
ABSTRACT
Natural polysaccharides have shown immune modulatory effects with low toxicity in both animal and human models. A previous study has shown that the polysaccharide from Codium fragile (CFP) promotes natural killer (NK) cell activation in mice. Since NK cell activation is mediated by dendritic cells (DCs), we examined the effect of CFP on DC activation and evaluated the subsequent induction of anti-cancer immunity in a murine model. Treatment with CFP induced activation of bone marrow-derived dendritic cells (BMDCs). Moreover, subcutaneous injection of CFP promoted the activation of spleen and lymph node DCs in vivo. CFP also induced activation of DCs in tumor-bearing mice, and combination treatment with CFP and ovalbumin (OVA) promoted OVA-specific T cell activation, which consequently promoted infiltration of IFN-γ-and TNF-α-producing OT-1 and OT-II cells into the tumors. Moreover, combination treatment using CFP and cancer self-antigen efficiently inhibited B16 tumor growth in the mouse model. Treatment with CFP also enhanced anti-PD-L1 antibody mediated anti-cancer immunity in the CT-26 carcinoma-bearing BALB/c mice. Taken together these data suggest that CFP may function as an adjuvant in the treatment of cancer by enhancing immune activation.
Abbreviations
CFP: Codium fragile polysaccharide; NK: natural killer; IFN: interferon; TNF: tumor necrosis factor; IL: interleukin; tdLN: tumor draining lymph node; BMDC: bone marrow-derived dendritic cell; OVA: ovalbumin; Ab: antibody; Ag: antigen; DC: dendritic cell; CTL: cytotoxic T lymphocyte; APC: antigen-presenting cell; pDC: plasmacytoid dendritic cell; mDC: myeloid dendritic cell; MHC: major histocompatibility complex; CR3: complement receptor type 3; TLR: Toll-like receptor; LPS: lipopolysaccharide; SP: sulfated polysaccharide; TRP2: tyrosinase-related protein 2; SR-A: scavenger receptor-A
1. Introduction
Recent studies have produced significant advances in the use of immunotherapies in cancer treatment, and we have seen the development of anti-cancer vaccines, immune checkpoint blockade strategies,Citation1–Citation3 and chimeric antigen (Ag) receptor T cell therapies.Citation4 Cancer immunotherapy is a promising treatment strategy which uses the immune cells of the body to combat the disease. The immune system has the ability to recall Ags and may contribute, as a memory response, to the prevention of secondary exposures to the same Ags.Citation5 As many patients with cancer have metastasis and relapse after tumor removal, cancer Ag-specific immune activation may serve as a strategy for preventing cancer metastasis. Therapeutic cancer vaccines rely on Ag-specific immune responses, including activation of cytotoxic T lymphocytes (CTLs) and helper T (Th) cells, as well as production of antibodies (Abs) by memory cells to prevent cancer regeneration.Citation6
Activation of CTLs and Th cells is controlled by the dendritic cells (DCs), Citation7 the most powerful of the Ag-presenting cells (APCs). DCs express pattern recognition receptors, which contribute to phagocytosis of pathogens and activation of DCs. Pathogens express immunostimulatory molecules that upregulate the expression of co-stimulators, production of pro-inflammatory cytokines, and migration of DCs to lymphoid organs. DCs also present Ags to T cells in lymphoid organs, resulting in the differentiation of naïve T cells into effector T cells. Although pathogens promote immune activation, cancer cells may not stimulate the immune system in the same way as some cancer Ags are not immunogenic. Therefore, induction of cancer Ag-specific immune responses demands additional stimulatory molecules and adjuvants.
DCs can be split into two subtypes: plasmacytoid DCs (pDCs) and myeloid DCs (mDCs). The mDCs are further classified into two sub-populations that differ in function with respect to T-cell activation. In mice, CD8α+ DCs present cytosolic Ags on the major histocompatibility complex (MHC) class I, which induces CD8 T-cell activation and differentiation into CTLs.Citation8,Citation9 On the other hand, CD8α− DCs process extracellular Ags presenting them via MHC class II receptors to induce CD4 T cell activation to Th cells. As in vitro-differentiated DCs, including bone marrow-derived DCs (BMDCs), are unable to function as DC subsets, we used a murine model to evaluate whether an adjuvant may induce CD8α+ and CD8α− DC activation in vivo.Citation9–Citation11
Marine algae polysaccharides such as alginate, fucoidan and ascophyllan, are known to promote immune modulation in humans and mice. It has been reported that the sulfated polysaccharide (SP) extracted from Codium fragile mainly consists of carbohydrates (54.6%), proteins (15.7%), sulfates (13.0%) and a small amount of uronic acid (1.4%).Citation12 In our previous study, we fractionated crude SP using ion-exchange chromatography and obtained two fractions (F1 and F2) which demonstrated a stimulatory effect for natural killer (NK) cells, which produce interferon (IFN)-γ, perforin and granzyme-B.Citation13 Although NK cell activation is controlled by DCs, the stimulatory effect of C. fragile polysaccharide (CFP) in DCs was not studied.
Most immunostimulatory adjuvants are extracted from pathogens or chemical products.Citation14 Although these adjuvants induce strong immune activation, they may also elicit undesirable side-effects, including inflammation in healthy tissues owing to toxicity.Citation15 Moreover, some adjuvants produced from chemical compounds may not completely induce T cell-mediated immune responses, but may elicit an Ab-mediated humoral immune response instead.Citation16 Lipopolysaccharide (LPS), the outer membrane component of gram-negative bacteria, is the most well-known and strongest type of immune stimulatory molecule, however LPS promotes inflammation in both human and animal models, which may result in sepsis.Citation17 The LPS contains O antigen, outer core, inner core and lipid-A.Citation18 It has been shown that lipid-A is main contributor in immune activation and that the O antigen promotes cytotoxicity.Citation18,Citation19 Unlike chemical- or pathogen-derived adjuvants, natural polysaccharide adjuvants like fucoidan, ascophyllan, and chitosan induce effective immune stimulation in animals and humans without strong cytotoxicity.Citation20–Citation23 In this study, we extracted a polysaccharide from C. fragile and evaluated its adjuvant properties in mice for future application in cancer treatment.
2. Materials and methods
2.1. Mice and cell lines
C57BL/6, C57BL/6-Ly5.1 (CD45.1) congenic, OT-I, and OT-II T cell receptor transgenic mice were purchased from Shanghai Public Health Clinical Center (SPHCC). Some C57BL/6 mice were obtained from Orient Bio (Gyeonggi, Korea). The mice were housed under pathogen-free conditions with 50–60% humidity at 22–24°C temperature. The study was carried out in accordance with the guidelines established by the Institutional Animal Care and Use committee at the SPHCC and Yeungnam University animal facility and was approved by the Ethics for Animal Experiments committee of the SPHCC (2018-A049-01) and Yeungnam University (2019–008). B16-F10 (murine melanoma cell line; ATCC, CRL-6475) and OVA-expressing B16-F10 (B16-OVA) cells were cultured in Roswell Park Memorial Institute (RPMI)-1640 medium supplemented with 1 M HEPES, 100 μg/mL streptomycin, 2 mM glutamine, 10% fetal bovine serum (FBS), and 100 U/mL penicillin in a humidified 5% CO2 incubator at 37°C.
2.2. Reagents
The polysaccharide from C. fragile was prepared as previously described.Citation12 Briefly, dried and milled C. fragile sample was treated with 90% ethanol at room temperature overnight. After removing the ethanol, the sample was extracted using distilled water at 65°C for 2 h. The water-soluble crude sample was precipitated in ethanol and subjected to filtration. The crude sample was re-dissolved in distilled water, and free proteins were removed using the Sevag method. The crude sample was fractionated using an ion-exchange chromatography system equipped with a DEAE Sepharose fast flow column (17–0709-01, GE Healthcare Bio-Science AB, Uppsala, Sweden). Chromatographic separation resulted in three fractions (F1, F2, and F3). The most immunostimulatory polysaccharide, fraction F2, was chosen for further study and designated as CFP. The endotoxin levels in CFP were measured using a Limulus amebocyte lysate (LAL) kit (Lonza, Basel, Switzerland). The CFP and OVA used in all experiments contained less than 0.05 endotoxin unit/mL. Escherichia Coli (E. coli) O111:B4 LPS was purchased from Sigma-Aldrich (St. Louis, Missouri, US; #L4130).
2.3. Antibodies
Isotype control Abs (IgG1, IgG2a, or IgG2b), CD11c-Brilliant Violet 785 (#117335, N418), CD8α-APC/Cyanine7 (#100713, 53–6.7), CD40-Alexa Fluor® 647 (#124614, 3/23), CD80-Brilliant Violet 605™ (#104729, 16–10A1) and CD86-PE/Cy7 (#105013, GL-1) were procured from BioLegend (San Diego, California, US). Anti-MHC class I-PE (#114620, 28-8-6) and anti-MHC class II-PE/Cyanine7 (#107607, M5/114.15.2) were also obtained from BioLegend (San Diego, CA, US).
2.4. BMDC generation
Bone Marrow (BM) was extracted from C57BL/6 mice, and BM cells (1 × 106 cells/mL) were cultured in 100 U/mL recombinant mouse granulocyte macrophage colony-stimulating factor (rmGM-CSF) and 100 U/mL recombinant mouse interleukin-4 (rmIL-4) in 2 mL of RPMI-1640 in 24-well plates.Citation24 Unless otherwise stated, the BM cells were cultured at 37°C under 10% CO2 for 6 days. The rates of BMDC differentiation were determined based on the expression CD11 c, and more than 95% of the cells were found to express CD11 c.
2.5. Flow cytometry analysis
After washing with phosphate-buffered saline (PBS), cells were pre-incubated with unlabeled isotype control Abs and Fc-blocked Abs for 15 min. This was followed by staining with fluorescence-conjugated Abs on ice for 30 min. After washing, the cells were treated with 4ʹ,6-diamidino-2-phenylindole (DAPI) (Sigma-Aldrich, St. Louis, Missouri, US; #D8417) and 1 × 105 cells were analyzed using a FACS Aria II (Becton Dickinson, Franklin Lakes, New Jersey, US) or NovoCyte flow cytometer (ACEA Biosciences Inc., San Diego, California, US).
2.6. Splenic DC analysis
The spleen was cut into small pieces and digested with a digestion buffer (10 mg of DNase I and 100 mg of collagenase IV). Cells were filtered through a nylon mesh (100 nm pore size) and washed with PBS. Pellets were re-suspended in 5 mL of 1077 Histopaque (Sigma-Aldrich, St. Louis, Missouri, US; #10771) and overlaid with an additional 5 mL of 1077 Histopaque. Cells were then centrifuged at 3000 rpm for 10 min, and splenocytes with a density higher than 1.077 g/mL were obtained and stained with FITC-conjugated lineage markers and CD11c monoclonal antibodies (mAbs), including CD3(#100204), CD90.1 (#202503), B220(#103205), Gr-1 (#108406), CD49 (#103503), TER-119 (#116205), and anti-CD11c-Brilliant Violet 785 (#117335, N418) Abs, obtained from BioLegend (San Diego, California, US). Spleen DCs were defined as CD11c+ lineage− cells. Spleen DCs were further divided into CD8α+ and CD8α− DCs, as described by previous studies.Citation15,Citation25
2.7. Tumor treatment
C57BL/6 mice were subcutaneously injected with 1 × 106/100 µL B16 or B16-OVA melanoma cells. Once tumors were well established the animals were intraperitoneally treated with PBS alone, 2.5 mg/kg Ag, 50 mg/kg CFP, or a mixture of Ag and CFP at days 7 and 14. Tumor growth was monitored until day 21 post challenge. Mice were then sacrificed, and the spleen was harvested for further analysis.
2.8. OT-I and OT-II T cell proliferation and tumor infiltration
CD4 or CD8 T cells were isolated from OT-II and OT-I mice using CD4 T or CD8 T cell isolation kits (Miltenyi Biotec, Bergisch Gladbach, Germany; #130-104-454; #130-104-075), respectively. The isolated OT-I and OT-II cells were suspended in culture medium supplemented with 10 μM CFSE (Invitrogen, San Diego, California, US; #C34554) and incubated for 10 min. The labeled cells (1 × 106) were intravenously injected into B16-OVA tumor-bearing CD45.1-expressing congenic mice. The mice were treated with PBS alone, 2.5 mg/kg OVA, 50 mg/kg CFP, or a mixture of OVA and CFP 24 h after cell transfer. Three days after treatment, spleen and tumor tissues were harvested. The proliferation of OT-I and OT-II cells in the spleen was evaluated by the dilution of CFSE fluorescence intensity in CD45.2+ cells. The tumors were digested using a digestion buffer (2% FBS and collagenase IV) and washed with PBS. The pellets were re-suspended in PBS and stained with anti-CD3, anti-CD4, anti-CD8, and anti-CD45.2 Abs. CD45.2+CD8+ and CD45.2+CD4+ cells with CD3+ cells being defined as the infiltrated OT-1 and OT-II cells, respectively.Citation15
2.9. Ex vivo T cell stimulation and intracellular cytokine staining
As described in previous studies, tumor-infiltrated cells were stimulated with 2 μg/mL Ag for 6 h and treated with 1 μg/mL Monensin Solution (Biolegend, San Diego, California, US; #420701) over the final 3 h.Citation5 Cells were harvested and stained with surface Abs for 15 min, followed by incubation in fixation buffer (Biolegend, San Diego, California, US; #420801) and permeabilization with Cytofix/Cytoperm buffer (eBioscience, San Diego, California, US; #426803). Cells were incubated with anti-cytokine Abs in Perm/Wash buffer (eBioscience, San Diego, California, US; #421002) for 30 min. Dead cells were excluded using zombie-violet staining.
2.10. Enzyme-linked immunosorbent assay (ELISA)
Concentrations of IL-6 (#431307), IL-12p70 (#433606), and TNF-α (#430904) in sera and cultured medium were analyzed in triplicate using standard ELISA kits (Biolegend, California, US;;).
2.11. ELISPOT assay
Single cells from tdLNs were seeded at a density of 50 × 103 cells/well in a pre-coated plate. The cells were stimulated with 2 μg/mL of Ag peptides or a negative control peptide at 37°C for 24 h. The plates were then dried, and the spots were counted using a CTL ELISPOT reader (CTL Europe GmbH, Bonn, Germany).
2.12. In vivo cytotoxicity assay
C57BL/6 mice were immunized with PBS, 2.5 mg/kg OVA, 50 mg/kg CFP, or a combination of OVA and CFP on day 7 and 14. Five days after the last treatment, target cells were transferred into the immunized mice. The splenocytes from naïve mice were extracted and 10 × 106 splenocytes were labeled with 10 mM Cell Tracker Orange CMTMR (Life Technologies, Carlsbad, California, US; #M7510) and loaded with control peptide. Another 10 × 106 splenocytes were labeled with 200 nM CFSE (Invitrogen, San Diego, California, US; #C34554) and 100 nM SIINFEKL peptide was loaded. A mixture containing each target cell population (a total of 10 × 106 cells) was intravenously injected to immunized mice. Percentage killing was calculated using the formula described by FACS Fortessa (Becton Dickinson, Franklin Lakes, New Jersey, US).
2.13. Statistical analysis
All data are expressed as the mean ± standard deviation (SD). One- or two-way analysis of variance (ANOVA) with Tukey multiple comparison and Mann-Whitney t-tests were used to analyze the data sets. Values of p < 0.05 were considered statistically significant.
3. Results
3.1. CFP activates BMDCs
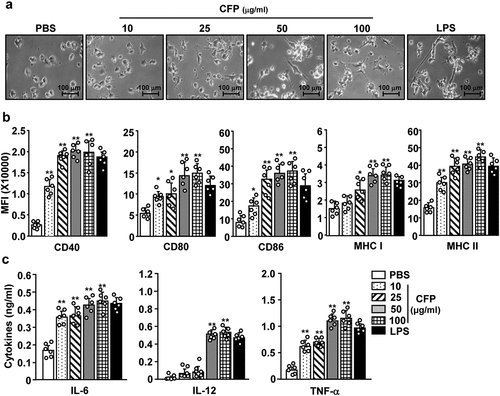
To evaluate the immunostimulatory effects of CFP in DCs, BMDCs were treated with different concentrations of this compound. BMDCs were differentiated from bone marrow cells using a combination of 100 U/mL GM-CSF and 100 U/mL IL-4 for 6 days. Immature BMDCs were treated with 10, 25, 50, and 100 μg/mL CFP for 24 h. The BMDCs were also treated with 0.1 μg/mL LPS as a positive control. Treatment with CFP induced substantial changes in the morphology of the BMDCs with these changes exhibiting dose-dependent behaviors (). The expression levels of co-stimulators were dose-dependently upregulated after CFP treatment (). Concentrations of IL-6, IL-12, and TNF-α in the culture medium of BMDCs were substantially elevated after CFP treatment, and all exhibited dose-dependent behaviors (). Thus, CFP induces BMDC activation; and the optimum concentration of CFP for activation was 50 μg/mL based on the expression of various co-stimulatory molecules.
Figure 1. Codium fragile polysaccharide (CFP) induced activation of bone marrow-derived dendritic cells (BMDCs). BMDCs were differentiated from bone marrow cells following incubation with 100 U/mL IL-4 and 100 U/mL GM-CSF for 6 days. BMDCs were treated with various concentrations of CFP and 0.1 μg/mL of LPS for 24 h. (a) Morphological changes in BMDCs. The images were obtained using a microscope (X 60 of magnification). (b) Mean fluorescence intensity (MFI) of co-stimulators and MHC class I and II measured by flow cytometry. (c) Concentrations of IL-6, IL-12, and TNF-α in culture medium analyzed by ELISA. Data are representative of a larger dataset or are the average of six independent samples (Two samples for three independent experiments). A significant difference from treatment with PBS is indicated by *p < .05, **p < 0.01.
3.2. CFP induces splenic DC activation in mice
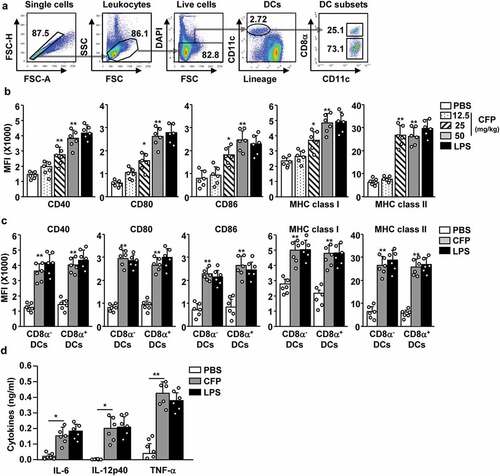
Because CFP promoted the activation of BMDCs in vitro, we went on to evaluate the effect of CFP on DC induction in vivo. C57BL/6 mice were intraperitoneally injected with 12.5, 25, or 50 mg/kg of CFP. Six hours after injection, the spleen was harvested, and DC activation was analyzed. Spleen DCs were defined as lineage− CD11c+ cells among live leukocytes and identified by flow cytometry (). Treatment with CFP induced a substantial increase in the levels of co-stimulators and class I and II MHCs in splenic DCs ( and S1). Moreover, CD8α+ and CD8α− subsets of splenic DCs also significantly increased their expression of co-stimulators and class I and II MHCs (). Serum concentrations of pro-inflammatory cytokines were significantly upregulated in mice (). The stimulatory effects of CFP were similar to those of LPSLPS). Thus, CFP can induce splenic DC activation in mice.
Figure 2. CFP promotes the activation of splenic DCs in mice. C57BL/6 mice were intraperitoneally injected with the indicated doses of CFP. The mice were intraperitoneally injected with 1 mg/kg LPS as a positive control. Six hours after treatment, the spleen was harvested and the activation of splenic DCs was analyzed by flow cytometry. (a) Strategy of spleen DC analysis by flow cytometry. Spleen DCs were defined as CD11c+ lineage− cells among live leukocytes. (b) MFI of co-stimulator and MHC class I and II. (c) MFI of co-stimulator and MHC class I and II in CD8α+ and CD8α− DCs. (d) Serum concentrations of pro-inflammatory cytokines measured by ELISA. Data are the average of six independent samples (two independent experiments performed with n = 3/group). A significant difference from the PBS control group compared to CFP-treated group is indicated by *p < .05, **p < 0.01.
3.3. CFP enhances OVA-specific immune responses
We went on to examine the effect of CFP in the tumor environment. B16-OVA tumor-bearing C57BL/6 mice were treated with CFP and the DC population in the tumor-draining lymph nodes (tdLN) was evaluated by flow cytometry (). The expression of co-stimulators and class I and II MHC molecules increased in tdLN DCs (). The production of pro-inflammatory cytokines also significantly increased after CFP treatment in tumor-bearing mice when compared with that in untreated tumor-bearing mice ().
Figure 3. CFP elicited antigen-specific T cell immunity in tumor-bearing mice. C57BL/6 mice were subcutaneously injected with B16-OVA melanoma cells. (a) Strategy behind tdLN DC analysis by flow cytometry. (b) Seven days after tumor injection, mice were intraperitoneally treated with 50 mg/kg CFP, and levels of co-stimulator and MHC class I and II were measured. (c) Serum concentrations of pro-inflammatory cytokines were determined. (E to G) OT-I and OT-II cells were transferred into B16-OVA tumor-bearing CD45.1 congenic mice. One day after cell transfer, mice were intraperitoneally injected with PBS, 2.5 mg/kg OVA, 50 mg/kg CFP, or a combination of OVA and CFP. (d) OT-I and OT-II cell proliferation was measured in tdLN 3 days after treatment. (e) Mean cell numbers of tumor-infiltrated OT-I and OT-II cells. (f) Intracellular IFN-γ and TNF-α levels in tumor-infiltrating OT-I and OT-II cells measured by flow cytometry. (g) Mean IFN-γ-producing (left panel) and TNF-α-producing (right panel) OT-I and OT-II cells in tumor tissues. All data are representative or show the average of six independent samples (two independent experiments performed with n = 3/group). A significant difference from the PBS treatment and CFP treatment is indicated by **p < 0.01.
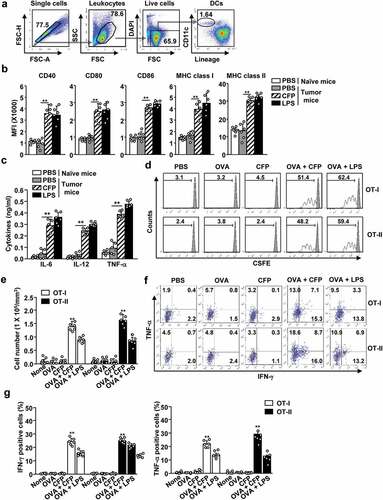
Specific Ag reaction is needed to target tumor cells without producing undesired side-effects.Citation26,Citation27 We used an experimental Ag, and OVA and evaluated the induction of Ag-specific T-cell activation. We isolated OT-I and OT-II cells and transferred them to B16-OVA tumor-bearing CD45.1 congenic C57BL/6 mice. Treatment with a combination of CFP and OVA promoted a substantial increase in the population of OT-I and OT-II cells in the tdLN from B16-OVA tumor-bearing mice (). Furthermore, infiltration of OT-I and OT-II cells into tumors markedly increased after combined treatment with CFP and OVA when compared with OVA or CFP treatment alone (). Tumor-infiltrating OT-I and OT-II cells subjected to the combination treatment showed a substantial increase in the levels of IFN-γ and TNF-α, while OVA or CFP treatment alone had no effect on these cytokines (). Thus, CFP can function as an adjuvant to enhance Ag-specific T cell activation in tumor-bearing mice.
3.4. CFP-induced OVA-specific immune activation suppresses OVA-B16 growth
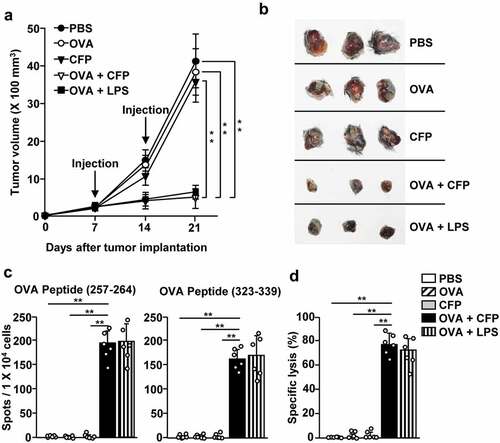
As CFP enhanced the Ag-specific activation of T cells, we examined the anti-cancer effects of CFP treatment in tumor-bearing mice. B16-OVA tumor-bearing mice were treated with a combination of CFP and OVA on days 7 and 14. B16-OVA tumor growth was suppressed in the combination treatment group when compared with the control groups (). The size of the tumor mass on day 21 after tumor injection was much smaller in the combination treatment group than in the PBS and OVA or CFP only groups (). Moreover, tdLN cells from mice receiving the combination treatment produced significantly higher levels of IFN-γ in response to OVA peptides than those from PBS, OVA, or CFP treated animals, which showed no IFN-γ production (). In addition, the combination of CFP and OVA increased the specific killing of OVA peptide-coated splenocytes (). These data indicate that a combination treatment using CFP and OVA could promote anti-cancer immunity in OVA-expressing tumors.
Figure 4. Treatment with a combination of CFP and OVA suppressed B16-OVA tumor growth. C57BL/6 mice were subcutaneously inoculated with B16-OVA. The mice received treatment with PBS, 2.5 mg/kg OVA, 50 mg/kg CFP, or a combination of OVA and CFP on days 7 and 14 after tumor injection. (a) B16-OVA tumor growth curves. (b) Images of tumor mass on day 21 after tumor injection. (c) IFN-γ production by tdLN cells in response to MHC class I-restricted epitope OVA peptide (257–264) (left panel) and MHC class II epitope (323–339) measured by ELISPOT. (d) OVA-specific cytotoxic activity. Data are representative or show the average of six independent samples (two independent experiments performed with n = 3/group). A significant difference from the PBS treatment compared to CFP treated group is indicated by **p < 0.01.
3.5. CFP enhances melanoma Ag-specific immunity
We then examined the adjuvant effect of CFP by investigating whether it could induce melanoma self-Ag-specific immune activation and anti-melanoma effects. To evaluate CFP treatment in enhancing melanoma self Ag-specific immune activation, we used tyrosinase-related protein 2 (TRP-2), which is expressed in murine B16 melanoma, as the antigen. C57BL/6 mice were treated with a combination of 50 mg/kg CFP and 2.5 mg/kg TRP2 on days 7 and 14 after B16 melanoma tumor inoculation. The combination treatment with CFP and TRP2 inhibited B16 melanoma growth (); the size of the tumor mass was substantially smaller in the combination treatment group than in the control groups (). In addition, IFN-γ production by tdLN cells was significantly higher after combination treatment with TRP2 and CFP than in mice from the other treatment groups ().
Figure 5. CFP induced melanoma antigen-specific immunity and inhibited melanoma tumor growth. C57BL/6 mice were subcutaneously injected with B16 cells and intraperitoneally treated with PBS, 2.5 mg/kg TRP2, 50 mg/kg CFP, or a combination of TRP2 and CFP on days 7 and 14 after tumor injection. (a) Tumor growth curves. (b) Size of tumor masses was determined on day 21 after tumor injection. (c) IFN-γ production levels in response to TRP2. (d and e) Once B16 tumors were well established, by day 7, the mice were depleted of CD4 or CD8 expressing cells and treated with a combination of TRP2 and CFP. (d) Tumor growth curves after CD4 (d) or CD8 T (e) depletion. Each condition had three mice and each experiment was conducted twice (n = 6), **p < 0.01.
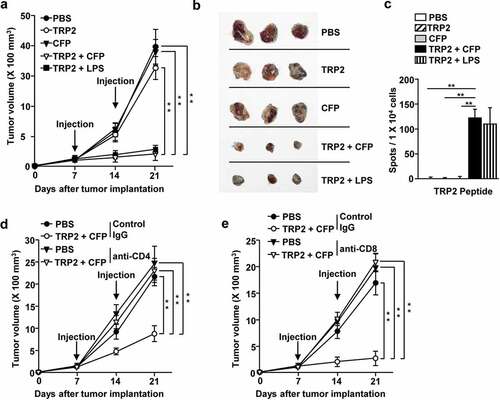
To evaluate the anti-cancer effects of CFP, we investigated T-cell immunity. We depleted CD4 or CD8 expressing cells during treatment with CFP and TRP2 in B16 melanoma-bearing mice. The suppressive effect of this combination treatment in B16 melanoma was completely diminished in CD4 or CD8 depleted mice (). This indicates that the anti-cancer effect of CFP and Ag treatment is mediated by T-cell immunity.
3.6. CFP enhances the anti-cancer effect of anti-PD-L1 Abs
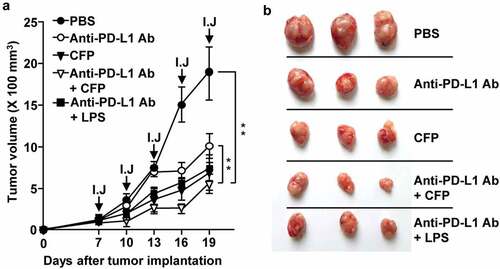
Immunostimulatory molecules are known to enhance anti-cancer effects of immune checkpoint blockade Ags.Citation28–Citation30 We examined whether CFP exhibits any anti-PD-L1 Ab-mediated anti-cancer effects in CT-26-bearing BALB/c mice. After the tumor was well established, day 7 after tumor cell inoculation, mice were treated with 10 mg/kg anti-PD-L1 Ab, 50 mg/kg CFP, or a combination of anti-PD-L1 Ab and CFP for 3 days. Treatment with anti-PD-L1 Ab suppressed CT-26 tumor growth, and the anti-cancer effects were markedly enhanced with the addition of CFP (). CFP alone had suppressive effects on the growth of CT-26 carcinoma (). These data suggest that CFP enhances the anti-cancer effects of anti-PD-L1 Ab in CT-26 carcinoma.
Figure 6. CFP enhanced the antitumor effect of an anti-PD-L1 antibody. BALB/c mice were subcutaneously injected with CT-26 carcinoma cells. The mice were intraperitoneally treated with 10 mg/kg anti-PD-L1 antibody, 50 mg/kg CFP, or a combination of anti-PD-L1 and CFP for 3 days. I.J indicates injection. (a) Tumor growth curves (b) Size of tumor mass on day 19 after tumor injection. Three mice for each condition and a total of two experiments means that n = 6 for these evaluations, **p < 0.01.
4. Discussion
Polysaccharides extracted from natural products are known to exert immunomodulatory effects in humans and animals. Fucoidans are well-known marine polysaccharides that activate immune cells, including DCs, macrophages, NK cells, and T cells in mice and humans.Citation31–Citation34 Chitosan, another polysaccharide from marine products, is known for its immunomodulatory effects and has been approved for human use by the Food and Drug Administration (FDA) of the USA and by the EU.Citation35,Citation36 Here, we found a marine natural polysaccharide, CFP, which induced activation of DC and T cells in mice. CFP elicited a cancer Ag-specific immune response, which consequently inhibited tumor growth in mice. Fucoidan, chitosan, and CFP have different components and structures.Citation34,Citation35,Citation37 Although the origin of fucoidan (brown algae) is different from that of CFP (green algae), both these compounds contain sulfate groups in their backbones.Citation12,Citation38,Citation39 The main backbone of fucoidan contains fucose molecules, while CFP is made up of mannose. Chitosan is extracted from the exoskeletons of crustaceans, and CFP is extracted from marine plants.Citation35 Molecular compositional analysis revealed that CFP consists of carbohydrates (80.5%), proteins (8.2%) and sulfates (3.2%) and a small amount of uronic acid (1.1%). The major sugar unit of CFP is mannose (91.3%) with a small amount of glucose (8.6%). The HPSEC chromatogram of CFP exhibits a single peak with an average molecular weight of 148 × 103 (g/mol).Citation12 According to our previous study, Citation12 the yield of CFP is approximately 41.13 g per Kg of dried raw material. While chitosan is water-insoluble, CFP is soluble in water. In addition, CFP includes small amounts of proteins that promote RAW 264.7 cell activation.Citation39 However, the exact structural feature necessary for initiating an immune response by these polymers remains unclear. Therefore, further studies should evaluate whether structural differences in these polysaccharides influence immune cell activation.
Induction of cancer Ag-specific immune activation is one of the immunotherapy strategies used to treat cancer.Citation26,Citation27,Citation40 Although cancer cells over-express Ag candidate proteins, these proteins are not immunogenic as they are produced from the somatic cells without mutation. Therefore, immune stimulatory molecules or adjuvants are required to elicit cancer Ag-specific immune activation.Citation41 Adjuvants activate APCs including DCs, macrophages, and B cells, which promote Ag-specific T-cell immunity.Citation27,Citation41 DCs are the most potent population involved in the induction of T-cell proliferation and activation.Citation27 Therefore, many types of DC-targeting adjuvants have been developed and tested. Alum is one of the adjuvants approved by the US FDA that is known to enhance Ag-specific immune-response activation in humans.Citation16 Many types of vaccines contain alum as an adjuvant.Citation42 However, alum-induced immune activation is limited to Ab production or humoral immunity.Citation16,Citation42,Citation43 In addition, monophosphoryl lipid A (MPLA), a toll-like receptor 4 (TLR4) ligand, is used as an adjuvant to enhance anti-cancer immunity, but MPLA is not water soluble and remains toxic to humans. The MPLA was modified for reducing toxicity from LPS.Citation44 In this study, we found a novel adjuvant CFP which promotes immune activation including Th1 and CTL responses. CFP is a natural product and water soluble. As shown the conversion of LPS into less cytotoxic MPLA, the CFP will further study for modify to reduce cytotoxicity in the animal and human by composition analysis and development of derivatives.
Efficient treatment of cancer demands preferential activation of CTL-mediated cytotoxicity against cancer cells.Citation26,Citation27 LPS, an endotoxin produced by gram-negative bacteria, is a well-known agonist of TLR4, which induces activation of DCs in humans and mice. Although LPS promotes strong immune activation, it may cause sepsis as humans are highly susceptible to endotoxins.Citation45 Novel adjuvants that promote cancer Ag-specific CTL activation, while having low cytotoxicity to healthy somatic cells, are desirable for effective cancer immunotherapy. LPS treatment with OVA promotes OVA-specific immune responses, as indicated by IFN-γ production and induction of cytotoxic activity against OVA-coated splenocytes. However, LPS is not suitable for use in humans, as human sensitivity to endotoxins is much higher than animals.Citation17 CFP is a natural polysaccharide that has low cytotoxicity in humans and mice. CFP promotes activation of cancer Ag-specific T-cell immunity and exerts anti-cancer effects. Therefore, CFP may be useful in enhancing immune responses against foreign Ags in humans.
Mouse spleen and LN DCs are made up of two main subsets: pDCs and mDCs.Citation7,Citation9,Citation46 pDCs contribute to the production of type I IFN in response to viral infection, while mDCs activate humoral and cellular immunities in response to exogenous protein Ags.Citation47 mDCs are further divided into two subtypes: CD8α+ and CD8α− DCs. These subsets of DCs differentially process Ags. While CD8α+ DCs are the main cell type promoting CTL activation, CD8α− DCs induce Th-cell activation.Citation8,Citation9,Citation11 The treatment of CFP stimulates both CD8α+ and CD8α− DC activation in mouse spleens. These activated DCs promoted T cell-mediated anti-cancer immunity, which was confirmed by the loss of activity when CD4 and CD8 cells were depleted. Thus, the observed T cell activation can be contributed to pDCs, mDCs and macrophages. In future studies, we will examine whether the pDCs and macrophages contribute to the induction of the T cell activation and anti-cancer immunity reported here in response to combination treatment with CFP and Ag.
We have previously shown that CFP-induced NK cell activation is mediated by the stimulation of complement receptor type 3 (CR3). Mannose or N-acetyl-D-glucosamine from CFP directly binds and stimulates CR3.Citation37 CR3 is expressed on DCs and macrophages, and its stimulation promotes their activation. although ligation of CR3 failed to induce production of pro-inflammatory cytokines.Citation48 Here we found that treatment with CFP induced production of pro-inflammatory cytokines in BMDCs, an effect that is different from that produced by CR3 stimulation. Moreover, administration of CFP increased the production of pro-inflammatory cytokines. Therefore, CFP may target not only CR3, but also other receptors, and contribute to DC activation. Fucoidan and ascophyllan bind to scavenger receptor-A (SR-A) and TLR4, respectively, Citation5,Citation20,Citation27 the CFP may target other pattern recognition receptors (PRRs). Further studies will investigate the properties of CFP in binding to these receptors on DCs using receptor knockout (KO) mice.
The tumor microenvironment has immunosuppressive properties including the expression of immune checkpoint proteins on tumor-cell surfaces.Citation29,Citation30 Expression of these proteins promotes the evasion of the immune system, while inhibition or blockade of these proteins may serve as a strategy to target immune cells in cancer.Citation29,Citation30 In an animal study, the blockade of immune checkpoints via the administration of monoclonal Abs was shown to suppress tumor growth, although this effect was considerably lower in humans.Citation49 To enhance the therapeutic effects of immune checkpoint Abs, additional treatment with adjuvants is needed to provide efficient therapeutic response.Citation29,Citation30,Citation49 In this study, we found that a combination of CFP and anti-PD-L1 Ab suppressed CT-26 tumor growth, and the effect was stronger than that observed for anti-PD-L1 Ab alone. Therefore, CFP can act as an immunostimulatory adjuvant and may be useful in enhancing the therapeutic effects of immune checkpoint blockade agents.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank the animal facility in Shanghai Public Health Clinical Center and Yeungnam University for maintaining the animals used in this study. We would like to thank Editage (www.editage.co.kr) for English language editing.
Additional information
Funding
References
- Postow MA, Callahan MK, Wolchok JD. Immune checkpoint blockade in cancer therapy. J clin oncol. 2015;33:1974–1982. doi: 10.1200/JCO.2014.59.4358.
- Kyi C, Postow MA. Immune checkpoint inhibitor combinations in solid tumors: opportunities and challenges. Immunotherapy. 2016;8:821–11. doi:10.2217/imt-2016-0002.
- Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348:56–61. doi:10.1126/science.aaa8172.
- Neelapu SS, Tummala S, Kebriaei P, Wierda W, Gutierrez C, Locke FL, Komanduri KV, Lin Y, Jain N, Daver N, et al. Chimeric antigen receptor T-cell therapy - assessment and management of toxicities. Nat Rev Clin Oncol. 2018;15:47–62. doi: 10.1038/nrclinonc.2017.148.
- Xu L, Kwak M, Zhang W, Zeng L, Lee PC, Jin JO. Rehmannia glutinosa polysaccharide induces toll-like receptor 4 dependent spleen dendritic cell maturation and anti-cancer immunity. Oncoimmunology. 2017;6:e1325981. doi:10.1080/2162402X.2017.1325981.
- Miconnet I, Coste I, Beermann F, Haeuw JF, Cerottini JC, Bonnefoy JY, Romero P, Renno T. Cancer vaccine design: a novel bacterial adjuvant for peptide-specific CTL induction. J Immunol. 2001;166:4612–4619. doi:10.4049/jimmunol.166.7.4612.
- Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi:10.1038/32588.
- Naik SH, Proietto AI, Wilson NS, Dakic A, Schnorrer P, Fuchsberger M, Lahoud MH, O’Keeffe M, Shao QX, Chen WF, et al. Cutting edge: generation of splenic CD8+ and CD8- dendritic cell equivalents in Fms-like tyrosine kinase 3 ligand bone marrow cultures. J Immunol. 2005;174:6592–6597. doi:10.4049/jimmunol.174.11.6592.
- Schnorrer P, Behrens GM, Wilson NS, Pooley JL, Smith CM, El-Sukkari D, Davey G, Kupresanin F, Li M, Maraskovsky E, et al. The dominant role of CD8+ dendritic cells in cross-presentation is not dictated by antigen capture. Proc Natl Acad Sci U S A. 2006;103:10729–10734. doi:10.1073/pnas.0601956103.
- Pooley JL, Heath WR, Shortman K. Cutting edge: intravenous soluble antigen is presented to CD4 T cells by CD8- dendritic cells, but cross-presented to CD8 T cells by CD8+ dendritic cells. J Immunol. 2001;166:5327–5330. doi:10.4049/jimmunol.166.9.5327.
- Jongbloed SL, Kassianos AJ, McDonald KJ, Clark GJ, Ju X, Angel CE, Chen CJ, Dunbar PR, Wadley RB, Jeet V, et al. Human CD141+ (BDCA-3)+ dendritic cells (DCs) represent a unique myeloid DC subset that cross-presents necrotic cell antigens. J Exp Med. 2010;207:1247–1260. doi:10.1084/jem.20092140.
- Tabarsa M, Karnjanapratum S, Cho M, Kim J-K, You S. Molecular characteristics and biological activities of anionic macromolecules from Codium fragile. Int J Biol Macromol. 2013;59:1–12.
- Tabarsa M, You S, Dabaghian EH, Surayot U. Water-soluble polysaccharides from Ulva intestinalis: molecular properties, structural elucidation and immunomodulatory activities. J Food Drug Anal. 2018;26:599–608. doi:10.1016/j.jfda.2017.07.016.
- Cui W, Joshi NS, Liu Y, Meng H, Kleinstein SH, Kaech SM. TLR4 ligands lipopolysaccharide and monophosphoryl lipid a differentially regulate effector and memory CD8+ T cell differentiation. J Immunol. 2014;192:4221–4232. doi:10.4049/jimmunol.1302569.
- Zhang W, Xu L, Park H-B, Hwang J, Kwak M, Lee PC, Liang G, Zhang X, Xu J, Jin J-O. Escherichia coli adhesion portion FimH functions as an adjuvant for cancer immunotherapy. Nat Commun. 2020;11:1–14. doi:10.1038/s41467-019-13993-7.
- Kool M, Soullié T, van Nimwegen M, Willart MA, Muskens F, Jung S, Hoogsteden HC, Hammad H, Lambrecht BN. Alum adjuvant boosts adaptive immunity by inducing uric acid and activating inflammatory dendritic cells. J Exp Med. 2008;205:869–882. doi:10.1084/jem.20071087.
- Kajiwara Y, Schiff T, Voloudakis G, Sosa MAG, Elder G, Bozdagi O, Buxbaum JD. A critical role for human caspase-4 in endotoxin sensitivity. J Immunol. 2014;193:335–343. doi:10.4049/jimmunol.1303424.
- Meredith TC, Aggarwal P, Mamat U, Lindner B, Woodard RW. Redefining the requisite lipopolysaccharide structure in Escherichia coli. ACS Chem Biol. 2006;1:33–42. doi:10.1021/cb0500015.
- Rhee SH. Lipopolysaccharide: basic biochemistry, intracellular signaling, and physiological impacts in the gut. Intestinal Res. 2014;12:90–95. doi:10.5217/ir.2014.12.2.90.
- Coffman RL, Sher A, Seder RA. Vaccine adjuvants: putting innate immunity to work. Immunity. 2010;33:492–503. doi:10.1016/j.immuni.2010.10.002.
- Zhang W, Okimura T, Xu L, Zhang L, Oda T, Kwak M, Yu Q, Jin JO. Ascophyllan functions as an adjuvant to promote anti-cancer effect by dendritic cell activation. Oncotarget. 2016;7:19284–19298. doi:10.18632/oncotarget.8200.
- Ale MT, Meyer AS. Fucoidans from brown seaweeds: an update on structures, extraction techniques and use of enzymes as tools for structural elucidation. RSC Adv. 2013;3:8131–8141. doi:10.1039/C3RA23373A.
- Zaharoff DA, Rogers CJ, Hance KW, Schlom J, Greiner JWJV. Chitosan solution enhances both humoral and cell-mediated immune responses to subcutaneous vaccination. Vaccine. 2007;25:2085–2094.
- Zhang W, Du JY, Jiang Z, Okimura T, Oda T, Yu Q, Jin JO. Ascophyllan purified from Ascophyllum nodosum induces Th1 and Tc1 immune responses by promoting dendritic cell maturation. Mar Drugs. 2014;12:4148–4164. doi:10.3390/md12074148.
- Xu L, Kwak M, Zhang W, Lee PC, Jin JO. Time-dependent effect of E. coli LPS in spleen DC activation in vivo: alteration of numbers, expression of co-stimulatory molecules, production of pro-inflammatory cytokines, and presentation of antigens. Mol Immunol. 2017;85:205–213. doi:10.1016/j.molimm.2017.02.017.
- Shang N, Figini M, Shangguan J, Wang B, Sun C, Pan L, Ma Q, Zhang Z. Dendritic cells based immunotherapy. Am J Cancer Res. 2017;7:2091–2102.
- Cohn L, Delamarre L. Dendritic cell-targeted vaccines. Front Immunol. 2014;5:255. doi:10.3389/fimmu.2014.00255.
- Wilkinson RW, Leishman AJ. Further advances in cancer immunotherapy: going beyond checkpoint blockade. Front Immunol. 2018;9:1082. doi:10.3389/fimmu.2018.01082.
- Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018;8:1069–1086. doi:10.1158/2159-8290.CD-18-0367.
- Sato-Kaneko F, Yao S, Ahmadi A, Zhang SS, Hosoya T, Kaneda MM, Varner JA, Pu M, Messer KS, Guiducci C, et al. Combination immunotherapy with TLR agonists and checkpoint inhibitors suppresses head and neck cancer. JCI Insight. 2017;2. doi:10.1172/jci.insight.93397.
- Zhang W, Oda T, Yu Q, Jin J-O. Fucoidan from Macrocystis pyrifera has powerful immune-modulatory effects compared to three other fucoidans. Mar Drugs. 2015;13:1084–1104. doi:10.3390/md13031084.
- Senthilkumar K, Kim S-K. Anticancer effects of fucoidan. Adv Food Nutr Res. 2014;72:195–213. doi: 10.1016/B978-0-12-800269-8.00011-7.
- Jin J-O, Zhang W, Du J-Y, Wong K-W, Oda T, Yu Q. Fucoidan can function as an adjuvant in vivo to enhance dendritic cell maturation and function and promote antigen-specific T cell immune responses. PLoS One. 2014;9:e99396. doi:10.1371/journal.pone.0099396.
- Li B, Lu F, Wei X, Zhao R. Fucoidan: structure and bioactivity. Molecules. 2008;13:1671–1695.
- Ahmed TA, Aljaeid BM. Preparation, characterization, and potential application of chitosan, chitosan derivatives, and chitosan metal nanoparticles in pharmaceutical drug delivery. Drug Des Devel Ther. 2016;10:483–507. doi:10.2147/DDDT.S99651.
- Mohammed MA, Syeda JTM, Wasan KM, Wasan EK. An overview of chitosan nanoparticles and its application in non-parenteral drug delivery. Pharmaceutics. 2017;9:53. doi:10.3390/pharmaceutics9040053.
- Surayot U, You S. Structural effects of sulfated polysaccharides from Codium fragile on NK cell activation and cytotoxicity. Int J Biol Macromol. 2017;98:117–124. doi:10.1016/j.ijbiomac.2017.01.108.
- You S, Yang C, Lee H, Lee B-Y. Molecular characteristics of partially hydrolyzed fucoidans from sporophyll of Undaria Pinnatifida and their in vitro anticancer activity. Food Chem. 2010;119:554–559. doi:10.1016/j.foodchem.2009.06.054.
- Tabarsa M, Park G-M, Shin I-S, Lee E, Kim J-K, You SJMB. Structure-activity relationships of sulfated glycoproteins from Codium fragile on nitric oxide releasing capacity from RAW264. 7 cells. Mar Biotechnol. 2015;17:266–276.
- Xu L, Zhang W, Park HB, Kwak M, Oh J, Lee PCW, Jin JO. Indocyanine green and poly I:C containing thermo-responsive liposomes used in immune-photothermal therapy prevent cancer growth and metastasis. J Immunother Cancer. 2019;7:220. doi:10.1186/s40425-019-0702-1.
- Dubensky TW Jr, Reed SG. Adjuvants for cancer vaccines, seminars in immunology. Semin Immunol. 2010 Jun;22(3):155–161. doi: 10.1016/j.smim.2010.04.007.
- Ebensen T, Delandre S, Prochnow B, Guzman CA, Schulze K. The combination vaccine adjuvant system alum/c-di-AMP results in quantitative and qualitative enhanced immune responses post immunization. Front Cell Infect Microbiol. 2019;9:31. doi:10.3389/fcimb.2019.00031.
- Kool M, Fierens K, Lambrecht BN. Alum adjuvant: some of the tricks of the oldest adjuvant. J Med Microbiol. 2012;61:927–934. doi:10.1099/jmm.0.038943-0.
- Mata-Haro V, Cekic C, Martin M, Chilton PM, Casella CR, Mitchell TC. The vaccine adjuvant monophosphoryl lipid A as a TRIF-biased agonist of TLR4. Science. 2007;316:1628–1632. doi:10.1126/science.1138963.
- Casella CR, Mitchell TC. Putting endotoxin to work for us: monophosphoryl lipid A as a safe and effective vaccine adjuvant. Cell Mol Life Sci. 2008;65:3231. doi:10.1007/s00018-008-8228-6.
- Rothenfusser S, Tuma E, Endres S, Hartmann G. Plasmacytoid dendritic cells: the key to CpG. Hum Immunol. 2002;63:1111–1119. doi:10.1016/S0198-8859(02)00749-8.
- Mo JH, Chung YJ, Hayashi T, Lee J, Raz E. The role of plasmacytoid and myeloid dendritic cells in induction of asthma in a mouse model and the effect of a TLR9 agonist on dendritic cells. Allergy Asthma Immunol Res. 2011;3:199–204. doi:10.4168/aair.2011.3.3.199.
- Behrens EM, Sriram U, Shivers DK, Gallucci M, Ma Z, Finkel TH, Gallucci S. Complement receptor 3 ligation of dendritic cells suppresses their stimulatory capacity. J Immunol. 2007;178:6268–6279. doi:10.4049/jimmunol.178.10.6268.
- Capasso A, Lang J, Pitts TM, Jordan KR, Lieu CH, Davis SL, Diamond JR, Kopetz S, Barbee J, Peterson J, et al. Characterization of immune responses to anti-PD-1 mono and combination immunotherapy in hematopoietic humanized mice implanted with tumor xenografts. J Immunother Cancer. 2019;7:37. doi:10.1186/s40425-019-0518-z.