expressing IL-2 from each tumor in an experimental set was normalized to average for all controls of that same set. Pre-normalized data can be found in Figure S4A. N ≥ 9, *p < .05, Student’s t-test. (h-k) Percentage of CD8+ T cells (h-l) or CD8+ effector T cells (m) expressing surface markers for (h) effector cells, (i) naïve T cells, (j) memory T cells, (k) the exhausted phenotype, (l) CTLA4 expression, or (m) PD-1 expression without Tim3 (activated). N ≥ 4, *p < .05, Mann-Whitney test.
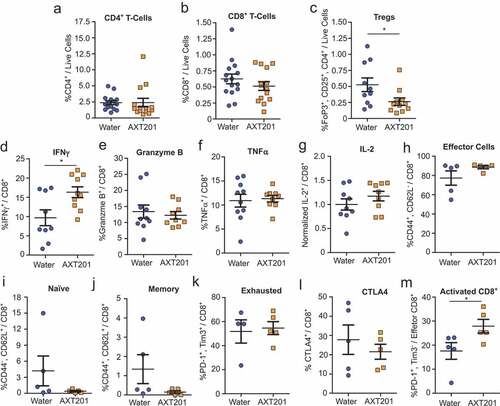
Given the importance of CD8+ T cells in immune-based tumor therapies we further investigated how AXT201 influences the expression of various surface proteins and secreted factors in isolated CD8+ T cells that were stimulated overnight (see Supplementary S2 for flow cytometry gating strategies). IFNγ levels were significantly (p = .0149) increased by 68% in AXT201-treated samples relative to control tumors ()). No significant changes were observed for the other secreted factors, even after normalization of IL-2 to account for large baseline differences between experimental sets (, pre-normalized IL-2 data can be found in Figure S4A). Subsequently we investigated changes in functional surface markers but observed no significant differences in the numbers of CD44+/CD62L− effector T cells (), CD44−/CD62L+ naïve T cells (), CD44+/CD62L+ memory T cells ()), exhausted T cells (defined as PD-1+/Tim3+ effector T cells) (), or the expression of CTLA4 ()). However, a significant 59% increase in PD-1+/Tim3− effector T cells, which corresponds to an activated, non-exhausted subpopulation, was observed in AXT201-treated animals (p = .0317; )).
AXT201 treatment alters myeloid cell populations within 4T1 tumors
While T cells remain the primary focus of most immunotherapies, other cell types strongly influence treatment responses. Notably, cells of myeloid lineages are recruited to the tumor in response to hypoxia and angiogenic factorsCitation31,Citation49 and can alter the functions and populations of T cells within the tumor through numerous mechanisms described in detail elsewhere.Citation16,Citation50,Citation51 As such, agents that reduce hypoxia and angiogenic signaling would be expected to alter myeloid cell recruitment and function. We therefore investigated the effects of AXT201 on the populations of various myeloid cell populations within the TIME using the same whole-tumor dissociation protocol as described for the lymphoid populations above (see Figure S3 for flow cytometry gating strategies). Total myeloid cell populations, as defined by CD11b expression, were significantly decreased by 13% in mice treated with AXT201 (p = .0301; )). We subsequently investigated changes to various subsets of myeloid cells. Macrophages accounted for 37% and 35% of CD11b+ cells () and 12% and 10% of live cells for control and AXT201-treated cells respectively (). The effects of macrophages on the TIME are complex and depend on different factors, including the polarization between the pro-inflammation, classically activated M1-like subtypes and the immunosuppressive, pro-angiogenic, non-classically activated M2-like subtype.Citation52 Therefore, we investigated if AXT201 would influence the expression of Arg1, a marker of immunosuppressive macrophages. However, no significant differences or trends were observed ().
Figure 5. The effects of AXT201 on myeloid cell populations in 4T1 tumors. (a-f) Subpopulations of various CD11b+ (myeloid) cells isolated from whole 4T1 tumors treated with water or AXT201 relative to (a, c, and f) total live cell, (b) total CD11b+ cells, and (d) total macrophages. Targets include (a) total myeloid cells, (b and c) macrophages, (d) Arginase-1 expressing macrophages, (e) normalized M-MDSCs, and (f) PMN-MDSCs. N ≥ 11, *p < .05, Student’s t-test or Mann-Whitney test.
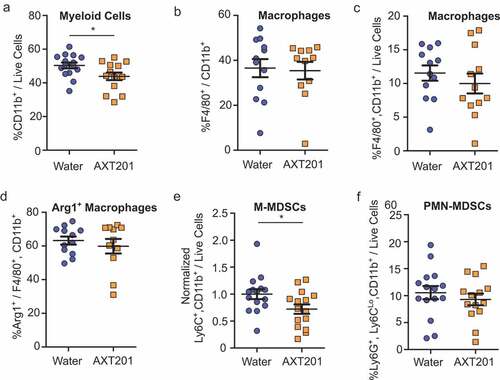
Another myeloid subpopulation, MDSCs, represents a heterogeneous, immature subset of cells that are loosely connected by their strong suppression of immune responses. These cells are further subdivided based on their morphological and phenotypic similarities to monocytic cells, known as monocytic MDSCs (M-MDSCs) and polymorphonuclear or granulocytic MDSCs (PMN-MDSC or G-MDSC). Baseline levels of M-MDSCs varied between experimental sets, but a significant (p = .0421) 28% reduction was observed following AXT201 treatment when the data from each experimental set was normalized to the average for all controls in the corresponding set (, pre-normalized data can be found in Figure S4B). PMN-MDSCs did not change significantly with treatment ().
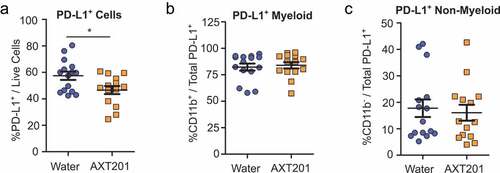
AXT201 treatment reduces PD-L1 expression in 4T1 tumors
Given the importance of the PD-L1/PD-1 pathway in many immunotherapy treatments we decided to investigate the effects of AXT201-treatment on the expression of PD-L1 (See Figure S5 for flow cytometry gating strategies). When compared to all living cells, a significant (p = .0201) 19% reduction of PD-L1+ cells was observed in AXT201-treated mice (). The majority of these, 82% and 84% for water and AXT201-treated cells respectively, were CD11b+ myeloid cells (). However, the ratio of CD11b+ to CD11b− cells among PD-L1+ cells did not change with AXT201 treatment, suggesting that the peptide’s effects on PD-L1 expression was not specific to the myeloid lineage ().
Figure 6. Effect of AXT201 treatment on PD-L1 expression in 4T1 tumors. (a-c) Subpopulations of PD-L1+ cells isolated from whole 4T1 tumors treated with water or AXT201. Subpopulations include (a) total PD-L1+ cells out of all live cells and (b) CD11b+ and (c) CD11b− cells out of total PD-L1+ cells. N ≥ 14, *p < .05, Student’s t-test.
Discussion
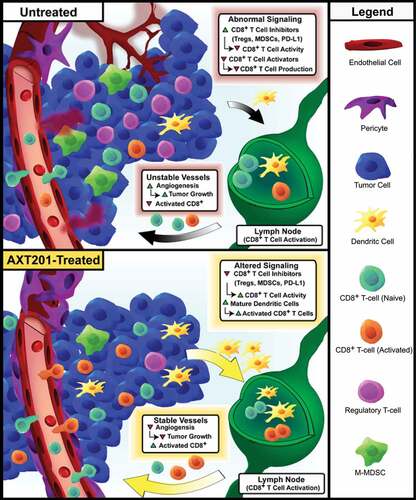
The data presented in this paper represent the first investigation into the immune-modulating functions of either AXT201 or its related class of peptides as well as the first time one of these peptides was used in an immune-competent model of TNBC. Our proposed model for these effects is shown in . Other peptides from the same class have been investigated in other models of TNBC and hepatocellular carcinoma and their common origins would suggest that their properties are likely similar.Citation36,Citation39–Citation42 Notably, all members of this class of peptides appear to inhibit multiple growth factor receptors through interactions with their integrin co-receptors.Citation34,Citation36,Citation39 In support of this hypothesis, we have demonstrated that AXT201 inhibits VEGF, HGF, and IGF1 signaling in vitro and significantly decreases tumor vascular density in vivo.
Figure 7. Model for the effects of AXT201 on the TIME. Top – Model of untreated 4T1 tumors. Unregulated growth factor signaling, including VEGF and HGF, stimulates angiogenesis and disrupts vessel maturation, resulting in aberrant, leaky tumor vessels that support a heterogeneous, pro-tumor microenvironment with hypoxic and acidic regions. VEGF and HGF signaling further contribute to the immunosuppressive environment by recruiting cells that inhibit cytotoxic T cells, such as M-MDSCs and Tregs, and stimulating the expression of checkpoints molecules, like PD-L1, on various cell types. VEGF also inhibits dendritic cell maturation, which can lead to a reduction in antigen presentation and CD8+ T-cell activation. Bottom – Treatment with AXT201 normalizes the tumor vasculature, resulting in stable vessels with substantial pericyte coverage. AXT201 also blocks signaling by several growth factors, such as VEGF and HGF, reducing the presence of immunosuppressive cell types and increasing the activation of tumor targeting cytotoxic T cells.
In our studies, AXT201 significantly inhibited tumor growth in both the 4T1 and NT4 tumor models. However, this significance was obtained earlier in the 4T1 model (day 15 vs day 38), suggesting that the 4T1 model was more susceptible to peptide treatment. Superficially, both cell lines behave similarly in tumor models, each showing rapid growth and necrosis starting within two weeks followed by the eventual formation of lung metastases, indicating that the differences likely lie at the level of signaling. The origins of the two tumors may provide a possible reason for this difference. While both 4T1 and NT4 tumors are considered triple-negative by histological assessment, the 4T1 line was derived from a triple-negative tumor while the NT4 line was derived from Her2 positive Neu/N mice. As such, it may be plausible that, while NT4 does not overexpress Her2 protein, there may some amount of Her2 signaling associated with these cells. Notably, another peptide from the same class as AXT201 was found not to inhibit the signaling of the epidermal growth factor receptor (EGFR),Citation36 suggesting that the EGFR family of receptors, including Her2, could be resistant to AXT201. As such, any expression of Her2 within the NT4 cell line could provide a survival advantage against AXT201 treatment that is not available to 4T1 tumors.
Treatment of tumors with low doses of anti-angiogenic agents has been previously associated with the normalization of tumor vasculature. Normalized vessels are commonly associated with increased tumor lymphocyte infiltration owing to reduced leakage and improved perivascular coverage, improved tissue perfusion and distribution of immune cell adhesion molecules.Citation24,Citation25,Citation27,Citation31 However, despite the increased pericyte coverage suggesting vessel normalization, our data show no differences in the total number of either CD4+ or CD8+ T lymphocytes between control and AXT201-treated samples. Possible explanations for this discrepancy could be related to the timing and dose of the peptide treatment. 4T1 tumors grow very rapidly and are often necrotic after 2 weeks. Therefore, peptide administration was started before palpable tumors were observed to allow for sufficient treatment duration prior to the two-week harvest date. However, this early treatment schedule may have inhibited the formation of vessels over normalizing existing ones, thereby reducing the transport of TILs into regions of the tumor as well as increasing hypoxia-mediated immunosuppressive changes. Additionally, the dose of peptide administered was based on previous studies but may be suboptimal for TIME changes. Nonetheless, hypoxia would be expected to increase the number of tumor-infiltrating M-MDSCs, as has been previously observed in mouse 4T1 and TS/A breast cancer models treated with the anti-VEGFR2 antibody DC101.Citation53 As we observed a significant decrease in M-MDSC populations following peptide treatment, our data suggest that hypoxia alone cannot account for these results.
As an alternative property to consider, the integrin targets of AXT201 are co-receptors that regulate the activities of several different pathways, as evidenced by the simultaneous inhibition of VEGFR2, cMet, and IGF1R, and the full effect of their inhibition by AXT201 may be difficult to predict. While inhibition of these several growth factor receptors is consistent in all members of this class of peptides, the effects on total receptor levels and downstream pathways can change with cell type. In blood endothelial cells, for instance, total receptor levels were reduced in peptide treated cells but not in tumor cells or fibroblasts. Similarly, the downstream phosphorylation of Akt appeared to increase with peptide treatment in blood endothelial cells but decreased in tumor cells, fibroblasts, and lymphatic endothelial cells.Citation36,Citation39,Citation54 In addition to this variability in known pathways, the fact that integrins are common to many receptor complexes suggests that the effects of AXT201 could extend into signaling pathways that have not yet been considered. Therefore, the fact that changes to T-cell infiltration in response to AXT201 treatment were insignificant, could be due to potential and unexpected disruption by AXT201 on the interactions between T-cell integrins and endothelial cell adhesion molecules, like VCAM-1 and ICAM-1, and may offset any benefits provided by the vessel normalization.
In addition to their roles in the formation of new vessels, many angiogenic signaling pathways can directly contribute to the immunosuppressive environment of the tumor. Notably, both HGF and VEGF are reported to influence dendritic cell (DC) activity or maturation, upregulate the expression of PD-L1 and/or PD-1, and stimulate immunosuppressive phenotypes in tumor-associated macrophages.Citation45–Citation48,Citation55,Citation56 Moreover, anti-VEGF treatments have also been observed to reduce the numbers of Tregs and MDSCs in tumor-bearing mice.Citation46,Citation57,Citation58 Consistent with these previous observations, our data show significant decreases in the overall number of PD-L1-expressing cells by 19% and the numbers of Treg and M-MDSCs by 50% and 28% respectively in the tumor following AXT201 treatment. In comparison, these responses were comparable or superior to small molecule, multiple kinase inhibitors like sorafenib, which reduced Tregs populations by 40% in tumors from renal cell carcinoma patientsCitation59 but also increased PD-L1 expression in liver tumors through hypoxia,Citation60 and sunitinib, which reduced MDSCs (defined as CD11b+,GR1+) by 33% in a 4T1 tumor model.Citation61 Conversely, Arg1 expression in TAMs did not change with AXT201 treatment and PD-1 expression was found to increase significantly in effector T cells. However, the role of PD-1 in cancer immunology is complicated and, while recognized as a major suppressor of T cell responses, it is also an early sign of T cell activation and represents the fraction of T cells responsive to neoantigens.Citation62,Citation63 Notably, in our study this increased expression is not associated with a similar upregulation of Tim3, thereby distinguishing this population of T cells from an exhausted phenotype, which did not change.
IFNγ was one of several CD8+-secreted factors that were investigated in this study and the only one to show a significant change in expression from AXT201 treatment. While considered an important immune factor, IFNγ has a complicated association with tumor growth and development. In terms of tumor elimination, IFNγ signaling plays key roles in the inflammatory, Th1-like T cell functions by upregulating processes associated with antigen presentation as well as inducing cytotoxic T cell differentiation and recruitment and the proliferation of precursor cells. Moreover, IFNγ indirectly promotes anti-tumor T cell functions by inhibiting the functions of immunosuppressive cells, including the induction of fragility in Tregs and the skewing of TAMs toward a pro-inflammatory phenotype.Citation64,Citation65 Conversely, several pro-tumorigenic activities have been reported for IFNγ, including the induction of immune checkpoint molecules, such as PD-L1 and CTLA4, and immunosuppressive factors, such as iNOS and IDO.Citation66–Citation69 IFNγ also exhibits conflicting influences on tumor vascular density, with reports showing both pro- and anti-angiogenic processes.Citation70,Citation71 Comparatively, our data demonstrate a reduction of Tregs in AXT201-treated tumors, which would be consistent with the increase in IFNγ levels. Similarly, the reported inhibition of endothelial cell proliferation by IFNγ also fits with our observations showing a significant reduction of tumor vascular density. Nonetheless, AXT201 inhibits several angiogenic signaling pathways, as shown in our data regarding growth factor inhibition, making it difficult to determine the specific contributions of increased IFNγ, if any, to these results. However, it is also possible that blocking these pathways precludes the pro-angiogenic function of IFNγ as well. Furthermore, despite reports of IFNγ-mediated induction of immune checkpoint molecules, treatment with AXT201 significantly reduced PD-L1 expression in the tumor and has no effect on CTLA4. As a possible explanation, one of the known integrin targets for this class of peptides, integrin αvβ3,Citation34 has been reported to function as a co-receptor in IFNγ-mediated stimulation of PD-L1 expression and, thus, some signaling by IFNγ may be directly inhibited by AXT201.Citation72 Nonetheless, the extent to which AXT201 inhibits pro-immune functions of IFNγ remains to be determined.
Our data indicate that AXT201 treatment is able to improve the TIME through the reduction of immunosuppressive cell types, such as Tregs and M-MDSCs, and increasing pro-immune signaling pathways of IFNγ while avoiding its negative effects by inhibiting PD-L1 expression and angiogenesis. Currently only one ICI, atezolizumab, in combination with nab-paclitaxel, has been approved by the FDA to treat TNBC. However, reports for this combination from the Impassion130 phase 3 clinical trial showed no statistical benefit to overall survival in intention-to-treat patients although possible benefits were observed in the PD-L1 positive subgroup (formal statistical analysis was not conducted).Citation8 As such, the timely development of new therapies able to benefit a broader set of TNBC patients is an urgent clinical need. The data in this study suggest that treatment with AXT201 would create a more immune-permissive microenvironment so that combinations with other immunotherapies could potentially induce potent inhibition of tumor growth. Notably, as AXT201 already reduces the numbers of PD-L1 expressing cells on its own, treatments that increase CD8+T cells may provide a greater benefit than targeting the PD-1/PD-L1 pathway. Therefore, immune checkpoint inhibitors that block CTLA4 or activate OX40 could be used to increase the number of active T cells and complement the effects of AXT201.
Disclosure of Potential Conflicts of Interest
NBP is the Vice President of R&D, ASP is Founder and the Chief Scientific Advisor, of AsclepiX Therapeutics, Inc. VS received research grants through Johns Hopkins University from Abbvie, Biocept, Pfizer, Novartis, Medimmune, and Puma Biotechnology, served as a consultant to Iridium Therapeutics, Inc., and is a member of the Data Safety Monitoring Board for Immunomedics. Inc. The terms of these arrangements are being managed by the Johns Hopkins University in accordance with its conflict-of-interest policies.
Supplemental Material
Download ()Acknowledgments
We thank the Johns Hopkins University Ross Flow Cytometry facility for the use of their LSR II with NCI support under P30CA006973 and School of Medicine Microscopy Facility (MicFac) for the use of the Zeiss LSM700 confocal microscope, supported by the Office of the Director of the National Institutes of Health under the award S10OD016374 and Olympus BX51TF microscope. This research was supported by the NIH under grants F32CA210482 (ACM), R01CA138264 (ASP), and P30CA006973 (Sidney Kimmel Comprehensive Cancer Center core grant).
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
References
- Guo F, Kuo Y, Berenson AB. Breast cancer incidence by stage before and after change in screening guidelines. Am J Prev Med. 2019;56(1):100–13. doi:10.1016/j.amepre.2018.08.018.
- Malorni L, Shetty PB, De Angelis C, Hilsenbeck S, Rimawi MF, Elledge R, Osborne CK, De Placido S, Arpino G. Clinical and biologic features of triple-negative breast cancers in a large cohort of patients with long-term follow-up. Breast Cancer Res Treat. 2012;136(3):795–804. doi:10.1007/s10549-012-2315-y.
- Bianchini G, Balko JM, Mayer IA, Sanders ME, Gianni L. Triple-negative breast cancer: challenges and opportunities of a heterogeneous disease. Nat Rev Clin Oncol. 2016;13(11):674–690. doi:10.1038/nrclinonc.2016.66.
- Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018;8(9):1069–1086. doi:10.1158/2159-8290.CD-18-0367.
- Hargadon KM, Johnson CE, Williams CJ. Immune checkpoint blockade therapy for cancer: an overview of FDA-approved immune checkpoint inhibitors. Int Immunopharmacol. 2018;62:29–39. doi:10.1016/j.intimp.2018.06.001.
- Li Z, Qiu Y, Lu W, Jiang Y, Wang J. Immunotherapeutic interventions of Triple Negative Breast Cancer. J Transl Med. 2018;16. doi:10.1186/s12967-018-1514-7.
- Vikas P, Borcherding N, Zhang W. The clinical promise of immunotherapy in triple-negative breast cancer. Cancer Manag Res. 2018;10:6823–6833. doi:10.2147/CMAR.S185176.
- Schmid P, Rugo HS, Adams S, Schneeweiss A, Barrios CH, Iwata H, Diéras V, Henschel V, Molinero L, Chui SY, et al. Atezolizumab plus nab-paclitaxel as first-line treatment for unresectable, locally advanced or metastatic triple-negative breast cancer (IMpassion130): updated efficacy results from a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019 Nov 27. doi:10.1016/S1470-2045(19)30689-8.
- Rodig N, Ryan T, Allen JA, Pang H, Grabie N, Chernova T, Greenfield EA, Liang SC, Sharpe AH, Lichtman AH, et al. Endothelial expression of PD-L1 and PD-L2 down-regulates CD8+ T cell activation and cytolysis. Eur J Immunol. 2003;33(11):3117–3126. doi:10.1002/eji.200324270.
- García-Teijido P, Cabal ML, Fernández IP, Pérez YF. Tumor-infiltrating lymphocytes in triple negative breast cancer: the future of immune targeting. Clin Med Insights Oncol. 2016;10(Suppl 1):31–39. doi:10.4137/CMO.S34540.
- Niu M, Valdes S, Naguib YW, Hursting SD, Cui Z. Tumor-associated macrophage-mediated targeted therapy of triple-negative breast cancer. Mol Pharm. 2016;13(6):1833–1842. doi:10.1021/acs.molpharmaceut.5b00987.
- Yu T, Di G. Role of tumor microenvironment in triple-negative breast cancer and its prognostic significance. Chin J Cancer Res. 2017;29(3):237–252. doi:10.21147/j.1000-9604.2017.03.10.
- Miyashita M, Sasano H, Tamaki K, Hirakawa H, Takahashi Y, Nakagawa S, Watanabe G, Tada H, Suzuki A, Ohuchi N, et al. Prognostic significance of tumor-infiltrating CD8+ and FOXP3+ lymphocytes in residual tumors and alterations in these parameters after neoadjuvant chemotherapy in triple-negative breast cancer: a retrospective multicenter study. Breast Cancer Res. 2015;17:124. doi:10.1186/s13058-015-0632-x.
- Stovgaard ES, Nielsen D, Hogdall E, Balslev E. Triple negative breast cancer – prognostic role of immune-related factors: a systematic review. Acta Oncol. 2018;57(1):74–82. doi:10.1080/0284186X.2017.1400180.
- Ouzounova M, Lee E, Piranlioglu R, El Andaloussi A, Kolhe R, Demirci MF, Marasco D, Asm I, Chadli A, Hassan KA, et al. Monocytic and granulocytic myeloid derived suppressor cells differentially regulate spatiotemporal tumour plasticity during metastatic cascade. Nat Commun. 2017;8:14979. doi:10.1038/ncomms14979.
- Christmas BJ, Rafie CI, Hopkins AC, Scott BA, Ma HS, Cruz KA, Woolman S, Armstrong TD, Connolly RM, Azad NA, et al. Entinostat converts immune-resistant breast and pancreatic cancers into checkpoint-responsive tumors by reprogramming tumor-infiltrating MDSCs. Cancer Immunol Res. 2018;6(12):1561–1577. doi:10.1158/2326-6066.CIR-18-0070.
- Qian B-Z, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141(1):39–51. doi:10.1016/j.cell.2010.03.014.
- Yuan Z-Y, Luo R-Z, Peng R-J, Wang -S-S, Xue C. High infiltration of tumor-associated macrophages in triple-negative breast cancer is associated with a higher risk of distant metastasis. OncoTargets Ther. 2014;7:1475–1480. doi:10.2147/OTT.S61838.
- Diaz-Montero CM, Salem ML, Nishimura MI, Garrett-Mayer E, Cole DJ, Montero AJ. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin–cyclophosphamide chemotherapy. Cancer Immunol Immunother. 2009;58(1):49–59. doi:10.1007/s00262-008-0523-4.
- Danza K, Pilato B, Lacalamita R, Addati T, Giotta F, Bruno A, Paradiso A, Tommasi S. Angiogenetic axis angiopoietins/Tie2 and VEGF in familial breast cancer. Eur J Hum Genet. 2013;21(8):824–830. doi:10.1038/ejhg.2012.273.
- Ramanathan R, Olex AL, Dozmorov M, Bear HD, Fernandez LJ, Takabe K. Angiopoietin pathway gene expression associated with poor breast cancer survival. Breast Cancer Res Treat. 2017;162(1):191–198. doi:10.1007/s10549-017-4102-2.
- Linderholm BK, Hellborg H, Johansson U, Elmberger G, Skoog L, Lehtiö J, Lewensohn R. Significantly higher levels of vascular endothelial growth factor (VEGF) and shorter survival times for patients with primary operable triple-negative breast cancer. Ann Oncol. 2009;20(10):1639–1646. doi:10.1093/annonc/mdp062.
- Huang D, Lan H, Liu F, Wang S, Chen X, Jin K, Mou X. Anti-angiogenesis or pro-angiogenesis for cancer treatment: focus on drug distribution. Int J Clin Exp Med. 2015;8:8369–8376.
- Kim J, de Sampaio PC, Lundy DM, Peng Q, Evans KW, Sugimoto H, Gagea M, Kienast Y, Do Amaral NS, Rocha RM, et al. Heterogeneous perivascular cell coverage affects breast cancer metastasis and response to chemotherapy. JCI Insight. 2016;1(21). doi:10.1172/jci.insight.90733
- Fukumura D, Kloepper J, Amoozgar Z, Duda DG, Jain RK. Enhancing cancer immunotherapy using antiangiogenics: opportunities and challenges. Nat Rev Clin Oncol. 2018;15(5):325–340. doi:10.1038/nrclinonc.2018.29.
- Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307(5706):58–62. doi:10.1126/science.1104819.
- Datta M, Coussens LM, Nishikawa H, Hodi FS, Jain RK. Reprogramming the tumor microenvironment to improve immunotherapy: emerging strategies and combination therapies. Am Soc Clin Oncol Educ Book. 2019;39:165–174. doi:10.1200/EDBK_237987.
- Hato T, Zhu AX, Duda DG. Rationally combining anti-VEGF therapy with checkpoint inhibitors in hepatocellular carcinoma. Immunotherapy. 2016;8(3):299–313. doi:10.2217/imt.15.126.
- Motz GT, Santoro SP, Wang L-P, Garrabrant T, Lastra RR, Hagemann IS, Lal P, Feldman MD, Benencia F, Coukos G. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat Med. 2014;20(6):607–615. doi:10.1038/nm.3541.
- Tian L, Goldstein A, Wang H, Ching Lo H, Sun Kim I, Welte T, Sheng K, Dobrolecki LE, Zhang X, Putluri N, et al. Mutual regulation of tumour vessel normalization and immunostimulatory reprogramming. Nature. 2017;544(7649):250–254. doi:10.1038/nature21724.
- Schaaf MB, Garg AD, Agostinis P. Defining the role of the tumor vasculature in antitumor immunity and immunotherapy. Cell Death Dis. 2018;9(2):1–14. doi:10.1038/s41419-017-0061-0.
- Goel S, Duda DG, Xu L, Munn LL, Boucher Y, Fukumura D, Jain RK. Normalization of the vasculature for treatment of cancer and other diseases. Physiol Rev. 2011;91(3):1071–1121. doi:10.1152/physrev.00038.2010.
- Munn LL, Jain RK. Vascular regulation of antitumor immunity. Science. 2019;365(6453):544–545. doi:10.1126/science.aaw7875.
- Karagiannis ED, Popel AS. A systematic methodology for proteome-wide identification of peptides inhibiting the proliferation and migration of endothelial cells. Proc Natl Acad Sci U S A. 2008;105(37):13775–13780. doi:10.1073/pnas.0803241105.
- Rosca EV, Koskimaki JE, Pandey NB, Tamiz AP, Popel AS. Structure-activity relationship study of collagen-derived anti-angiogenic biomimetic peptides. Chem Biol Drug Des. 2012;80(1):27–37. doi:10.1111/j.1747-0285.2012.01376.x.
- Lee E, Lee SJ, Koskimaki JE, Han Z, Pandey NB, Popel AS. Inhibition of breast cancer growth and metastasis by a biomimetic peptide. Sci Rep. 2014;4:7139. doi:10.1038/srep07139.
- Kim E, Lee E, Plummer C, Gil S, Popel AS, Pathak AP. Vasculature-specific MRI reveals differential anti-angiogenic effects of a biomimetic peptide in an orthotopic breast cancer model. Angiogenesis. 2015;18(2):125–136. doi:10.1007/s10456-014-9450-5.
- Lima eSilva R, Kanan Y, Mirando AC, Kim J, Shmueli RB, Lorenc VE, Fortmann SD, Sciamanna J, Pandey NB, Green JJ, et al. Tyrosine kinase blocking collagen IV-derived peptide suppresses ocular neovascularization and vascular leakage. Sci Transl Med. 2017;9(373):eaai8030. doi:10.1126/scitranslmed.aai8030.
- Barbhuiya MA, Mirando AC, Simons BW, Lemtiri-Chlieh G, Green JJ, Popel AS, Pandey NB, Tran PT. Therapeutic potential of an anti-angiogenic multimodal biomimetic peptide in hepatocellular carcinoma. Oncotarget. 2017;8(60):101520–101534. doi:10.18632/oncotarget.21148.
- Koskimaki JE, Karagiannis ED, Rosca EV, Vesuna F, Winnard PT, Raman V, Bhujwalla ZM, Popel AS. Peptides derived from type IV collagen, CXC chemokines, and thrombospondin-1 domain-containing proteins inhibit neovascularization and suppress tumor growth in MDA-MB-231 breast cancer xenografts. Neoplasia N Y N. 2009;11(12):1285–1291. doi:10.1593/neo.09620.
- Rosca EV, Koskimaki JE, Pandey NB, Wolff AC, Popel AS. Development of a biomimetic peptide derived from collagen IV with anti-angiogenic activity in breast cancer. Cancer Biol Ther. 2011;12(9):808–817. doi:10.4161/cbt.12.9.17677.
- Rosca EV, Penet M-F, Mori N, Koskimaki JE, Lee E, Pandey NB, Bhujwalla ZM, Popel AS. A biomimetic collagen derived peptide exhibits anti-angiogenic activity in triple negative breast cancer. PLoS One. 2014;9(11):e111901. doi:10.1371/journal.pone.0111901.
- Pulaski BA, Ostrand-Rosenberg S. Mouse 4T1 breast tumor model. Curr Protoc Immunol. 2001;Chapter 20:Unit20.2. doi:10.1002/0471142735.im2002s39.
- Reilly RT, Gottlieb MB, Ercolini AM, Machiels JP, Kane CE, Okoye FI, Muller WJ, Dixon KH, Jaffee EM. HER-2/neu is a tumor rejection target in tolerized HER-2/neu transgenic mice. Cancer Res. 2000;60:3569–3576.
- Okunishi K, Dohi M, Nakagome K, Tanaka R, Mizuno S, Matsumoto K, Miyazaki J, Nakamura T, Yamamoto K. A novel role of hepatocyte growth factor as an immune regulator through suppressing dendritic cell function. J Immunol. 2005;175(7):4745–4753. doi: 10.4049/jimmunol.175.7.4745
- Gabrilovich D, Ishida T, Oyama T, Ran S, Kravtsov V, Nadaf S, Carbone DP. Vascular Endothelial Growth Factor Inhibits the Development of Dendritic Cells and Dramatically Affects the Differentiation of Multiple Hematopoietic Lineages In VivoPresented in part at the Keystone Symposium “Cellular and Molecular Biology of Dendritic Cells,” Santa Fe, NM, March 3-9, 1998, and at the annual meeting of the American Association for Cancer Research, March 28-April 1, 1998. Blood. 1998;92(11):4150–4166. doi:10.1182/blood.V92.11.4150.423k45_4150_4166.
- Balan M, Mier Y Teran E, Waaga-Gasser AM, Gasser M, Choueiri TK, Freeman G, Pal S. Novel roles of c-Met in the survival of renal cancer cells through the regulation of HO-1 and PD-L1 expression. J Biol Chem. 2015;290(13):8110–8120. doi:10.1074/jbc.M114.612689.
- Lai Y-S, Wahyuningtyas R, Aui S-P, Chang K-T. Autocrine VEGF signalling on M2 macrophages regulates PD-L1 expression for immunomodulation of T cells. J Cell Mol Med. 2019;23(2):1257–1267. doi:10.1111/jcmm.14027.
- Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H, Diéras V, Hegg R, Im S-A, Shaw Wright G, et al. Atezolizumab and nab-paclitaxel in advanced triple-negative breast cancer. N Engl J Med. 2018;379(22):2108–2121. doi:10.1056/NEJMoa1809615.
- Nakamura K, Smyth MJ. Myeloid immunosuppression and immune checkpoints in the tumor microenvironment. Cell Mol Immunol. 2019 Oct 14:1–12. doi:10.1038/s41423-019-0306-1.
- Awad RM, De Vlaeminck Y, Maebe J, Goyvaerts C, Breckpot K. Turn back the TIMe: targeting tumor infiltrating myeloid cells to revert cancer progression. Front Immunol. 2018;9. doi:10.3389/fimmu.2018.01977.
- Zhao C, Mirando AC, Sové RJ, Medeiros TX, Annex BH, Popel AS. A mechanistic integrative computational model of macrophage polarization: implications in human pathophysiology. PLOS Comput Biol. 2019;15(11):e1007468. doi:10.1371/journal.pcbi.1007468.
- Bauer R, Udonta F, Wroblewski M, Ben-Batalla I, Santos IM, Taverna F, Kuhlencord M, Gensch V, Päsler S, Vinckier S, et al. Blockade of myeloid-derived suppressor cell expansion with all-trans retinoic acid increases the efficacy of anti-angiogenic therapy. Cancer Res. 2018 Apr 19:canres.3415.2017. doi:10.1158/0008-5472.CAN-17-3415.
- Koskimaki JE, Lee E, Chen W, Rivera CG, Rosca EV, Pandey NB, Popel AS. Synergy between a collagen IV mimetic peptide and a somatotropin-domain derived peptide as angiogenesis and lymphangiogenesis inhibitors. Angiogenesis. 2013;16(1):159–170. doi:10.1007/s10456-012-9308-7.
- Voron T, Colussi O, Marcheteau E, Pernot S, Nizard M, Pointet A-L, Latreche S, Bergaya S, Benhamouda N, Tanchot C, et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J Exp Med. 2015;212(2):139–148. doi:10.1084/jem.20140559.
- Linde N, Lederle W, Depner S, van Rooijen N, Gutschalk CM, Mueller MM. Vascular endothelial growth factor-induced skin carcinogenesis depends on recruitment and alternative activation of macrophages. J Pathol. 2012;227(1):17–28. doi:10.1002/path.3989.
- Nefedova Y, Huang M, Kusmartsev S, Bhattacharya R, Cheng P, Salup R, Jove R, Gabrilovich D. Hyperactivation of STAT3 is involved in abnormal differentiation of dendritic cells in cancer. J Immunol. 2004;172(1):464–474. doi:10.4049/jimmunol.172.1.464.
- Wada J, Suzuki H, Fuchino R, Yamasaki A, Nagai S, Yanai K, Koga K, Nakamura M, Tanaka M, Morisaki T, et al. The contribution of vascular endothelial growth factor to the induction of regulatory T-cells in malignant effusions. Anticancer Res. 2009;29(3):881–888.
- Desar IME, Jacobs JFM, Hulsbergen‐vandeKaa CA, Oyen WJG, Mulders PFA, van der Graaf WTA, Adema GJ, van Herpen CML, de Vries IJM. Sorafenib reduces the percentage of tumour infiltrating regulatory T cells in renal cell carcinoma patients. Int J Cancer. 2011;129(2):507–512. doi:10.1002/ijc.25674.
- Chen Y, Ramjiawan RR, Reiberger T, Ng MR, Hato T, Huang Y, Ochiai H, Kitahara S, Unan EC, Reddy TP, et al. CXCR4 inhibition in tumor microenvironment facilitates anti-PD-1 immunotherapy in sorafenib-treated HCC in mice. Hepatol Baltim Md. 2015;61(5):1591–1602. doi:10.1002/hep.27665.
- Ko JS, Rayman P, Ireland J, Swaidani S, Li G, Bunting KD, Rini B, Finke JH, Cohen PA. Direct and differential suppression of myeloid-derived suppressor cell subsets by sunitinib is compartmentally constrained. Cancer Res. 2010;70(9):3526–3536. doi:10.1158/0008-5472.CAN-09-3278.
- Simon S, Vignard V, Florenceau L, Dreno B, Khammari A, Lang F, Labarriere N. PD-1 expression conditions T cell avidity within an antigen-specific repertoire. Oncoimmunology. 2016;5(1):e1104448. doi:10.1080/2162402X.2015.1104448.
- Gros A, Robbins PF, Yao X, Li YF, Turcotte S, Tran E, Wunderlich JR, Mixon A, Farid S, Dudley ME, et al. PD-1 identifies the patient-specific CD8+ tumor-reactive repertoire infiltrating human tumors. J Clin Invest. 2014;124(5):2246–2259. doi:10.1172/JCI73639.
- Overacre-Delgoffe AE, Chikina M, Dadey RE, Yano H, Brunazzi EA, Shayan G, Horne W, Moskovitz JM, Kolls JK, Sander C, et al. Interferon-γ drives treg fragility to promote anti-tumor immunity. Cell. 2017;169(6):1130–1141.e11. doi:10.1016/j.cell.2017.05.005.
- Castro F, Cardoso AP, Gonçalves RM, Serre K, Oliveira MJ. Interferon-gamma at the crossroads of tumor immune surveillance or evasion. Front Immunol. 2018;9. doi:10.3389/fimmu.2018.00847.
- Garcia-Diaz A, Shin DS, Moreno BH, Saco J, Escuin-Ordinas H, Rodriguez GA, Zaretsky JM, Sun L, Hugo W, Wang X, et al. Interferon receptor signaling pathways regulating PD-L1 and PD-L2 expression. Cell Rep. 2017;19(6):1189–1201. doi:10.1016/j.celrep.2017.04.031.
- Mo X, Zhang H, Preston S, Martin K, Zhou B, Vadalia N, Gamero AM, Soboloff J, Tempera I, Zaidi MR. Interferon-γ signaling in melanocytes and melanoma cells regulates expression of CTLA-4. Cancer Res. 2017 Jan 1;canres.1615.2017. doi:10.1158/0008-5472.CAN-17-1615.
- Brody JR, Costantino CL, Berger AC, Sato T, Lisanti MP, Yeo CJ, Emmons RV, Witkiewicz AK. Expression of indoleamine 2,3-dioxygenase in metastatic malignant melanoma recruits regulatory T cells to avoid immune detection and affects survival. Cell Cycle. 2009;8(12):1930–1934. doi:10.4161/cc.8.12.8745.
- Shime H, Maruyama A, Yoshida S, Takeda Y, Matsumoto M, Seya T. Toll-like receptor 2 ligand and interferon-γ suppress anti-tumor T cell responses by enhancing the immunosuppressive activity of monocytic myeloid-derived suppressor cells. Oncoimmunology. 2017;7(1):e1373231. doi:10.1080/2162402X.2017.1373231.
- Beatty GL, Paterson Y. IFN-γ-dependent inhibition of tumor angiogenesis by tumor-infiltrating CD4+ T cells requires tumor responsiveness to IFN-γ. J Immunol. 2001;166(4):2276–2282. doi:10.4049/jimmunol.166.4.2276.
- Briesemeister D, Sommermeyer D, Loddenkemper C, Loew R, Uckert W, Blankenstein T, Kammertoens T. Tumor rejection by local interferon gamma induction in established tumors is associated with blood vessel destruction and necrosis. Int J Cancer. 2011;128(2):371–378. doi:10.1002/ijc.25350.
- Vannini A, Leoni V, Barboni C, Sanapo M, Zaghini A, Malatesta P, Campadelli-Fiume G, Gianni T. αvβ3-integrin regulates PD-L1 expression and is involved in cancer immune evasion. Proc Natl Acad Sci. 2019;116(40):20141–20150. doi:10.1073/pnas.1901931116.