
ABSTRACT
The success of CD8+ T cell-based cancer immunotherapy emphasizes the importance of understanding the mechanisms of generation of MHC-I peptide ligands and the possible pathways of tumor cell escape from immunosurveillance. Recently, we showed that peptides generated in the nucleus during a pioneer round of mRNA translation (pioneer translation products, or PTPs) are an important source of tumor specific peptides which correlates with the aberrant splicing and transcription events associated with oncogenesis. Here we show that up-regulation of PSME3 proteasome activator in cancer cells results in increased destruction of PTP-derived peptides in the nucleus thus enabling cancer cell to subvert immunosurveillance. These findings unveil a previously unexpected role for PSME3 in antigen processing and identify PSME3 as a druggable target to improve the efficacy of cancer immunotherapy.
Introduction
Cellular immune responses against cancer cells expressing non-self epitopes require the activation of CD8+ T cells by professional antigen presenting cells (pAPCs) which take up external peptide material and present it on their major histocompatibility complex class I (MHC-I) molecules as part of a process called cross-presentation.Citation1 The MHC-I direct and cross-presentation pathways are fundamental processes for the detection and elimination of cells that pose a threat to the host. In recent years, it has been suggested that peptides that are directly presented to the MHC-I restricted pathway are not derived from the degradation of full-length proteins but from so-called defective ribosomal products or DRiPs.Citation2 In addition, some MHC-I-bound peptides are generated by cryptic translation, which refers to polypeptides synthesized in the cell via non-conventional translational mechanisms. These are either peptides encoded by introns, intron/exon junctions, 5ʹ and 3ʹ untranslated regions (UTR), alternate translational reading frames or even fusion peptides generated by the proteasome.Citation3–Citation6 These observations have led to a shift in focus toward the notion that the degradation of full-length proteins is not the only critical process for antigen production. More recently, we have shown that the antigen presentation is equivalent whether the peptide is expressed from an intron or from an exon and is supported by so-called pioneer translation products (PTPs),Citation7 which are produced by a translation event distinct from canonical translation prior to mRNA splicing. The PTP model is appealing because it partly explains how the immune system “tolerates” tissue-dependent alternative splicing products. By first examining how direct presentation applies to viral mechanisms of immune evasion, we recently demonstrated that PTPs are also a source of peptides for the exogenous MHC-I pathway. Indeed, PTP-derived peptides are cross-presented by pAPCs in order to specifically activate naïve CD8+ T cells.Citation8 Moreover, PTPs are present in exosomes that are engulfed by bone marrow dendritic cells (BMDCs) for cross-presentation. Finally, PTPs purified from tumor cells (tumor-associated PTPs or TA-PTPs) have been used in combination with exosomes as a potent immune cancer vaccine.Citation8
Polypeptides such as PTPs that enter either the endogenous or the exogenous MHC-I pathway must be processed to fit the MHC-I molecule binding groove to elicit an immune response. A key player of this process is the proteolytic system of the eukaryotic cell, the ubiquitin-proteasome system.Citation9 Central to this system is the proteasome, a multicatalytic complex consisting of a 20S proteolytic core controlled by regulatory complexes that bind to it.Citation10,Citation11 One of these regulatory complexes is the 19S particle, which, along with the 20S proteolytic core, forms the 26S proteasome that degrades ubiquitylated and some non-ubiquitylated proteins in an ATP-dependent manner. Other regulatory complexes, including the REG/PA28 family, have been shown to associate with the 20S or with asymmetric (i.e. single 19S-capped) 26S proteasomes.Citation12,Citation13 The REG family consists of three related subunits which together form two proteasome regulatory complexes: (i) REGα/β, a heteroheptamer formed by REGα and REGβ subunits, located primarily in the cytoplasm; and (ii) PSME3, a homoheptamer formed by the PSME3 subunit, located in the nucleus.Citation14–Citation16 The exact functions and mechanisms of action of PSME3 remain elusive, as only a limited number of proteins whose degradation is mediated or controlled by this regulator have been described. Among them are cell cycle regulators, including the cyclin-dependent kinase inhibitors p21 and p16, the oncogene SRC-3 and the tumor suppressor p53.Citation17–Citation20 This paradigm aligns with the proliferation-promoting and anti-apoptotic properties of PSME3 deduced from an analysis of KO mice and the observation that PSME3 is overexpressed in many cancers and is often associated with a poor prognosis.Citation21 Moreover, other observations point to a central role for PSME3 in intranuclear dynamics through the regulation of (i) nuclear bodies (including nuclear speckles, Cajal and PML bodies) and (ii) nuclear trafficking of splicing factors.Citation22–Citation24 Evolutionary analyzes have shown that REGα and REGβ appeared much later in evolution than PSME3 and diverged concomitantly with the emergence of MHC.Citation25 PSME3 is important for responses to genotoxic and oxidative stress as well as the impairment of proteasome function, which is for example the case in neurodegenerative diseases.Citation26 Interestingly, it was shown that cellular PSME3 is massively recruited to proteasome after nontoxic treatment with proteasome inhibitors.Citation27 Furthermore, a specific interactor of PSME3 has been recently identified, namely PIP30, which positively modulates the interaction of PSME3 to the 20S proteasome complex and alters the selectivity of the PSME3/20S proteasome complex toward different peptides.Citation28 We have also recently reported that PTP levels inside the nuclear compartment increased when the proteasomal system was blocked using the highly specific proteasome inhibitor epoxomicin.Citation7
Although many studies have demonstrated a strong link between REGα/β and antigen production in the immune system,Citation29 no data are currently available suggesting a negative or positive role for PSME3 in this process, although PSME3-knockout MEF cells and PSME3-deficient animal models have been produced and challenged to assess specific immune responses.Citation30,Citation31 Here, we show i) an inverse correlation between the expression of PSME3 and the presentation of different MHC-I antigenic peptides, ii) PSME3 involvement in the processing of different PTPs and the negative regulation of CD8+ T cell responses against cancer, iii) an increased presentation of MHC-I PTP-derived antigens due to a downregulation of PSME3 and iv) a tumor growth defect in-vivo of PSME3-knockout tumors. These findings describe a mechanism by which PSME3 negatively influences cancer immune responses in an opposite manner compared to the other members of the REG family.
Materials and methods
T cell hybridomas, cell culture and transfection
The SIINFEKL:Kb-specific (B3Z), the MBP:Kk-specific (MBP CD8+) and the gp100(25-33):Kk-specific T cell reporter hybridomas were described previously.Citation32–Citation34 Human cell lines were obtained from the American Type Culture Collection (ATCC) and cultured according to standard culture protocols and sterile technique. MRC5 (ATCC, n° CCL-171), A375 (ATCC, n° CRL-1619), A549 (ATCC, n° CCL-185), HT29 (ATCC, n° HTB-38) and T84 (ATCC, n° CCL-248) were cultured according to ATCC’s protocol. The WM3526 and WM3682 were supplied by Dr. Meenhard Herlyn and cultured as accordingly.
Once a month, mycoplasma contamination in cell cultures was assessed using the Venor®GeM OneStep mycoplasma detection kit (Minerva biolabs). All cells were used within four weeks after thawing (≈10 passages). All cells were transfected with different increasing amount of plasmid DNA with a final total concentration of 1 µg of plasmid DNA along with 2 μL of JetPrime according to the manufacturer’s protocol (Ozyme). Each plasmid is detail in the supplementary data. No difference in growth rates was observed between WT cells and cells overexpressing or downregulating PSME3.
Drugs
Cells were treated with different drugs: epoxomicin (Peptides International) was used at 300 nM and cisplastin (Sigma) at 5 and 10 mg/mL.
T cell assay
Human cell lines were cotransfected with different plasmids expressing the SL8 epitope and the Kb, Kk or Kd expression vectors depending on the epitope tested. All CD8+T cell hybridomas express LacZ in response to the activation of T cell receptors specific for the SIINFEKL peptide (Ova-immunodominant peptide) in the context of H-2Kb MHC class I molecules, the MBP peptide (myelin basic protein-immunodominant peptide) in the context of H2-Kk MHC class I molecules or the gp100(25-33) peptide in the context of H2-Kd MHC class I molecules. For the minigene antigen presentation assays, all cell lines were co-transfected with 0.5 µg of SL8-minigene construct and 0.5 µg of H-2Kb construct for 48 h. Cancer cells were then washed twice in 1X PBS and 105 cells were co-cultured with either 105 SL8-specific B3Z T cell hybridoma or 105 MBP-specific T cell hybridoma for 16–20 h. Free peptide was added to cells to ensure that T-cell assays were carried out at non-saturated conditions and that the expression of MHC class I molecules was not affected. Next, cells were centrifuged at 1,200 rpm for 5 min. The cells were washed twice with 1X PBS and lysed for 5 min at room temperature (RT) in the following buffer: 0.2% Triton X-100, 0.5 M K2HPO4, 0.5 M KH2PO4. The lysates were centrifuged at 3,000 rpm for 10 min to pellet cell debris. Next, the supernatant was transferred into an optiplate (Packard Bioscience) and a revelation buffer containing 40 µM methylumbelliferyl β-D-galactopyranoside (MUG) was then added. The plate was incubated for 3 h at RT. The activity of β-galactosidase (luminescence) was measured with FLUOstar OPTIMA (BMG LABTECH Gmbh). The values from mock-transfected cells were subtracted as in all the other reported T cell assay experiments.
CRISPR/Cas9 transfection and selection
After the transfection of 1 µg of CRISPR plasmid vector, cells were sorted 2 days later (1 cell/well). PCR and Western blotting were performed using the clones; selected clones were sent for sequencing. TOPO TA cloning (Life Technologies) was carried out for selected clones. The CRISPR/Cas9 system was applied to the A375 cell line, and A375 clone number 11 was generated and designated A375c.XI.
FACS analysis for H-2Kb expression and recovery at the cell surface
To study the kinetics of endogenous surface Kb recovery cells were treated with ice-cold citric acid buffer (0.13 M citric acid, 0.061 M Na2HPO4, 0.15 M NaCl [pH 3]) at 1 × 107 cells per milliliter for 120 s, washed three times with PBS, and resuspended in culture medium. At the indicated time point, a cell aliquot (generally 1.5 × 106 cells) was removed and stained with anti-mouse 25-D1.16 antibody, used for the detection of specific MHC-I/peptide-complexes (H-2Kb+SIINFEKL) at the cell surface. All flow cytometry experiments were conducted using the BD LSRII flow cytometer (BD Biosciences) and data were analyzed with the FlowJow software (V10).
In vitro peptide degradation
Reconstitution of REG-20S complexes and the degradation of short fluorogenic substrates, the MP-45 and the KH-52 polypeptides (containing the SIINFEKL epitope in their middle) or the MP-46 and the KH-53 polypeptides (containing the MBP epitope in their middle), were performed according to previously described methods.Citation35–Citation37 Briefly, PSME3- and REGα/β-20S proteasomes were reconstituted by preincubating human 20S constitutive or immuno particles (BostonBiochem, USA) with a 6-fold molar excess of PSME3 (BostonBiochem, USA) or REGαβCitation36 at 37°C for 30 min in 20 mM HEPES, pH 7.6, and 2 mM NaCl and were immediately used for degradation experiments. For kinetic analysis and to generate peptide products for MS/MS studies, all polypeptides (50 µM) were incubated with 20S, PSME3-20S or REGα/β-20S (20 nM) for 8 h at 37°C in 20 mM HEPES, pH 7.6, 2 mM NaCl. To assay the peptides generated during protein degradation, we measured the appearance of new amino groups using fluorescamine as previously described.Citation37 In brief, at the end of the incubation, the peptide products were separated from undegraded polypeptides by ultrafiltration through a membrane with a 3-kDa cutoff (Nanosep, Pall, USA), and these samples were assessed with fluorescamine (Sigma) and used for MS/MS analysis.
Liquid chromatography–tandem MS (LC–MS/MS) analysis
Five-microliter samples from the 20S -/+ PSME3 8 hour degradation experiments containing approximately 130 pmol NH2/µl were loaded onto StageTipsµC18;Citation38,Citation39 peptides were eluted in 40 μl of 80% acetonitrile in 0.1% formic acid. The acetonitrile was allowed to evaporate in a Speed-Vac, and then the samples were resuspended in 6 µl of eluent A (see the composition below) for nLC-MS/MS analysis. Two microliters of each sample was injected as technical replicates into a nLC–ESI–MS/MS quadrupole Orbitrap QExactive-HF mass spectrometer (Thermo Fisher Scientific). Peptide separation was achieved using a linear gradient from 95% solvent A (2% ACN, 0.1% formic acid) to 50% solvent B (80% acetonitrile, 0.1% formic acid) for 23 min and from 60 to 100% solvent B for 2 min at a constant flow rate of 0.25 µl/min on a UHPLC Easy-nLC 1000 (Thermo Scientific) connected to a 25-cm fused-silica emitter with an inner diameter of 75 µm (New Objective, Inc. Woburn, MA, USA), packed in-house with ReproSil-Pur C18-AQ 1.9-µm beads (Dr. Maisch Gmbh, Ammerbuch, Germany) using a high-pressure bomb loader (Proxeon, Odense, Denmark). MS data were acquired using a data-dependent top-15 method for HCD fragmentation. Survey full-scan MS spectra (300–1750 Th) were acquired in the Orbitrap at a resolution of 60,000, an AGC target of 1e6, and an IT of 120 ms. For the HCD spectra, the resolution was set to 15,000 at m/z 200, with an AGC target of 1e5, an IT of 120 ms, an NCE of 28% and an isolation width of 3.0 m/z.
Data processing and analysis
For quantitative proteomic analysis, raw data were processed with MaxQuant (ver. 1.5.2.8) and searched against a database containing the sequence of the KH-52 intron SIINFEKL+ contaminant fasta included in MaxQuant. No enzyme specificity was selected, and there were no differences between I and L. The mass deviation for MS-MS peaks was set at 20 ppm, and the peptide false discovery rate (FDR) was set at 0.01; the minimal length required for a peptide identification was eight amino acids. The list of identified peptides was filtered to eliminate contaminants. Statistical analyses were performed with Perseus (ver. 1.5.1.6) considering the peptide intensity; normalization based on the Z-score and imputation was applied. Significant peptides were determined with a t-test, Benjamini Hochberg correction and FDR<0.05 (more stringent assignments), or a t-test with a p-value of 0.05 (less stringent assignments). Only significant peptides were used for supervised hierarchical clustering analysis. MS data as raw files, peptides identified with relative intensities, peptide score, PEP, etc. and search parameters have been loaded into Peptide Atlas repository (ftp://PASS01235:[email protected]/).
Tumor challenge in-vivo
C57Bl/6J female mice were obtained from Harlan Laboratories. NU/NU nude mice were obtained from Charles River. 7-week-old mice were injected subcutaneously into the right flank with 1 × 105 MCA205 wild type (WT) cells or 1 × 105 Cas9-PSME3 MCA205 clones. Area of the tumor was recorded every 3 to 4 days until ethical limit points are reached. All animal experiments were authorized and validated by the ethics committee n°26 of University Paris-Saclay in compliance with the European directive 2010/63 UE and its transposition into French law.
Results
The expression of PSME3 is inversely correlated with the level of antigen presentation in multiple cancer cell lines
A series of studies have reported the overexpression of PSME3 in different cancer types.Citation40–Citation43 To clarify the specific role of PSME3 in MHC-I antigen presentation, we selected different cancer cell lines and examined PSME3 mRNA and protein expression levels. First, we analyzed PSME3 mRNA levels by real-time qRT-PCR in three human melanoma cell lines (A375, WM3526 and WM3682), one human lung cancer cell line (A549) and two colon cancer cell lines (HT29 and T84), and we observed that PSME3 was notably upregulated in each cell line compared to the control normal lung fibroblast cell line MRC5 ()). Next, we measured PSME3 protein expression and observed that it was overexpressed in all cancer cell lines compared with MRC5 normal cells, although levels of PSME3 overexpression varied between the different cell lines analyzed. Indeed, PSME3 was highly overexpressed in melanoma and colon cancer cell lines, expressed at low levels in the lung cancer cell line A549, and demonstrated very low expression in the normal cell line ()). Furthermore, we assessed the cellular distribution of PSME3 in these cancer cell lines. ) shows that endogenous PSME3 is localized in the nucleus of all cell lines analyzed independently of its expression levels.
Figure 1. Inverse correlation between the expression of PSME3 and antigen presentation in multiple cancer cell lines. (a) PSME3 mRNA levels were analyzed by RT-qPCR in different cell lines and normalized to β-actin mRNA levels. The MRC5 cell line was used as a reference. Experiments were performed in triplicate. Data are expressed as the mean ± SEM from three technical replicates. (b) Western blot analysis and quantification (relative to the housekeeping protein β-actin) of PSME3 expression. Protein levels are indicated below each gel. (c) PSME3 protein localization was determined by immunofluorescence in several tumor cell lines and in the MRC5 normal cell line. PSME3 was stained with Alexa Fluor 488, and nuclei were counterstained with DAPI. Cells were analyzed by confocal microscopy. As expected, the regulator PSME3 was localized in the nucleus in all tested cell lines. (d, e) All cell types were transfected in vitro with Glob-intron-SL8 (d, left panel) or Glob-intron-MBP(79–87) (d, right panel), β-Glob-exon-SL8 (e, left panel) and OVA cDNA (e, right panel) expression constructs. The cells were incubated with the SL8-specific CD8+T cell hybridoma (B3Z) for 16h. The data show the average of at least three independent experiments ± SD minus the values from mock-transfected cells. Free SL8 peptides were added to the cells to ensure that the T cell assays were performed under non-saturation conditions and that the expression of MHC-I molecules was not affected.
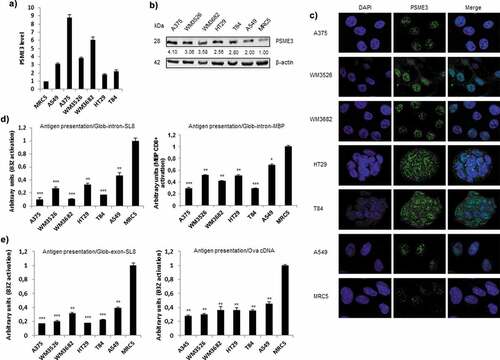
To evaluate whether the observed differences in PSME3 expressions in cancer and normal cell lines contribute to changes in antigen production and presentation, we examined the presentation of PTP-derived antigens at the cell surface of these cell lines. To achieve this goal, we expressed the mouse MHC-I H-2Kb molecule and the Glob-intron-SL8 constructs in these human cell lines to examine the production of specific SL8-comprising PTPs as described previously.Citation6,Citation7 Using a SIINFEKL:Kb (B3Z) T cell hybridomaCitation32 that specifically detects the SL8 epitope presented on Kb molecules, we first found that the antigen presentation was lower in all cancer cell lines tested than in the normal MRC5 cell line (), left panel and Fig. S1D). Interestingly, we observed a complete inverse correlation between PTP-derived-SL8 presentation and PSME3 expression in all cell lines tested. This result suggested that PSME3 overexpression negatively affected PTP-derived antigen presentation (compare ,)). Crucially, this result was not merely due to differences in the cellular level of the β-globin construct, which in fact were higher in tumor cell lines overexpressing PSME3 (Fig. S1A). Moreover, FACS analysis showed that the overall H-2Kb expression differs among the cancer cell lines tested but the variations observed do not correlate with the expression of SL8 as determined with the B3Z assay. In fact, we observed that MHC-I molecules were more abundant at the cell surface of tumor cell lines overexpressing PSME3 and exhibiting a decrease in PTP-dependent antigen presentation (Fig. S1B).
In order to rule out the possibility that the negative role of PSME3 on PTP dependent antigen presentation could be restricted to the SL8 epitope or the Kb molecule, we determined whether also the presentation of the MBP(79–87) epitope, which is derived from the Myelin Basic Protein (MBP) and is presented on H-2Kk molecules was affected by the overexpression of PSME3 in different cancer cell lines. Using the specific MBP CD8+T cell hybridomaCitation33 we could obtain results similar to those previously observed with the PTP-derived SL8, that is to say a complete inverse correlation between PTP-derived MBP presentation and PSME3 expression in all cancer cell lines tested (), right panel). All together, these results indicate: i) a negative role of PSME3 in the MHC-I antigen presentation pathway since antigen presentation was reduced in all cancer lines overexpressing the PSME3 regulator while the β-globin protein and the MHC-I molecules were more abundant in the same cell lines and ii) that the inverse correlation between PSME3 expression and antigen presentation is not restricted to a specific epitope or MHC-I molecule.
We then wanted to determine whether the previously observed differences in SL8 antigen presentation were restricted to intron-derived tumor-associated antigens or if they were also observed with an exon-derived tumor-associated antigen. To this purpose, we analyzed cells expressing mouse MHC-I Kb molecules and the Glob-exon-SL8 construct or the ovalbumin cDNA, in which the SL8 epitope is found in its correct setting. As shown in ), antigen presentation showed an inverse correlation with PSME3 expression in all cell lines tested, independently of the position of the antigenic epitope in the exon sequence (left panel) or in cDNA constructs (right panel) even if with the cDNA construct the effect of PSME3 on antigen presentation was weaker.
Furthermore, we expressed in the different human cell lines the mouse MHC-I H-2Kb molecule and the SL8-minigene construct, which contains only the presented 8-amino-acid sequence of SL8 and does not require an additional processing step to be exported by the TAP system in to the ER and to be loaded on the MHC class I molecules.Citation44 Importantly, we observed that differences in PSME3 expression did not correlate with SL8 antigen presentation (Fig. S1C). Hence, the effects of PSME3 on MHC-I antigen presentation only occurs with longer polypeptides containing the presented MHC-I antigenic peptides, indicating that PSME3 directly acts on epitopes processing and not on subsequent steps of class-I presentation.
All together these results clearly support the hypothesis that the proteasome regulator PSME3 is involved in the inhibition of the MHC-I presentation of antigens from exon- as well as intron-derived PTPs, a mechanism that may allow tumor cells to avoid immune responses against conventional and unconventional epitopes.
Exogenous overexpression of PSME3 decreases MHC-I antigen presentation
In the rest of our study we decide to use the Glob-intron-SL8 construct since, due to its nuclear localization it is the most appropriate tool to investigate the role of a nuclear proteasomal regulator on PTP-dependent antigen presentation. To study the specific effects of PSME3 on PTP-dependent MHC-I antigen presentation, MRC5 lung fibroblast cells and A549 lung cancer cells which express PSME3 at very low levels, (see ,)), were therefore co-transfected with the mouse MHC-I Kb molecule, the Glob-intron-SL8 and the exogenous Flag-PSME3 constructs. Using Western blotting ()), immunofluorescence ()) or qRT-PCR (Fig. S2), we monitored the expression of the exogenous PSME3 construct and compared it to the expression of endogenous PSME3. Notably, the overexpressed exogenous PSME3 showed the same expression pattern and localization as the endogenous PSME3. Next, we examined the antigen presentation of the SL8 epitope in these cell lines. The introduction of increasing amounts of PSME3 in both A549 and MRC5 cell lines, which express fixed amounts of the Glob-intron-SL8 construct and MHC-I Kb molecules, resulted in a dose-dependent decrease in the activation of the B3Z hybridoma ()). This observation supports the hypothesis that the overexpression of PSME3 has a clear-cut negative effect on PTP-dependent antigen presentation.
Figure 2. Exogenous PSME3 overexpression decreases antigen presentation. (a) MRC5 and A549 were transfected with a construct expressing PSME3 WT from 0.0625 to 0.5 µg or a corresponding empty construct for 48 h. PSME3 protein levels were examined by Western blotting using β-actin as a loading control. Protein levels are indicated below each gel. (b) MRC5 and A549 were transfected with a construct expressing Flag-PSME3 WT (0.25 µg). The Flag tag was stained with Alexa Fluor 488, and the nuclei were stained with DAPI. Cells were analyzed by confocal microscopy. As expected, exogenous transfected PSME3 WT localized in the nuclei. (c) MRC5 cells (left panel) and A549 cells (right panel). The cells were incubated with the SL8-specific CD8+T cell hybridoma (B3Z) for 16 h. The data show the average of at least three independent experiments ± SD minus the values from mock-transfected cells. All cells were transfected with increasing amounts (from 0 to 0.5 μg) of Glob-intron-SL8 construct with a final total concentration of 1 µg of plasmids in each cell lines. (d) MRC5 cells were transfected with a PSME3 WT construct for 48 h. The increase in PSME3 protein expression had no effect on the expression of REGα protein. (e) MRC5 cells were transfected with increasing amounts (from 0 to 0.5 μg) of Flag-PSME3 construct for 48 h. At 36 h post-transfection, the cells were then treated overnight with epoxomicin (300 nM). Next, the cells were incubated with the B3Z T cell hybridoma for 16 h. The data show the average of at least three independent experiments ± SD. *** p <.001, * p <.05 (unpaired t-test). (f) Western blot analysis and quantification (relative to the housekeeping protein β-actin) of p21 expression in MRC5 cells treated overnight with epoxomicin at 300 nM. Protein levels are indicated below each gel. As expected, p21 protein levels increased after 12 h of epoxomicin treatment, even though the treated cells overexpressed the exogenous regulator PSME3.
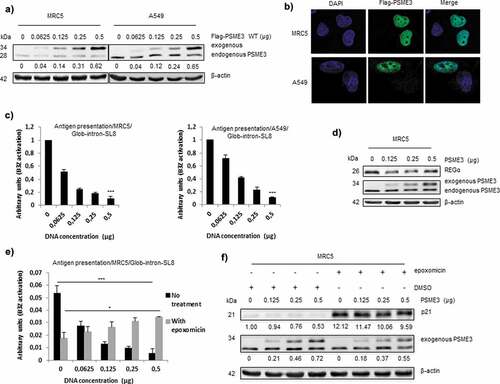
As mentioned earlier, PSME3 is not the sole regulator of the proteasomal pathway; 20S proteasome also binds to the REGα/β regulator, which has been shown to be implicated in the accurate processing of MHC-I antigenic epitopes.Citation14 To investigate in more detail at which exact level of the proteasomal pathway PSME3 contributes to PTP-dependent antigen presentation regulation, we determined whether the overexpression of PSME3 influenced the expression of endogenous REGα. ) shows that increasing the amount of exogenous PSME3 did not affect the expression of REGα. This result therefore demonstrates that the decrease in PTP-dependent antigen presentation caused by PSME3 in different cancer cell lines is not due to a decrease in the expression of the regulator REGα but more likely it results from a direct effect on the 20S proteasome. In fact, the presentation of antigenic peptides depends on proteolytic processing by proteasome complexes that are known to possess the capacity to process and destroy epitopes.Citation45 To ensure that differences in the production of SL8 peptides by the proteasome can be at least partially attributable to the overexpression of PSME3, we treated MRC5 cells expressing increasing amounts of PSME3 and a fixed amount of Glob-intron-SL8 and MHC-I Kb molecules with the 20S proteasome-specific inhibitor epoxomicin and examined SL8 presentation. Importantly, to perform these experiments, we utilized a dose of inhibitor (i.e. 300 nM) that does not completely annihilate all three peptidase activities of proteasomeCitation46 as demonstrated by the only partial reduction of SIINFEKL presentation in ) and by the partial stabilization of p21 levels in ). Interestingly, while non-inhibited cells displayed a decrease in antigen presentation with PSME3 overexpression, a partial increase in MHC-I antigen presentation was observed when cells were treated with epoxomicin ()). Although we cannot exclude that PSME3 may also exert some positive proteasome-independent activity on antigen presentation, this observation supports the idea that PSME3 exerts a direct activity on 20S core particle, consisting in a fine tuning of the balance between proteolytic activities that generate and destroy epitopes (and that are not equally sensitive to epoxomicin), with the final result that the generation of SIINFEKL is increased by this partially inhibited proteasome. Accordingly, previous reports clearly demonstrated that proteasome inhibitors are able to block generation of some antigenic peptides and enhance production of others.Citation47 Moreover, as a control for the efficacy of the proteasomal partial inhibition under the condition used in the treated cell lines, we examined p21 protein levels, which has been reported to be regulated specifically by the PSME3-20S proteasome complex in an ubiquitin and ATP-independent process.Citation18 As shown in ), p21 protein levels increased following 18 h of epoxomicin treatment, but this increase was still reduced in cells overexpressing the exogenous PSME3 regulator, which showed the presence of residual proteasome activity.
Therefore, all these results confirm that the overexpression of PSME3 does not induce (i) a decrease in MHC-I expression or export or (ii) a decrease in the expression of the other members of the REG family, but does directly affect proteasomal proteolytic activities.
Knockdown and knockout of the regulator PSME3 promote antigen presentation
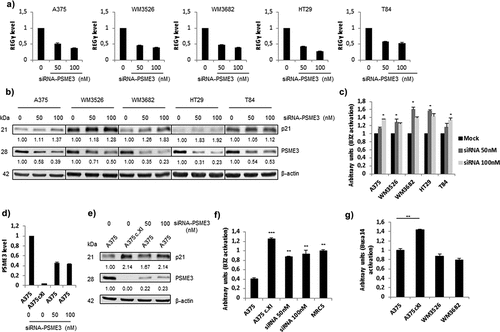
Considering our finding showing that endogenous overexpression of the PSME3 regulator negatively affects MHC-I antigen presentation in cancer cells, we speculated whether PSME3 knockdown might restore PTP-dependent antigen presentation. To test this hypothesis, we silenced PSME3 by transient siRNA treatment in human melanoma and colon cancer cell lines. As expected, PSME3 knockdown ()) enhanced p21 protein levels in all cancer cell lines compared to scrambled siRNA ()). More importantly in terms of PTP-dependent antigen presentation, we observed a close correlation between PSME3 knockdown and the increase of the SL8 epitope at the cell surface ()).
Figure 3. Knockdown and knockout of the expression of the regulator PSME3 promotes antigen presentation. The A375, WM3526, WM3682, HT29 and T84 cell lines were transfected with siRNA specific for PSME3 (50 and 100 nM) or siRNA negative control (0) for 48 h and then analyzed by RT-qPCR (a), Western blotting (b) and a T cell assay (c). (a) Experiments were performed in triplicate. Data are expressed as the mean ± SEM of three technical replicates. (b) Western blotting was performed to analyze and quantify PSME3 and p21 expression, and β-actin was used as a reference protein. The relative protein level is indicated below each gel. (c) Cell lines were transfected with the Glob-intron-SL8 construct (0.5 μg) and treated with different concentrations of siRNA specific for PSME3 (0, 50 and 100 nM). After 48 h, the different cell lines were incubated with the B3Z T cell hybridoma for 16 h. The data show the average of at least three independent experiments ± SD minus the values from mock-transfected cells. (d) qPCR analysis of A375 cells transfected with siRNA specific for PSME3 (50 and 100 nM) was performed. The inhibition of PSME3 mRNA levels was quantified and normalized to β-actin mRNA levels. Experiments were performed in triplicate. Data are expressed as the mean ± SEM from three technical replicates. (e) Western blot analysis and quantification (relative to the housekeeping protein β-actin) of PSME3 and p21 expression. Protein levels are indicated below each gel. (f) The A375cXI and A375 cell lines were both transfected with the Glob-intron-SL8 construct (0.5 μg) for 48 h, and PSME3-siRNA (50 or 100 nM) was only added to A375 cells. The cells were incubated with the B3Z T cell hybridoma for 16 h. The data show the average of at least three independent experiments ± SD minus the values from mock-transfected cells. *** p <.001, ** p <.01 (unpaired t-test). (g) The A375, A375cXI, WM3526 and WM3682 cell lines were co-transfected with the Glob-intron-SL8 (0.5 μg) and the H2-Kd (0.5 μg) constructs for 48 h. The different cell types were then incubated with the Busa14 T cell hybridoma for 16 h. The data show the average of at least three independent experiments ± SD minus the values from mock-transfected cells. ** p <.01 (unpaired t-test). Note that panel C refers to the experiment shown in panels A and B, while panel F refers to the experiment of panel E. Differences in the magnitude of B3Z activation correlate with the extent of PSME3 reduction obtained by siRNA in the different experiments.
The expression of PSME3 is controlled by several mechanisms inside the cell. miRNA-7, which has been reported as a tumor suppressorCitation48,Citation49 and is downregulatedCitation50,Citation51 in several human cancers, also negatively controls the expression of the regulator PSME3 in lung cancer.Citation52 The A375 melanoma cancer cell line, which in our hands expressed the highest levels of PSME3, was therefore transfected with increasing amounts of miRNA-7. Of great interest, we observed that miRNA-7 inhibited PSME3 expression (Fig. S3A) and led to an increase in PTP-SL8-dependent antigen presentation (Fig. S3B). In addition, treatment of cancer cells with the chemotherapeutic molecule cisplatin has been shown to induce a decrease in mRNA and protein levels of PSME3Citation53 (and Fig. S3C). Accordingly, treatment of A375 cells with cisplatin restores the expression of PTP-dependent antigens at the cell surface (Fig. S3D). All together, these results support the idea that PSME3 downregulation restores PTP-derived antigen presentation and indicate a possible chemotherapeutic approach to achieve this goal.
To clarify in more detail the functional importance of PSME3 in cancer immune escape, we used the CRISPR/Cas9 system to create a melanoma Cas9-A375 PSME3-knockout cell line (A375cXI). The heterozygous knockout A375cXI was confirmed by Sanger sequencing, qRT-PCR and Western blot analysis. The A375cXI cell line did not express PSME3 mRNA ()) or its corresponding protein ()). As a control for the loss of PSME3 expression, the p21 protein accumulates in this cell line as in cells that were knocked down with specific PSME3 siRNAs ()). CruciallyA375cXI cells showed increased PTP-dependent antigen presentation compared to A375WT or A375 cell lines that are only knocked down for PSME3 expression ()). Moreover, the level of SL8 antigen presentation in these cell lines was even higher than the control MRC5 line which weakly expresses PSME3 ()). Differences in PSME3 levels are likely to account for this result, since both the tested cell lines (A375cXI and A375WT) expressed equal amounts of exogenous Glob-intron-SL8 protein (Fig. S3E, left panel) as well as equal amounts of the mouse MHC class I molecule at their cell surface (Fig. S3E, right panel).
In order to rule out the possibility that the negative role of PSME3 on PTP dependent antigen presentation could be restricted to the SL8 epitope or the Kb molecule, we also determined whether the presentation of an endogenous epitope from the melanoma cell line, the gp100(25-33) epitope, which is derived from the Pmel17/gp100 protein, was affected by the overexpression of PSME3 in the different melanoma cancer cell lines tested in . Using the specific Busa14 CD8+ T cell hybridoma that has been shown to specifically recognize the gp100(25-33) epitope on H2-Kd molecules,Citation33 we observed that the presentation of the endogenous epitope is enhanced at the surface of A375cXI cells compared to its parental cell line or other melanoma cell lines where PSME3 is overexpressed ()). This result demonstrates that PSME3 also has the capacity to negatively regulate the presentation of an endogenous MHC class I antigen.
All together these results clearly confirm that there is an inverse correlation between the expression of PSME3 and PTP-dependent antigen presentation in cancer cell lines. This effect is likely due to a close control of the proteasomal degradation pathway and not to an overall effect of PSME3 on MHC-I molecule expression and export to the cell surface.
PSME3 regulates the nuclear proteasomal pathway
In our previous studies, we demonstrated that PTPs are produced by a translation event that is distinct from canonical translation and occurs prior to mRNA splicing, supporting the idea that PTPs are generated by a nuclear translation event. Furthermore, we have also reported that treatment with the proteasome inhibitor epoxomicin increases the amount of PTPs within the nuclear compartment, indicating that the nuclear proteasome may be involved in the processing and/or degradation of PTP-antigenic epitopes.Citation7 Based on these studies and all our results so far, we next sought to understand how PSME3 contributes to the specific inhibition of PTP-dependent cancer immune responses and in which cell compartment this inhibition occurs. Antigenic epitopes derived from PTP processing are generated either by a cytosolic or nuclear proteasomal complex, in which the REG family plays an essential role. As proteasomal regulator, PSME3 has to bind to the 20S proteasome to control protein or polypeptide degradation and this interaction should occur in the nucleus. A PSME3 gene sequence mutation that leads to the replacement of Asn 151 by Tyr (N151Y) has been reported to impair the ability of the regulator to activate the trypsin-like activity of the 20S proteasome.Citation54 A375cXI cells were transfected with increasing amounts of Flag-PSME3 WT or mutated Flag-PSME3-N151Y. First, we showed by western-blotting that the mutated regulator was no longer able to degrade the p21 protein likely as a direct consequence of its inability to activate the trypsin-like activity of the 20S proteasome. Moreover, the expression of the other members of the REG family was not impacted under all the experimental conditions used ()). More importantly, PSME3-N151Y inhibited SL8 antigen production and presentation at the cell surface from Glob-intron-SL8 less efficiently than PSME3-WT ()). This inhibition of SL8 presentation again was not attributable to a decrease in expression of the other REG family members shown to be involved in antigen presentation. Two explanations are therefore consistent with this result: i) exogenous mutated PSME3-N151Y may no longer be localized in the nucleus; or ii) exogenous mutated PSME3-N151Y may have lost its capacity to bind to the 20S proteasome and, therefore, contrary to PSME3-WT it is no longer able to inhibit proteasome activity. To discriminate between these two alternative hypotheses, we first examined the distribution of the mutated PSME3-N151Y as well as its capacity to bind to the 20S proteasome in cellulo. As shown in ) and Fig. S4A, the mutated and WT exogenous PSME3 regulators are both localized in the nucleus. Since we did not observe any differences in the localization that could explain the differences in the production of MHC class I epitopes between the cell lines expressing the mutated or the WT regulators, we determined whether mutated Flag-PSME3-N151Y retained the capacity to bind to the nuclear 20S proteasome in cellulo. To achieve this goal, we used proximal ligation assay (PLA)-labeled secondary antibodies, which allow the detection of two primary antibodies in close proximity. Using a combination of PLA-labeled anti-Flag and anti-proteasomal α4 antibodies, we observed a specific signal in the nuclear compartments of cells expressing the mutated and PSME3-WT constructs, revealing the co-localization of both form of PSME3 with the 20S proteasome () and Fig. S4B). Furthermore, as a control, anti-Flag alone under these conditions produced no PLA reaction, and no positive signals were detected in non-transfected cells (), top panel). We confirmed further the interaction between PSME3 WT and mutated PSME3-N151Y with the 20S core proteasome by coimmunoprecipitation using an antibody directed against the PSME3 regulator ()). Localization and capacity to bind to the 20S core were equivalent between exogenous PSME3 WT and mutated PSME3-N151Y, again demonstrating that the PSME3-20S proteasome complex is involved in the processing/degradation of PTP-derived antigenic peptides.
Figure 4. PSME3 regulates the nuclear proteasomal pathway. (a) Western blotting was performed to analyze and quantify PSME3, REGα and p21 protein expression in A375cXI cells (relative to the housekeeping protein β-actin). The cells were transfected with a construct expressing PSME3 WT or the mutated PSME3 N151Y at different concentrations or with a corresponding empty construct for 48 h. Protein levels are indicated below each gel. (b) A375cXI cells transiently expressing the mouse H-2Kb molecules and the intron-derived SL8 epitope. The same cells were co-transfected with increasing amounts (from 0 to 0.5 μg) of constructs expressing Flag-PSME3 WT or the mutated Flag-PSME3-N151Y at different concentrations and then incubated with the B3Z T cell hybridoma for 16 h. Data are the average of at least three independent experiments ± SD minus the values from mock-transfected cells. (c) A375cXI cells were transfected with a construct expressing a Flag-PSME3 WT or a Flag-PSME3 N151Y or a Flag-PSME3-ΔNLS. The Flag tag was stained with Alexa Fluor 488, and the nuclei were stained with DAPI. Cells were analyzed by confocal microscopy. As expected, the exogenous PSME3 WT and PSME3-N151Y proteins were localized in the nuclei, when PSME3ΔNLS was localized in the nucleus and in the cytoplasm. (d) The interactions of the 20S proteasome with the exogenous Flag-PSME3 WT or a Flag-PSME3-N151Y or a Flag-PSME3-ΔNLS constructs were analyzed using DuoLink. PLA was performed using an antibody against the Flag tag and α4 subunit – of the 20S proteasome. Staining was analyzed by confocal microscopy. (e) Immunoprecipitation was performed using the PSME3 antibody, and Western blotting was carried out using an antibody against the 20S α6 subunit. For that purpose the A375cXI CRISPR cell line was transfected with the different Flag-PSME3 constructs and a comparison was made with the endogenous PSME3 in the A375 for the capacity of the Flag construct to bind to the 20S core compare to the endogenous PSME3. This experiment confirmed the interaction of the 20S proteasome and exogenous PSME3 WT or PSME3-N151Y. (f) Western blot analysis and quantification (relative to the housekeeping protein β-actin) of PSME3, REGα and p21 protein levels in A375c.XI cells transfected with a construct expressing PSME3-WT or PSME3-ΔNLS at different concentrations or with a corresponding empty construct for 48 h. Protein levels are indicated below each gel. (g) A375cXI cells transiently expressing the mouse H-2Kb molecules and intron-derived SL8 epitope. The same cells were co-transfected with increasing amounts (from 0 to 0.5 μg) of constructs expressing Flag-PSME3 WT or the mutated Flag- PSME3-ΔNLS at different concentrations and were incubated with the B3Z T cell hybridoma for 16 h. Data are the average of at least three independent experiments ± SD minus the values from mock-transfected cells.
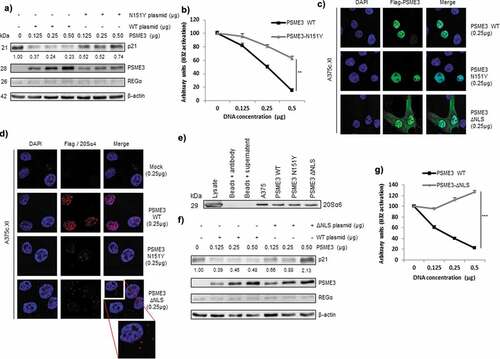
To further define the compartment hosting PSME3-dependent PTP degradation, we generated an exogenous Flag-PSME3 construct that did not contain a nuclear localization signal (NLS). Unlike PSME3-WT, PSME3-ΔNLS was expressed throughout the cell and was not anymore confined to the nucleus () and Fig. S4A). The combination of PLA-labeled anti-Flag and anti-proteasomal α4 antibodies produced a specific signal in the cytoplasmic compartments of cells expressing the PSME3-ΔNLS and PSME3 WT constructs. We also observed staining corresponding to an interaction between PSME3-ΔNLS and the 20S core in the nucleus. This result indicates that PSME3-ΔNLS, which was localized throughout the cell, retained the capacity to bind to the 20S proteasome in both the cytoplasm and the nucleus (), left bottom panel and Fig. S4B). We also confirmed the interaction between PSME3 WT and PSME3-ΔNLS with the 20S core proteasome by coimmunoprecipitation using an antibody directed against the PSME3 regulator ()). Moreover, Cas9-A375 PSME3-knockout cell line (A375cXI) cells transfected with PSME3-ΔNLS slightly prevented nuclear 20S proteasome degradation of the p21 protein in a PSME3-dependent manner compared to cells transfected with PSME3 WT ()). This result suggests that the low levels of PSME3-ΔNLS in the nucleus retained the capacity to partially activate the nuclear 20S proteasome and, furthermore, demonstrate that the degradation of the p21 protein is a nuclear and not a cytoplasmic event ()). In parallel, when we examined the effect of the partial relocalization of PSME3-ΔNLS in the cytoplasm on the antigenic presentation pathway, we observed an increase in PTP-dependent antigen presentation compared to the loss of antigen presentation in cells expressing exogenous nuclear PSME3 WT ()).
All these results hence demonstrate that PSME3 is involved in PTP-dependent antigen presentation by binding to the nuclear 20S proteasome.
PSME3 promotes the degradation of MHC class I PTP-derived antigenic epitopes
The above results raised at least two alternative hypothesis regarding the role of PSME3 in the PTP-dependent proteasomal degradation pathway: i) PSME3 may inhibit 20S proteasomal peptidase activities responsible for producing antigenic peptides with the correct characteristics (i.e. length and anchor residues) to serve in MHC-I pathway, with a consequent decrease in production of PTP-dependent epitopes; or ii) PSME3 may stimulate specific 20S proteasomal activities that cause further degradation of the correct size MHC-I antigenic epitopes. Recently, we showed that inhibition of the proteasomal pathway with epoxomicin leads to increased amounts of unprocessed PTPs in cells and promotes the cross-presentation of longer polypeptides compared with untreated cells.Citation8 To test the first hypothesis, we transfected different cancer cell lines, normal MRC5 fibroblasts and the Cas9-A375 PSME3-knockout cell line with the Glob-intron-SL8 construct and incubated them with mouse BMDCs for 24 h. We observed a reduction in B3Z activation when the tumor cell lines were incubated with BMDCs compared to normal MRC5 cells while A3cXI showed enhanced antigen presentation ()). The decrease in cross-presentation in the different cell lines overexpressing PSME3 suggested that PSME3 did not inhibit PTP processing since the accumulation of unprocessed polypeptides would have been a source of material for the exogenous MHC-I pathway. To confirm that PSME3 did not inhibit the capacity of 20S proteasome to hydrolyze long polypeptides, we assessed the effects of this regulator on the 20S core particle cleaving properties by using an in-vitro degradation assay. For that purpose, a 45-mer (MP-45) and a 52-mer (KH-52) precursor peptides containing the SIINFEKL epitope as well as a 46-mer (MP-46) and a 53-mer (KH-53) precursor peptides containing the MBP(79–87) epitope were synthesized (Fig. S5A). The 45-mer and the 46-mer precursor peptides correspond to the amino acid sequence of the β-globin exon region in which the SL8 or the MBP(79–87) epitopes are introduced in the construct used in ). The 52-mer and the 53-mer precursor peptides correspond to the amino acid sequence of the β-globin intron region in which the SL8 and the MBP(79–87) epitope are introduced in the same construct used in ). Importantly, preliminary studies demonstrated that the commercial enzymes used in these experiments were active and free of contaminant proteases, as shown by the ability of epoxomicin to completely inhibit the chymotrypsin- and trypsin-like activities of these preparations (Table S1). Moreover, although it is well known that the free 20S particle is a relatively inactive protease, presumably because the N-terminal tails of its α subunits obstruct the two opposite axial pores through which substrates access the internal catalytic lumen,Citation55 the latency of unliganded 20S proteasome is not absolute. Indeed, even in the absence of artificial treatments that are known to activate it,Citation56 the 20S core particle degrades proteins at detectable and reproducible rates, probably by a mechanism involving transient and/or only partial channel opening.Citation57,Citation58 Furthermore, in line with these in vitro findings, several studies indicate that a significant fraction of cellular proteins are also degraded in vivo by 20S proteasomes in an ubiquitin- and ATP-independent process.Citation59–Citation61 Hence, KH-52 and KH-53 were incubated in vitro with human constitutive 20S proteasome at 37°C for several hours, and the rates of peptide bond hydrolysis were assessed by measuring the generation of new amino groups with fluorescamine.Citation37 Under these conditions, KH-52 and KH-53 were clearly degraded by h20Sc at linear rates over the entire time course of incubation ()). Moreover, the rates of peptide bond hydrolysis were not appreciably enhanced in the presence of PSME3 and appeared to be only slightly lower than the rates measured when the proteasome associated with REGα/β (), compare blue and red curves, left and right panels). Furthermore, indicative of absolute proteasome-dependent degradation, peptide hydrolysis was completely prevented in the presence of 20 µM epoxomicin (), green curve, left and right panels). Similar results were also obtained when MP-45 and MP-46 precursor peptides were incubated under the same experimental conditions ()) and when KH-52 was hydrolyzed by 20S immunoproteasomes alone or when associated with PSME3 (Fig. S5B).
Figure 5. PSME3 promotes the degradation of MHC-I PTP-derived antigenic epitopes. (a) A375, WM3526, WM3682, HT29, T84, A549, MRC5 and A375cXI cell lines expressing the Glob-intron-SL8 construct (0.5 μg) were cultured with BMDCs for 24 h. The BMDCs were then co-cultured with the SL8-specific CD8+T cell hybridoma (B3Z) for 16 h, and T cell activation was estimated by measuring β-galactosidase levels. The data show the average of at least three independent experiments ± SD minus the values from mock-transfected cells. *** p <.001, ** p <.01, * p <.05 (unpaired t-test). Hydrolysis rates of KH-52 and KH-53 (b) and MP-45 and MP-46 (c) peptides. Precursor substrates were incubated with human 20S constitutive proteasomes (h20Sc) alone or activated by PSME3 or REGα/β, and the amino groups released were measured with fluorescamine at the indicated time points. Data are representative of three independent experiments. (d) Heatmap comparison of the abundance of significant peptides generated after 8 hours from proteasomal degradation of KH-52 precursor peptide in the h20Sc (biological and technical replicates of samples designated A and C, respectively) and PSME3-h20Sc (biological and technical replicates of samples designated B and D, respectively) samples. Samples were analyzed by nLC-MS/MS and processed by MaxQuant against the database KH-52 Intron SIINKEL + contaminant sequences. Differences and similarities in peptide intensities (normalized to the Z-score) are shown; green indicates decreased levels, and red indicates increased levels. Data were obtained from supervised hierarchical clustering analysis by applying a t-test, the Benjamini Hochberg correction and a p-value of 0.05. (e) Box plot of SIINFKEL peptide intensity calculated by MaxQuant in the h20Sc (in green) and PSME3-h20Sc (in blue) samples. (f) Cross presentation assay of peptide products generated during hydrolysis by 20S proteasome of KH-52 peptide precursor in the presence or absence of PSME3. *** p <.001 (unpaired t-test).
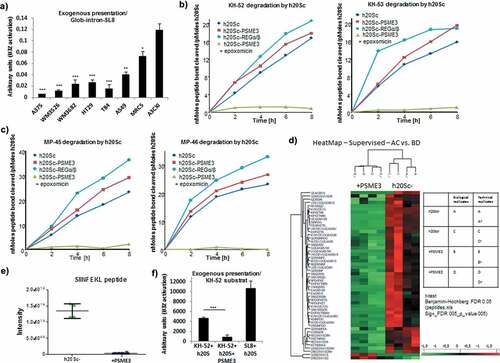
We then assessed the second hypothesis namely that PSME3-dependant stimulation of the 20S proteasome leads to complete degradation of MHC class I epitopes. In fact, although binding of PSME3 did not result in significantly increased rates of substrate hydrolysis (,)), it may modify proteasome cleavage specificities to generate different patterns of peptide products as already extensively demonstrated for REGα/βCitation36 Such a change in enzymatic properties may potentially affect the formation of specific antigenic peptides such as SIINFEKL. To test directly this possibility, peptides released during KH-52 hydrolysis by equimolar amounts of 20S with and without PSME3 were analyzed by tandem mass spectrometry (MS/MS). Importantly, shorter peptides (representing the great majority of proteasomal productsCitation36,Citation62,Citation63 were excluded to minimize false-positive identification and signals originating from small chemical compounds and therefore were not identified or quantified. In contrast, peptides with the correct size to bind to MHC-I heterodimers (i.e., 8–10 mers) were accurately analyzed. Using this approach, we identified 52 different peptides from KH-52, ranging in length from 8 to 16 residues and derived from the entire sequence of the substrate. Although MS/MS does not provide quantitative information regarding the absolute abundance of the peptides detected, it is possible to assess their relative amounts by comparing the corresponding ion intensities measured in sequential MS/MS analyses. Therefore, we used ion intensities to quantify the relative amounts of single fragments generated from KH-52 in both degradation reactions. Remarkably, this analysis demonstrated that different amounts of peptide products are released by 20Sc and PSME3-20S ()). Most importantly, we demonstrated that the generation of SIINFEKL was strikingly suppressed in the presence of PSME3, as inferred by a two-log value reduction in its ion intensity ()). This important finding was subsequently confirmed by a cross presentation assay which demonstrated that the mixture of peptide products generated during KH-52 peptide precursor hydrolysis by PSME3-20S proteasome was nearly unable to activate a SIINFEKL-specific T cell hybridoma unlike the mixture of peptides released by 20S alone ()).
All these results demonstrate that the regulator PSME3 has the capacity to modify the cleavage properties of the 20S proteasome in the nuclear compartment in such a way that PTP-derived MHC class I antigenic peptides are destroyed rather than being correctly processed and released.
Knockout of PSME3 gene causes tumor growth defect
Given the overexpression of PSME3 in human cancer cell lines compared to the human healthy MRC5 lung fibroblast cell line, we decided to look at the PSME3 mRNA and protein levels in the mouse MCA205 sarcoma cell line in comparison with the mouse normal B6 skin fibroblast cell line. We observed by qRT-PCR that PSME3 mRNA level was notably upregulated in MCA205 tumor cell line compared to the B6 cell line (Fig. S6A). Furthermore, we noticed an overexpression of PSME3 protein in MCA205 sarcoma cell line compared to B6 cell line (), upper panel). To evaluate whether the difference in PSME3 expression in murine cell lines contributes to antigen production and presentation, MCA205 murine sarcoma and B6 skin fibroblast cell lines were transiently expressing the Globin-SL8-intron construct and their ability to activate B3Z hybridoma was assessed by T-cell assay. We observed that antigen presentation in MCA205 sarcoma cancer cell line was lower than that in the normal fibroblast cell line supporting our previous results with the human cancer cell lines (), lower panel). To further depict the role of PSME3 in tumorigenicity and cancer immune response, we genetically deleted PSME3 in the MCA205 sarcoma cancer cell line using the CRISPR/Cas9 technology (), upper panel and Fig. S6B). The Cas9-PSME3 MCA205 clones MCA-c13, MCA-c19 and MCA-c21 and the MCA205 WT cells were then transiently transfected with the Globin-SL8-intron construct and their ability to activate B3Z hybridoma was assessed by T-cell assay. We observed that Cas9-PSME3 MCA205 clones induce a stronger activation of B3Z hybridoma compared to the parental murine cell line (), lower panel). Subsequently, we acid stripped cell surface class I molecules and measured the recovery of surface class I expression. The Cas9-PSME3 MCA205 clone MCA-c13 does not show any increase in MHC-I Kb molecules at the cell surface compare to the MCA205 WT cancer cell line (Fig. S6C). Rather, a decrease both in the steady-state level and in the recovery of MHC- I molecules is observed in the Cas9-PSME3 MCA205 clone while those cells elicit a better cancer immune response. This result convincingly supports the idea that the effect of PSME3 on antigen presentation takes place earlier than the loading of the antigenic peptides and the exporting steps of the MHC class I peptide complex.
Figure 6. Knockout of PSME3 gene causes tumor growth defect. (a) The B6 fibroblast and MCA205 sarcoma cell lines were transfected with β-Glob-intron-SL8 constructs. Cells were co-cultured with the SL8-specific CD8+T cell hybridoma (B3Z) for 16 h. Upper panel shows the protein level of PSME3 in the tumor cells assessed by Western Blot (quantification relative to the housekeeping protein β-actin). Lower panel shows the relative level of antigen presentation in the tumor cells. (b) The MCA205 sarcoma cell line and the Cas9-PSME3 MCA205 clones MCAc13, MCAc19 and MCAc21 were transfected with the β-Glob-intron-SL8 construct for 48 h. The cells were then co-cultured with the SL8-specific CD8 + T cell hybridoma (B3Z) for 16 h. Upper panel shows the protein level of PSME3 in the tumor cells assessed by Western Blot (quantification relative to the housekeeping protein β-actin). Lower panel shows the relative level of antigen presentation in the tumor cells. (c) Tumor size in area of sarcoma MCA205 WT and Cas9-PSME3 MCA205 clones subcutaneously inoculated into the right flank of immunocompetent C57BL/6 mice. Data are given as mean ± SEM of 12 mice per group. (d) Tumor size in area of sarcoma MCA205 WT and Cas9-PSME3 MCA205 clones subcutaneously inoculated into the right flank of immunodeficient Nu/Nu mice or immunocompetent C57BL/6 mice (left panel). Relative growth of MCA205 WT and Cas9-PSME3 MCA205 clones in immunodeficient Nu/Nu mice compared to immunocompetent C57BL/6 mice. Data are given as mean ± SEM of 6 mice per group. *p <.05, **p <.01 (ANOVA with Tukey’s multiple comparaison test comparing all groups).
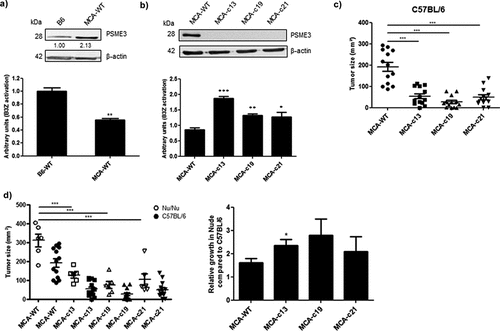
The proteasome regulator PSME3 has been reported to have a specific effect on tumorigenicity. In fact, it has been shown that PSME3 contributes to the proliferation and metastasis of different cancer types. To gauge the effect of PSME3 on cancer immune response and tumorigenicity in vivo, C57BL/6 mice were subcutaneously injected into the flank with MCA205 WT cells or with several Cas9-PSME3 MCA205 clones. As shown in ) and in figure S6D (upper panels), the Cas9-PSME3 MCA205 clones lead to tumors smaller than the parental cell line at day 20 after challenge. This result indicates that the loss of PSME3 induces a decrease in the tumorigenicity of sarcoma cells. To validate that this decrease in tumorigenicity was due, at least in part, to a specific activation of the cancer immune response, as we observed in vitro, nude mice were subcutaneously injected with MCA205 cells or several Cas9-PSME3 MCA205 clones. We observed that the relative tumor growth in Nude mice vs C57BL/6 mice was higher for the Cas9-PSME3 MCA205 clones than MCA205 WT () and Fig. S6D). This difference suggests that the enhanced eradication of Cas9-PSME3 MCA205 clones compared to MCA205 WT in immunocompetent mice is partly mediated by the adaptive immune system. PSME3 play therefore an important role in antigen presentation, tumorigenicity and antitumor immune responses.
Discussion
This study describes for the first time a distinct and unexpected role of the PSME3-proteasome complex, which proved to be responsible for the degradation of MHC-I peptides in cancer. Specifically, we demonstrate an inverse correlation between PSME3 expression and MHC-I antigen presentation in cancers, thus revealing a new function of this regulator in blunting immune responses by modulating the activity of the proteasome processing pathway in vitro and in vivo.
In contradiction with our results, Barton et al. demonstrated, approximately twenty years ago, that PSME3-/- mice did not show notable impairment in antigen presentation after viral infection.Citation30 The only observed phenotypes reported so far for PSME3-/- mice are a small reduction in the number of specific CD8+ T lymphocytes and growth retardation.Citation30,Citation31 In this study, we clearly demonstrate for the first time a specific role for PSME3 in MHC-I antigen processing and presentation. First, we show that PSME3 acts as a negative regulator of MHC-I antigen presentation in cancer by destroying in vitro and in cellulo MHC-I peptides generated during proteasomal degradation of PTPs. Furthermore, our data suggest that overexpression of PSME3 negatively affects cancer immune response in vivo. Considering our results, it is therefore not surprising that Barton et al. have not observed any effect on antigen presentation in mice lacking PSME3. Indeed, we observed a defect in MHC-I antigen presentation only in cancer cell lines which naturally overexpress PSME3. The knock-down of PSME3 in different cancer cell lines using either siRNA, miRNA-7 overexpression, cisplatin treatment or the CRISPR/Cas9 technology rescued the ability of the tumor cells to activate CD8+ T cells in-vitro and enhanced the antitumor response in vivo by inducing the presence of tumor associated antigen from different sequences.
It is generally believed that only REGα/β in association with the immunoproteasome can affect the production of MHC-I epitopes. Strikingly, our results demonstrate a close correlation between the expression of PSME3 and tumor immune evasion. Thus, not only the heteroheptamer REGα/β plays a role in antigen presentation but so does PSME3, in an opposite manner. In fact, we demonstrated that knocking out of PSME3 could reduce the growth of MCA205 sarcoma tumors in immunocompetent mice. However, when we performed the same experiment in nude mice which are deficient in T cells but not in B or innate immune cells, we did observe an increased tumor growth of several Cas9-PSME3 MCA205 clones compared to the MCA205 WT cells that overexpress PSME3. This experiment indicates that the knockout of PSME3 could induce the presentation of new PTP-MIPs that are recognized by CD8+T cells and thus delay tumor growth.
Interestingly, a recent study reported that PSME3 can affect MHC class I presentation by decreasing the expression levels of immunoproteasomes through a molecular mechanism involving modulation of the JAK-STAT pathway obtained via enhancement of proteasomal degradation of phosphorylated STAT-3 in bone marrow-derived and splenic DC, MEF and HeLa cells. However, in the mouse and human cell lines tested in our study, we could not detect any major effect of PSME3 on the expression levels of immunoproteasome in basal conditions or under INFγ stimulation (Fig. S7). These findings, therefore, suggest that different molecular mechanisms control immunoproteasome expression in cells of diverse origin and emphasize the importance of further studies to systematically investigate the exact role of the JAK-STAT pathway in modulating cell-mediated class I immune responses in different cell types and in physiological and pathological contexts. In this regard, it is worth noting that the expression of immunoproteasome in tumor cells is extremely variable, without a general rule but with large differences depending on the different types of tumors analyzed.Citation64,Citation65
As mentioned above, many studies have reported that PSME3 is overexpressed in several cancers.Citation40–Citation42 This overexpression constitutes a beneficial setting for tumors to proliferate and become metastatic. In fact, PSME3 was recently shown to be involved in the degradation of some regulatory proteins. For example, the cyclin-dependent kinase inhibitor p21 is specifically degraded in an ATP- and ubiquitin-independent process via the PSME3-proteasomal pathway.Citation18 Furthermore, two other cyclin-dependent kinases inhibitors, p16 and p14, are reportedly degraded via the same pathway.Citation17 Even more importantly, PSME3 has been shown to be involved in the MDM2-mediated p53 degradation process.Citation20 These findings demonstrate the important role of PSME3 in tumorigenicity by regulating cell proliferation and apoptosis. In this study, we demonstrate a new role for PSME3 in tumor growth and development. Indeed, PSME3 overexpression not only promotes cancer progression but it reduces the amount of polypeptides that can serve as MHC-I epitopes, thus impairing the recognition of cancer cells by the immune system.
The REGα/β complex is mainly located in the cytoplasm, while PSME3 is mostly a nuclear proteasomal regulator.Citation15 PSME3 has been shown to be associated with the 20S proteasome in nuclear speckles.Citation22 Moreover, many reports have demonstrated that the 20S proteasome and immunoproteasome are present in the nucleus and more specifically in clastosome structures.Citation66 Considering that PTPs are produced by a nuclear translation event, it is clearly more beneficial for cells to process the different PTPs in the same compartment where they are produced. Cancer cell stealth is also promoted if TA-PTPs are rapidly destroyed where they are produced and do not have a chance to be properly processed in the cytoplasm and loaded on MHC I molecules. Therefore, by overexpressing PSME3 in the nucleus where PTPs are located, cancer cells add another layer of regulation to the immune response. Indeed, we were able to restore a proper immune response against TA-PTPs by manipulating the localization of PSME3 with the ΔNLS construct. This identification of a specific site for TA-PTP degradation reinforced our recent hypothesis that under normal conditions peptides for the MHC-I presentation pathway are produced and processed in the nucleus while under abnormal conditions such as cancer development, the TA-PTPs produced in the nucleus are no longer processed by the standard 20S proteasome, the immunoproteasome or the REGα/β-proteasomal complex but rather by the PSME3-proteasome complex which minimizes the generation of MHC-I epitopes.Citation13,Citation67–Citation70
Although the heptameric REGα/β has been reported to stimulate all three proteasomal activities,Citation12 the effect of PSME3 on proteasomal cleaving specificities appear to be more complex and strictly related to the exact sequence of the fluorogenic short peptides used to assess them.Citation28 The heptameric REGα/β regulator binds to 20S proteasome and immunoproteasome and in this way enhances generation of some, but not all, MHC-I epitopes.Citation14 Two different explanations might be proposed to explain the ability of PSME3 to decrease PTP-dependent antigen presentation: PSME3 might (i) inhibit PTP processing via the proteasome complex or (ii) enhance the destruction by 20S proteasome of MHC-I epitopes embedded in PTPs. Here, we propose that the binding of PSME3 to the nuclear 20S proteasome stimulates the degradation, rather than the proper processing, of MHC-I antigenic peptides by changing the peptidase activities of the 20S core particle. In vitro degradation assays clearly showed that several polypeptides (ranging in length between 45 and 53 residues) with sizes and sequences corresponding to different PTPs are efficiently hydrolyzed by PSME3-20S proteasomes at rates only slightly lower than those obtained with REGα/β-20S particles. These experiments rule out the possibility that PSME3, by binding to 20S proteasome, may simply act as general inhibitor of proteasomal degradation of PTPs. Interestingly, the same kinetics of substrate hydrolysis were obtained when the polypeptide precursor was degraded by 20S immunoproteasomes in the presence or absence of PSME3, which suggests that the activator acts in a similar manner on the two proteasomal species, although further studies will be required to clarify this important point in greater detail. In line with the alternative hypothesis that PSME3 may induce destruction, rather than proper processing, of specific antigenic peptides, MS analysis demonstrated that 20S proteasome is able to efficiently process and release the immunodominant epitope SIINFEKL embedded in the precursor peptide KH-52, corresponding to the sequence of the β-globin intron region. Crucially, however, when the precursor peptide KH-52 was degraded by the 20S proteasome activated by PSME3, the generation of the immunodominant epitope SIINFEKL was completely abolished. Importantly, degradation assays were performed under conditions that have previously been shown to allow generation of individual peptides at linear rates for several hours.Citation71 Therefore, although our MS semiquantitative analysis precluded the recognition of small peptides generated following the fragmentation of SIINFEKL, the stimulation of proteasomal peptidase activities, resulting in the destruction of the antigenic peptide, appeared to be the more likely explanation for the inability to detect the immunodominant epitope in the presence of PSME3.
We recently reported that PTPs are a major source of MHC-I antigenic peptides for the endogenous and exogenous pathways. In fact, by inhibiting PTP degradation by the proteasome in tumor cell lines, we were able to increase the amounts of unprocessed PTPs entering the exogenous pathway, resulting in an increase in the activation of naïve CD8+ T cells.Citation7,Citation8 The role we unveiled for PSME3 in the degradation of PTPs and specifically its ability to inhibit the endogenous MHC-I antigen presentation from TA-PTPs in tumors, leads to the hypothesis that this mechanism might also affect PTP-dependent exogenous MHC-I presentation. Indeed, by overexpressing PSME3 in a CRISPR PSME3 knockout cell line, we showed that this cell line was no longer able to induce the proliferation of CD8+ T cells via the exogenous pathway. Moreover, mutated and ΔNLS-PSME3 constructs played a less active role in PTP degradation, indicating that the main function of PSME3 was to modulate the activity of the nuclear proteasomal complex, with strong inhibitory consequences not only on MHC-I endogenous antigen presentation but also on MHC-I exogenous pathway. Thus, we are driven to consider the PSME3-dependent downregulation of MHC-I presentation in cancer cells as evolutionary advantages allowing cancer cells to ensure that TA-PTPs have no chances to enter the MHC-I exogenous pathway and so to induce the proliferation of naïve CD8+ T cells.
Furthermore, it has been recently shown that retained intron can be an important source of tumor neo-antigens.Citation72 In a previous study we have reported that PTP-derived antigens can arise from intronic but also from exonic sequences, independently of mRNA stability or processing by the NMD pathway.Citation7 We demonstrated here that PSME3 affects the presentation of epitopes derived from both exonic and intronic sequences. This suggests that PSME3 could degrade antigens that arise from the canonical translation of exons and retained introns in the cytoplasm in addition to the PTP-derived antigens produced in the nuclear compartment. In a near future it will be interesting to conduct a proteogenomic analysis to gauge the effect of PSME3 downregulation on the presence of exon- and intron-derived antigens in the MHC-I immunopeptidome of cancer cells.
In conclusion, the results of the present study, together with those of previous studies investigating PSME3 functions, support the idea that tumor cells have acquired an established system to positively regulate PSME3 expression enabling them (i) to control the cell cycle arrest, (ii) to control their proliferation and survival and most importantly (iii) to inhibit the endogenous and exogenous MHC-I antigen presentation pathways by specifically degrading TA-PTP-encoded MHC-I epitopes. These results potentially serve as a starting point for the development of new therapies aiming at decreasing intracellular levels of PSME3 thus enhancing the production of tumor-associated antigens and stimulating specific immune responses against cancer.
Disclosure of potential conflicts of interest
The authors declare no potential conflicts of interest.
Supplemental Material
Download ()Acknowledgments
We thank Dr. Xiaotao Li (Department of Biochemistry and Molecular Biology, Institute of Biomedical Sciences, Shanghai, China) for the PSME3 expression plasmids. We would like to thanks Dr. Lea Eisenbach and Dr. Joan M. Goverman for supplying respectively the Busa14 hybridoma and the MBP(79-87) hybridoma. We would like to gratefully acknowledge Dr. Meenhard Herlyn for supplying several melanoma short-term cultures for these studies. This work was funded by The ATIP/Avenir program (INSERM), La Fondation ARC and Fondation Gustave Roussy. BM is supported by Fondation Gustave Roussy, PA is supported by the Université Paris Sud and DR is supported by the Philanthropia Fondation.
Supplementary Material
Supplemental data for this article can be accessed on the publisher’s websie.
Additional information
Funding
References
- Joffre OP, Segura E, Savina A, Amigorena S. Cross-presentation by dendritic cells. Nat Rev Immunol. 2012;12:557–16.
- Yewdell JW, Anton LC, Bennink JR. Defective ribosomal products (DRiPs): a major source of antigenic peptides for MHC class I molecules? J Immunol. 1996;157:1823–1826.
- Coulie PG, Lehmann F, Lethe B, Herman J, Lurquin C, Andrawiss M, Boon T. A mutated intron sequence codes for an antigenic peptide recognized by cytolytic T lymphocytes on a human melanoma. Proc Natl Acad Sci U S A. 1995;92:7976–7980. doi:10.1073/pnas.92.17.7976.
- Guilloux Y, Lucas S, Brichard VG, Van Pel A, Viret C, De Plaen E, Brasseur F, Lethé B, Jotereau F, Boon T, et al. A peptide recognized by human cytolytic T lymphocytes on HLA-A2 melanomas is encoded by an intron sequence of the N-acetylglucosaminyltransferase V gene. J Exp Med. 1996;183:1173–1183. doi:10.1084/jem.183.3.1173.
- Shastri N, Cardinaud S, Schwab SR, Serwold T, Kunisawa J. All the peptides that fit: the beginning, the middle, and the end of the MHC class I antigen-processing pathway. Immunol Rev. 2005;207:31–41. doi:10.1111/j.0105-2896.2005.00321.x.
- Apcher S, Daskalogianni C, Lejeune F, Manoury B, Imhoos G, Heslop L, Fahraeus R. Major source of antigenic peptides for the MHC class I pathway is produced during the pioneer round of mRNA translation. Proc Natl Acad Sci U S A. 2011;108:11572–11577. doi:10.1073/pnas.1104104108.
- Apcher S, Millot G, Daskalogianni C, Scherl A, Manoury B, Fahraeus R. Translation of pre-spliced RNAs in the nuclear compartment generates peptides for the MHC class I pathway. Proc Natl Acad Sci U S A. 2013;110:17951–17956. doi:10.1073/pnas.1309956110.
- Duvallet E, Boulpicante M, Yamazaki T, Daskalogianni C, Prado Martins R, Baconnais S, Manoury B, Fahraeus R, Apcher S. Exosome-driven transfer of tumor-associated Pioneer Translation Products (TA-PTPs) for the MHC class I cross-presentation pathway. Oncoimmunology. 2016;5(9):e1198865. doi:10.1080/2162402X.2016.1198865.
- Collins GA, Goldberg AL. The Logic of the 26S Proteasome. Cell. 2017;169:792–806. doi:10.1016/j.cell.2017.04.023.
- Kish-Trier E, Hill CP. Structural biology of the proteasome. Annu Rev Biophys. 2013;42:29–49. doi:10.1146/annurev-biophys-083012-130417.
- Leone P, Shin EC, Perosa F, Vacca A, Dammacco F, Racanelli V. MHC class I antigen processing and presenting machinery: organization, function, and defects in tumor cells. J Natl Cancer Inst. 2013;105:1172–1187. doi:10.1093/jnci/djt184.
- Whitby FG, Masters EI, Kramer L, Knowlton JR, Yao Y, Wang CC, Hill CP. Structural basis for the activation of 20S proteasomes by 11S regulators. Nature. 2000;408:115–120. doi:10.1038/35040607.
- Cascio P, Call M, Petre BM, Walz T, Goldberg AL. Properties of the hybrid form of the 26S proteasome containing both 19S and PA28 complexes. Embo J. 2002;21:2636–2645. doi:10.1093/emboj/21.11.2636.
- Cascio P. PA28alphabeta: the enigmatic magic ring of the proteasome? Biomolecules. 2014;4:566–584. doi:10.3390/biom4020566.
- Soza A, Knuehl C, Groettrup M, Henklein P, Tanaka K, Kloetzel PM. Expression and subcellular localization of mouse 20S proteasome activator complex PA28. FEBS Lett. 1997;413:27–34. doi:10.1016/S0014-5793(97)00864-8.
- Wojcik C, Tanaka K, Paweletz N, Naab U, Wilk S. Proteasome activator (PA28) subunits, alpha, beta and gamma (Ki antigen) in NT2 neuronal precursor cells and HeLa S3 cells. Eur J Cell Biol. 1998;77:151–160. doi:10.1016/S0171-9335(98)80083-6.
- Chen X, Barton LF, Chi Y, Clurman BE, Roberts JM. Ubiquitin-independent degradation of cell-cycle inhibitors by the REGgamma proteasome. Mol Cell. 2007;26:843–852. doi:10.1016/j.molcel.2007.05.022.
- Li X, Amazit L, Long W, Lonard DM, Monaco JJ, O’Malley BW. Ubiquitin- and ATP-independent proteolytic turnover of p21 by the REGgamma-proteasome pathway. Mol Cell. 2007;26:831–842. doi:10.1016/j.molcel.2007.05.028.
- Li X, Lonard DM, Jung SY, Malovannaya A, Feng Q, Qin J, Tsai SY, Tsai M-J, O’Malley BW. The SRC-3/AIB1 coactivator is degraded in a ubiquitin- and ATP-independent manner by the REGgamma proteasome. Cell. 2006;124:381–392. doi:10.1016/j.cell.2005.11.037.
- Zhang Z, Zhang R. Proteasome activator PA28 gamma regulates p53 by enhancing its MDM2-mediated degradation. Embo J. 2008;27:852–864. doi:10.1038/emboj.2008.25.
- Chai F, Liang Y, Bi J, Chen L, Zhang F, Cui Y, Bian X, Jiang J. High expression of REGy is associated with metastasis and poor prognosis of patients with breast cancer. Int J Clin Exp Pathol. 2014:7834–43 p.
- Baldin V, Militello M, Thomas Y, Doucet C, Fic W, Boireau S, Jariel-Encontre I, Piechaczyk M, Bertrand E, Tazi J, et al. A novel role for PA28gamma-proteasome in nuclear speckle organization and SR protein trafficking. Mol Biol Cell. 2008;19:1706–1716. doi:10.1091/mbc.e07-07-0637.
- Cioce M, Boulon S, Matera AG, Lamond AI. UV-induced fragmentation of Cajal bodies. J Cell Biol. 2006;175:401–413. doi:10.1083/jcb.200604099.
- Zannini L, Buscemi G, Fontanella E, Lisanti S, Delia D. REGgamma/PA28gamma proteasome activator interacts with PML and Chk2 and affects PML nuclear bodies number. Cell Cycle. 2009;8:2399–2407. doi:10.4161/cc.8.15.9084.
- Fort P, Kajava AV, Delsuc F, Coux O. Evolution of proteasome regulators in eukaryotes. Genome Biol Evol. 2015;7:1363–1379. doi:10.1093/gbe/evv068.
- Boulon S, Westman BJ, Hutten S, Boisvert FM, Lamond AI. The nucleolus under stress. Mol Cell. 2010;40:216–227. doi:10.1016/j.molcel.2010.09.024.
- Welk V, Coux O, Kleene V, Abeza C, Trumbach D, Eickelberg O, Meiners S. Inhibition of Proteasome Activity Induces Formation of Alternative Proteasome Complexes. J Biol Chem. 2016;291:13147–13159. doi:10.1074/jbc.M116.717652.
- Jonik-Nowak B, Menneteau T, Fesquet D, Baldin V, Bonne-Andrea C, Mechali F, Fabre B, Boisguerin P, de Rossi S, Henriquet C, et al. PIP30/FAM192A is a novel regulator of the nuclear proteasome activator PA28gamma. Proc Natl Acad Sci U S A. 2018;115:E6477–E86. doi:10.1073/pnas.1722299115.
- Groettrup M, Kirk CJ, Basler M. Proteasomes in immune cells: more than peptide producers? Nat Rev Immunol. 2010;10:73–78. doi:10.1038/nri2687.
- Barton LF, Runnels HA, Schell TD, Cho Y, Gibbons R, Tevethia SS, Deepe GS, Monaco JJ. Immune defects in 28-kDa proteasome activator gamma-deficient mice. J Immunol. 2004;172:3948–3954. doi:10.4049/jimmunol.172.6.3948.
- Murata S, Kawahara H, Tohma S, Yamamoto K, Kasahara M, Nabeshima Y, Tanaka K, Chiba T. Growth retardation in mice lacking the proteasome activator PA28gamma. J Biol Chem. 1999;274:38211–38215. doi:10.1074/jbc.274.53.38211.
- Shastri N, Gonzalez F. Endogenous generation and presentation of the ovalbumin peptide/Kb complex to T cells. J Immunol. 1993;150:2724–2736.
- Perchellet A, Stromnes I, Pang JM, Goverman J. CD8+ T cells maintain tolerance to myelin basic protein by ‘epitope theft’. Nat Immunol. 2004;5:606–614. doi:10.1038/ni1073.
- Cafri G, Sharbi-Yunger A, Tzehoval E, Eisenbach L. Production of LacZ inducible T cell hybridoma specific for human and mouse gp100(2)(5)(-)(3)(3) peptides. PLoS One. 2013;8:e55583. doi:10.1371/journal.pone.0055583.
- Cascio P, Goldberg AL. Preparation of hybrid (19S-20S-PA28) proteasome complexes and analysis of peptides generated during protein degradation. Methods Enzymol. 2005;398:336–352.
- Raule M, Cerruti F, Benaroudj N, Migotti R, Kikuchi J, Bachi A, Navon A, Dittmar G, Cascio P. PA28alphabeta reduces size and increases hydrophilicity of 20S immunoproteasome peptide products. Chem Biol. 2014;21:470–480. doi:10.1016/j.chembiol.2014.02.006.
- Raule M, Cerruti F, Cascio P. Enhanced rate of degradation of basic proteins by 26S immunoproteasomes. Biochim Biophys Acta. 2014;1843:1942–1947. doi:10.1016/j.bbamcr.2014.05.005.
- Rappsilber J, Ishihama Y, Mann M. Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal Chem. 2003;75:663–670. doi:10.1021/ac026117i.
- Rappsilber J, Mann M, Ishihama Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat Protoc. 2007;2:1896–1906. doi:10.1038/nprot.2007.261.
- Wang X, Tu S, Tan J, Tian T, Ran L, Rodier JF, Ren G. REG gamma: a potential marker in breast cancer and effect on cell cycle and proliferation of breast cancer cell. Med Oncol. 2011;28:31–41. doi:10.1007/s12032-010-9546-8.
- Chai F, Liang Y, Bi J, Chen L, Zhang F, Cui Y, Bian X, Jiang J. High expression of REGgamma is associated with metastasis and poor prognosis of patients with breast cancer. Int J Clin Exp Pathol. 2014;7:7834–7843.
- He J, Cui L, Zeng Y, Wang G, Zhou P, Yang Y, Ji L, Zhao Y, Chen J, Wang Z, et al. REGgamma is associated with multiple oncogenic pathways in human cancers. BMC Cancer. 2012;12:75. doi:10.1186/1471-2407-12-75.
- Yu G, Zhao Y, He J, Lonard DM, Mao CA, Wang G, Li M, Li X. Comparative analysis of REG{gamma} expression in mouse and human tissues. J Mol Cell Biol. 2010;2:192–198. doi:10.1093/jmcb/mjq009.
- Craiu A, Akopian T, Goldberg A, Rock KL. Two distinct proteolytic processes in the generation of a major histocompatibility complex class I-presented peptide. Proc Natl Acad Sci U S A. 1997;94:10850–10855. doi:10.1073/pnas.94.20.10850.
- Sijts EJ, Kloetzel PM. The role of the proteasome in the generation of MHC class I ligands and immune responses. Cell Mol Life Sci. 2011;68:1491–1502. doi:10.1007/s00018-011-0657-y.
- Kisselev AF, Goldberg AL. Proteasome inhibitors: from research tools to drug candidates. Chem Biol. 2001;8:739–758. doi:10.1016/S1074-5521(01)00056-4.
- Schwarz K, de Giuli R, Schmidtke G, Kostka S, van den Broek M, Kim KB, Crews CM, Kraft R, Groettrup M. The selective proteasome inhibitors lactacystin and epoxomicin can be used to either up- or down-regulate antigen presentation at nontoxic doses. J Immunol. 2000;164:6147–6157. doi:10.4049/jimmunol.164.12.6147.
- Giles KM, Brown RA, Ganda C, Podgorny MJ, Candy PA, Wintle LC, Richardson KL, Kalinowski FC, Stuart LM, Epis MR, et al. microRNA-7-5p inhibits melanoma cell proliferation and metastasis by suppressing RelA/NF-κB. Oncotarget. 2016;7:31663–31680. doi:10.18632/oncotarget.9421.
- Xiong S, Zheng Y, Jiang P, Liu R, Liu X, Chu Y. MicroRNA-7 inhibits the growth of human non-small cell lung cancer A549 cells through targeting BCL-2. Int J Biol Sci. 2011;7:805–814. doi:10.7150/ijbs.7.805.
- Kefas B, Godlewski J, Comeau L, Li Y, Abounader R, Hawkinson M, Lee J, Fine H, Chiocca EA, Lawler S, et al. microRNA-7 inhibits the epidermal growth factor receptor and the Akt pathway and is down-regulated in glioblastoma. Cancer Res. 2008;68:3566–3572. doi:10.1158/0008-5472.CAN-07-6639.
- Saydam O, Senol O, Wurdinger T, Mizrak A, Ozdener GB, Stemmer-Rachamimov AO, Yi M, Stephens RM, Krichevsky AM, Saydam N, et al. miRNA-7 attenuation in Schwannoma tumors stimulates growth by upregulating three oncogenic signaling pathways. Cancer Res. 2011;71(3):852–861. doi:10.1158/0008-5472.CAN-10-1219.
- Xiong S, Zheng Y, Jiang P, Liu R, Liu X, Qian J, Gu J, Chang L, Ge D, Chu Y, et al. PA28gamma emerges as a novel functional target of tumour suppressor microRNA-7 in non-small-cell lung cancer. Br J Cancer. 2014;110(2):353–362. doi:10.1038/bjc.2013.728.
- Ali A, Wang Z, Fu J, Ji L, Liu J, Li L, Wang H, Chen J, Caulin C, Myers JN, et al. Differential regulation of the REGgamma-proteasome pathway by p53/TGF-beta signalling and mutant p53 in cancer cells. Nat Commun. 2013;4:2667. doi:10.1038/ncomms3667.
- Kanai K, Aramata S, Katakami S, Yasuda K, Kataoka K. Proteasome activator PA28gamma stimulates degradation of GSK3-phosphorylated insulin transcription activator MAFA. J Mol Endocrinol. 2011;47:119–127. doi:10.1530/JME-11-0044.
- Groll M, Bajorek M, Kohler A, Moroder L, Rubin DM, Huber R, Rubin DM, Huber R. A gated channel into the proteasome core particle. Nat Struct Biol. 2000;7:1062–1067. doi:10.1038/80992.
- Coux O, Tanaka K, Goldberg AL. Structure and functions of the 20S and 26S proteasomes. Annu Rev Biochem. 1996;65:801–847. doi:10.1146/annurev.bi.65.070196.004101.
- Osmulski PA, Hochstrasser M, Gaczynska M. A tetrahedral transition state at the active sites of the 20S proteasome is coupled to opening of the alpha-ring channel. Structure. 2009;17:1137–1147. doi:10.1016/j.str.2009.06.011.
- Osmulski PA, Gaczynska M. Nanoenzymology of the 20S proteasome: proteasomal actions are controlled by the allosteric transition. Biochemistry. 2002;41:7047–7053. doi:10.1021/bi0159130.
- Baugh JM, Pilipenko EV. 20S proteasome differentially alters translation of different mRNAs via the cleavage of eIF4F and eIF3. Mol Cell. 2004;16:575–586. doi:10.1016/j.molcel.2004.10.017.
- Orlowski M, Wilk S. Ubiquitin-independent proteolytic functions of the proteasome. Arch Biochem Biophys. 2003;415:1–5. doi:10.1016/S0003-9861(03)00197-8.
- Davies KJ. Degradation of oxidized proteins by the 20S proteasome. Biochimie. 2001;83:301–310. doi:10.1016/S0300-9084(01)01250-0.
- Kisselev AF, Akopian TN, Woo KM, Goldberg AL. The sizes of peptides generated from protein by mammalian 26 and 20 S proteasomes. Implications for understanding the degradative mechanism and antigen presentation. J Biol Chem. 1999;274:3363–3371. doi:10.1074/jbc.274.6.3363.
- Nussbaum AK, Dick TP, Keilholz W, Schirle M, Stevanovic S, Dietz K, Heinemeyer W, Groll M, Wolf DH, Huber R, et al. Cleavage motifs of the yeast 20S proteasome beta subunits deduced from digests of enolase 1. Proc Natl Acad Sci U S A. 1998;95:12504–12509. doi:10.1073/pnas.95.21.12504.
- Seliger B. Molecular mechanisms of MHC class I abnormalities and APM components in human tumors. Cancer Immunol, Immunother. 2008;57:1719–1726. doi:10.1007/s00262-008-0515-4.
- Cerruti F, Martano M, Petterino C, Bollo E, Morello E, Bruno R, Buracco P, Cascio P. Enhanced expression of interferon-gamma-induced antigen-processing machinery components in a spontaneously occurring cancer. Neoplasia. 2007;9:960–969. doi:10.1593/neo.07649.
- Lafarga M, Berciano MT, Pena E, Mayo I, Castano JG, Bohmann D, Rodrigues JP, Tavanez JP, Carmo-Fonseca M. Clastosome: a subtype of nuclear body enriched in 19S and 20S proteasomes, ubiquitin, and protein substrates of proteasome. Mol Biol Cell. 2002;13:2771–2782. doi:10.1091/mbc.e02-03-0122.
- Guillaume B, Chapiro J, Stroobant V, Colau D, Van Holle B, Parvizi G, Bousquet-Dubouch M-P, Theate I, Parmentier N, Van den Eynde BJ, et al. Two abundant proteasome subtypes that uniquely process some antigens presented by HLA class I molecules. Proc Natl Acad Sci U S A. 2010;107:18599–18604. doi:10.1073/pnas.1009778107.
- Morel S, Levy F, Burlet-Schiltz O, Brasseur F, Probst-Kepper M, Peitrequin A-L, Monsarrat B, Van Velthoven R, Cerottini J-C, Boon T, et al. Processing of some antigens by the standard proteasome but not by the immunoproteasome results in poor presentation by dendritic cells. Immunity. 2000;12(1):107–117. doi:10.1016/S1074-7613(00)80163-6.
- Van den Eynde BJ, Morel S. Differential processing of class-I-restricted epitopes by the standard proteasome and the immunoproteasome. Curr Opin Immunol. 2001;13:147–153. doi:10.1016/S0952-7915(00)00197-7.
- Vigneron N, Van den Eynde BJ. Proteasome subtypes and the processing of tumor antigens: increasing antigenic diversity. Curr Opin Immunol. 2012;24:84–91. doi:10.1016/j.coi.2011.12.002.
- Cascio P, Hilton C, Kisselev AF, Rock KL, Goldberg AL. 26S proteasomes and immunoproteasomes produce mainly N-extended versions of an antigenic peptide. Embo J. 2001;20:2357–2366. doi:10.1093/emboj/20.10.2357.
- Smart AC, Margolis CA, Pimentel H, He MX, Miao D, Adeegbe D, Fugmann T, Wong -K-K, Van Allen EM. Intron retention is a source of neoepitopes in cancer. Nat Biotechnol. 2018;36:1056–1058. doi:10.1038/nbt.4239.