
ABSTRACT
Purpose
This was a phase I/II adoptive T cell trial in 7 locally advanced and metastatic pancreatic cancer patients using 3–8 infusions of anti-CD3 x anti-EGFR bispecific antibody armed activated T cells (BATs) to determine safety, the maximum tolerated dose (MTD), immune responses, time to progression (TTP), and overall survival (OS). Study Design: T cells obtained by apheresis were expanded and armed with EGFRBi, cryopreserved for infusions. In a phase I dose escalation, five patients received three weekly infusions of 10–40 × 109 BATs/infusion followed by a booster infusion 3 months later, and 2 patients received 8 infusions twice weekly for 4 weeks in a phase II. The trials were registered at http://www.clinicaltrials.gov, NCT01420874 and NCT02620865. Results: There were no dose-limiting toxicities (DLTs), and the targeted dose of 80 × 109 BATs was met. The median TTP is 7 months, and the median OS is 31 months. Two patients had stable disease for 6.5 and 25+ months, and two patients developed complete responses (CRs) after restarting chemotherapy. Infusions of BATs induced anti-pancreatic cancer cytotoxicity, innate immune responses, cytokine responses (IL-12, IP-10), and shifts in CD4 and CD8 Vβ repertoire with enhanced cytoplasmic IFN-γ staining in the Vβ repertoire of the CD8 subset that suggest specific clonal TCR responses. Conclusions: Infusions of BATs are safe, induce endogenous adaptive anti-tumor responses, and may have a potential to improve overall survival.
Introduction
Chemotherapy (chemo) for locally advanced pancreatic cancer (LAPC) and metastatic pancreatic cancer (MPC) is associated with poor response and grim survival rates. Progress has been dismal with incremental results since 1997 when gemcitabine (gem) was approved for first-line treatment.1 By 2013, nab-paclitaxel (nab) and gem had improved overall survival (OS) to 8.5 months from the 6.7 months for gem alone.Citation2 FOLFIRINOX (5-FU, leucovorin, irinotecan, and oxaliplatin) regimen further improved the median OS to 11.1 monthsCitation3 with increased toxicity. Other combinations with gem failed to provide benefit.Citation1,Citation4-Citation14 OS after second line therapy using various combinations ranges from 4.5 to 11.4 months.Citation15-Citation20 These dismal results drive the need to develop novel and innovative therapies.
Although cetuximab was approved for metastatic colon cancer, gem and cetuximab failed in clinical trials for PC.Citation21 Arming anti-CD3 activated T cells with anti-CD3 x anti-EGFR bispecific antibody (EGFRBi) turns each activated T cell into a specific cytotoxic T lymphocyte.Citation22 Our preclinical studies showed that EGFRBi-armed ATC (BATs) killed PC cancer cell line MiaPaCa-2, secreted cytokines, and inhibited tumor growth.Citation22 Engaging the T cell receptor on the redirected T cell with EGFRBi triggers activation, proliferation, cytokine/chemokine synthesis, and non-MHC restricted cytotoxicity while the anti-EGFR targets tumors.Citation22
This series asks whether BATs are safe and induce adaptive cellular responses that provide anti-tumor activity. Seven LAPC and PC patients from phase I and II trials highlight the exceptional clinical outcomes.
Materials and methods
Production of bispecific antibody
Bispecific antibodies (BiAbs) were produced by chemical heteroconjugation of muromonab which is, murine IgG2a anti-CD3 monoclonal antibody (OKT3) and cetuximab which is a IgG1 chimeric anti-epidermal growth factor receptor (EGFR) (Erbitux®, ImClone LLC., Branchburg, NJ) or OKT3 and Herceptin (a humanized anti-HER2 IgG1, Genentech Inc., South San Francisco, CA) using sulfo-succinimidyl 4-(N-Maleimidomethyl) cyclohexane-1-carboxylate (Sulfo-SMCC) and Traut’s reagents as described previously.Citation23
Expansion, generation, and arming of activated T cells (ATC)
T cells from the pheresis product were activated with 20 ng/ml of OKT3 and expanded in 100 IU/ml of IL-2 for 14 days in RPMI-1640 supplemented with 2% human serum as previously described.Citation24 Harvested ATC were armed with bispecific antibody anti-CD3 x anti-EGFR [EGFRBi] at a pre-optimized concentration of 50 ng/106 ATC as previously described.Citation22 Armed ATC were washed twice to remove unbound EGFRBi and cryopreserved in four or eight equal aliquots until they are thawed at the bedside. The EGFRBi armed ATC (EGFR BATs) were quality controlled to be pathogen and mycoplasma free with acceptable endotoxin levels as described.Citation22-Citation24
Study population, enrollment, and eligibility
The phase I protocol WSU 2011–025 entitled “Treatment of Advanced Colorectal or Pancreatic Cancer with anti-CD3 x anti-Erbitux® Armed Activated T cells (Phase Ib)” (NCT01420874) and a phase II protocol WSU 2015–100 entitled “Phase Ib/II Treatment of Advanced Pancreatic Cancer with anti-CD3 x anti-EGFR-Bispecific Antibody Armed Activated T-Cells (BATs) in Combination with Low Dose IL-2 and GM-CSF” (NCT02620865) were approved by Wayne State University Institutional Review Board and the FDA. The trials were conducted at Barbara Ann Karmanos Cancer Institute (KCI) and monitored by the KCI data safety monitoring committee. All patients enrolled had pathological confirmation of their disease at the time of diagnosis. The primary endpoint of the phase I study is safety and feasibility of multiple infusion of EGFR BATs in PC and CRC patients. The phase I trial targeting colorectal and pancreatic cancer with BATs included 5 PC and 6 colorectal cancer (CRC) pts who received up to 78.8 × 1010 (median of 43, range 20.4–78.8 × 109) BATs in 3–4 doses given once per week for 3 weeks. Three weeks before immunotherapy, one treatment of modified FOLFOX6 was given as bridging therapy. Immune depletion therapy followed a standard 3 + 3 dose-escalation of 10, 20, and 40 × 109 BATs/infusion (). The same dose was given after 3 months if the patient was stable or better. Patient IT20091 with PC in phase 1 was treated twice (IT20103).
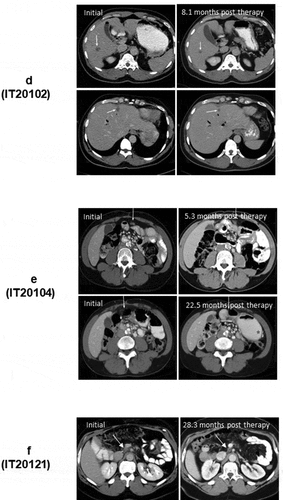
Figure 1. (a). The protocol schema for the phase I, which included colorectal cancer and pancreatic cancer patients, is shown with infusions of EGFRBi armed T cells (BATs) with restaging 4 weeks after the last infusion of BATs and a boost of BATs. 1b) Protocol schema for the phase II with 2 infusions per week for 4 weeks in combination with low dose IL-2 and GM-CSF. 1 c) The overall survival and time to progression for the 7 patients. 1d) CT scan of patient IT20102 before immunotherapy (upper and lower left panels) and after immunotherapy (upper and lower right panels) with stable disease and a decrease in size by 27%; 1e) CT scan of patient IT20104 before immunotherapy, persistence of the lesion after 5 months of immunotherapy, and complete response by 7 months post immunotherapy that persisted almost 2 years; 1 f) CT of patient IT20121 shows stable disease before IT to 25 months after enrollment.
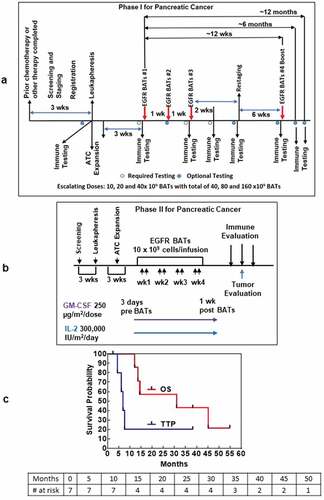
Figure 1. (Continued).
The primary endpoint in the phase II was to confirm safety and estimate clinical efficacy. In the phase 2, 2 MPC patients, after receiving bridging chemotherapy, received eight BATs infusions twice weekly in combination with low dose IL-2 (300,000 IU/m2/day) and granulocyte-macrophage colony-stimulating factor (GM-CSF) (250 µg/m2/twice per week) for 4 weeks (). The CRC patients were included only for evaluation of toxicities. Toxicity scoring was done with CTCAE v4.0.
Immune evaluations
Specific cytotoxicityCitation25 and IFN-γ EliSpots were assessed using fresh patient PBMC exposed to MiaPaCa-2 cells (PC-line) and K562 (NK activity).Citation24 Phenotyping, serum cytokine and chemokine levels, cytoplasmic IFN-γ staining Vβ repertoire, IgG antibodies to pooled CEA, HER2, and EGFR peptides were evaluated.Citation26
Statistical analyses
Immune responses were compared to each time point from pre-study IFN-γ Elispots and differences between cytotoxicity directed at PC cell line were tested using Wilcoxon signed-rank test. Log-rank tests GraphPad Prism was used to generate the Kaplan–Meier curves.
Results
Clinical toxicities
The common toxicities grade 1–3 side effects were chills, headache, hypertension, hypotension, and fatigue (Tables 1 and 2). There were no persistent grade 3 (>72 hours) or grade 4 and no DLTs as previously described for treatment of metastatic breast cancer.Citation24 The chemical heteroconjugation of OKT3 (anti-CD3 mAb) with Erbitux (anti-EGFR mAb) has been described.Citation22 There was no change in the manufacture of BATs between the two protocols. The infusions were performed in the outpatient setting without any dose reductions. Hospital admission or ICU supportive care was not required for any of the patients.
Clinical responses in phase I/phase II patients
The age, disease status, prior chemotherapy, BATs doses, time to progression (TTP), overall survival (OS), clinical status, and clinical course is shown for seven patients with LAPC or MPC in . The median OS is 31.0 months for 7 patients with TTP of 7.0 months ().
Table 1. Toxic reaction incidence and grade by dose level in phase I study.
Table 2. Toxic reaction incidence and grade by dose level in phase II study.
Table 3. Patient characteristics.
Patient IT20087 (#1) was a 58-year-old white man with T4 disease and multiple liver metastases, who was receiving FOLFIRINOX before immunotherapy (IT) with EGFR BATs. He progressed after IT and expired 13.6 months after enrollment.
Patient IT20091 (#2) was a 63-year-old white man with extensive metastases to liver post-Whipplee procedure. He was treated with only 9.3 × 109 total BATs in 8 divided doses after he had developed minimal residual disease after 26+ cycles of FU, Leukovorin/5 FU, and FOLFIRINOX. His disease had recurred, and he was restarted on FOLFIRINOX, which induced a CR at 9 months. He (IT20103) was given another course of 4 infusions of BATs totaling 78.8 × 109 BATs. Patient eventually progressed and expired at 31 months.
Patient IT20092 (#3) was a 64-year-old white man with T2b disease with abdominal lymph nodes, who received gemcitabine, 5 FU, and radiation. Patient was treated with 36 × 109 BATs. He developed an acute abdomen with an inflamed appendix that showed metastatic pancreatic cancer and expired at 14.5 months.
Patient IT20102 (#4) was a 57-year-old African American man presented with a pancreatic mass and multiple liver lesions that were positive for CK7, CK19, and CK20 associated with an elevated serum CA19-9. After 14 cycles of FOLFIRINOX, he was switched to FOLFOX. After receiving three infusions of 18.9 × 109 BATs/infusion, and received a boost 3 months later. Patient IT20102 had a minor response (↓27%) based on response of target lesions in the liver using sum of diameters compared to baseline that persisted for 6.5 months ().
Patient IT20104 (#5) is a 51-year-old African American woman with T4 disease and a mesenteric lymph node that was stable for 1 year on capecitabine. Her pretreatment scan (, upper left) shows a mesenteric node that increased in size after three infusions (, upper right) from 1.0 × 0.7 cm to 1.5 × 0.9 cm. Treatment was stopped due to “progression,” and the same dose of capecitabine was restarted. Follow-up scans shown in (lower left and right) showed no evidence of disease. She remained free of disease until 54 months after enrollment (24 months off all therapy) when disease recurred, she received radiation therapy.
In the phase II study, patient IT20120 (#6) was a 72-year-old white female with T4 disease with abdominal nodes, who had received FOLFIRINOX and radiation. He received 80 × 109 BATs but progressed and expired at 12.2 months. Patient IT20121 is a 54-year-old African American man with stable disease for 25+ months at his last scan at 22+ months. He is clinically well at 38.5 months ().
Immune evaluations
The specific cytotoxicity shown in (upper left panel) for all seven patients increased significantly (p < 0.01) from baseline mean±SD of 11.5 ± 6.5 (range 3–22%) to 24 ± 6 (range 13.9–31.9%) after immunotherapy at an effector:target (E/T) ratio of 25:1. Similarly, the specific IFN-γ EliSpots directed at MiaPaCa-2 cells for all seven patients increased significantly (p < 0.03) from a baseline mean±SD of 364 ± 122 (range 93–1033) to 1408 ± 376 (range 310–3203) EliSpots/106 PBMC after IT (, lower left panel). Innate immune responses measured by cytotoxicity directed at the NK target K562 by specific 51Cr release increased significantly (p < 0.04) from baseline mean±SD of 5.3 ± 5.0 (range 0–12.4%) to 13 ± 6.8 (range 1.6–18.2%) after IT at an E/T of 25:1 and IFN-γ EliSpots increased from pretreatment mean of 273 EliSpots/106 PBMC (158–542) to a mean level of 686 EliSpots/106 PBMC (342–1208) after IT (p < 0.07). compares the cytotoxicity and IFN-γ Elispot responses against PC specific MiaPaCa-2 and NK target K562 cells at preIT and postIT time points (postIT time point that showed the highest anti-tumor response).
Figure 2. (a).PBMC from all seven PC patients were tested for direct cytotoxicity and IFN-γ Elispots responses against MiaPaCa-2 and five phase I patients were tested against NK cell targets K562 for cytotoxicity and IFN-γ Elispots responses; 2b) Shows the highest specific (MiaPaCa-2) and innate (K562) cytotoxic and IFN-γ Elispots responses postIT compared to preIT base line responses; 2 c) IP-10 and IL-8 tested before and after immunotherapy in 7 patients; 2d) PBMC of patient IT20091 obtained at various time points were left unstimulated (background), stimulated with MiaPaCa-2, or K562. Data are expressed as number of IFN-γ Elispots per million PBMC plated at pre-IT; post BATs infusion #1, #3, and #4; 2 and 4 weeks after infusion #3; pre infusion #4; and post boost infusion at 2 and 4 weeks.
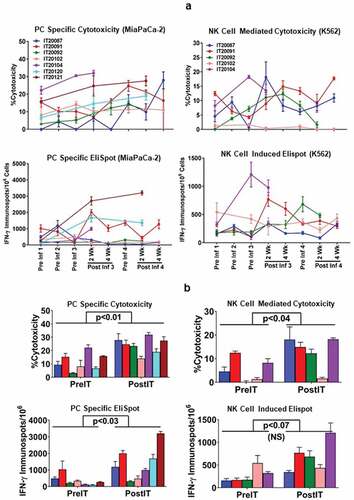
Figure 2. (Continued).
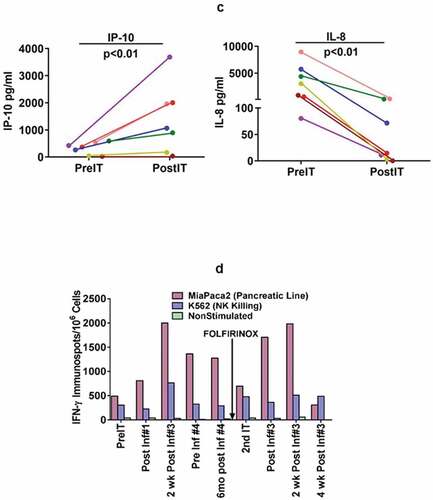
Interestingly, IFN-γ induced chemokine IP-10 [CXCL10] levels (, left panel) increased significantly (p < 0.04) from a mean preIT of 434 ± 130 pg/ml (range 261–590) to a mean post IT of 1992 ± 1108 pg/ml (range 896–3687). IL-8 levels decreased significantly (p < 0.01) from a mean level preIT of 3369 ± 3239 pg/ml (range 80–8956) for IL-8 (, right panel) to a mean level post IT of 59.9 ± 73.1 pg/ml (range 0–180) 2 weeks after the 3rd infusion (p < 0.01). There were no significant changes from baseline at the designated time points for serum levels of IL-2, IL-2R, IL-6, IL-12p40p70, TNFα, IFNα, IFN-γ, GM-CSF, MIP1α, MIP1β, Eotaxin, RANTES, and MCP-1 (data not shown).
Phenotyping of the clinical product and pre- and postIT PBMC from seven patients at the designated time points are summarized in and . Phenotyping for pre- and post IT PBMC was performed for the proportion of CD4+/CD8+ T cells, effector memory T cells (CD45RO+/CD45RA−), T regulatory cells (CD4+/CD25+/CD127−) and MDSC (CD11b+/CD33+/HLA-DR−) (). IT20102 and IT20104 appear to have higher proportions of CD8+ T cells in ATC and higher proportions of CD8+ T cells in PBMC after IT.
Table 4. Phenotype and characteristics of the apheresis and harvest product.
Table 5. Phenotype of pre and post IT T cell subpopulations and myeloid-derivedd suppressor cells.
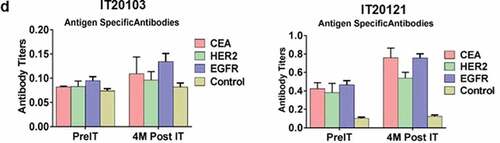
In a pilot screen of the Vβ repertoire of PBMC from IT20091 before and after IT, there was a suggestion that there were shifts in the repertoire that could be detected (). In a subsequent Vβ repertoire study on IT20121, we asked what members of the Vβ clonal repertoire in CD4+ T cells () and CD8+ T cells () would respond to stimulation with MiaPaCa-2 by producing cytoplasmic IFN-γ before IT, in the middle of IT, and after IT. There were no changes seen in the CD4+ T cell Vβ repertoire; however, there were enhanced cytoplasmic IFN-γ producing clones (Vβ1, 2, 4, 5.1, 13.6, 17, 22) in responses to MiaPaCa-2 cells in the CD8+ T cells after IT. Other Vβ clones were either decreased (Vβ 13.2) or there were no change in Vβ clones after IT. Specific antibody levels measured by antigen-specific antibody ELISAs to CEA, HER2, and EGFR peptides in IT20103 (serum collected after second BATs treatment of IT20091) and IT20121 increased after treatment from levels seen prior to IT (). These results complement the high anti-MiaPaCa-2 IFN-γ EliSpot responses seen for IT20121.
Figure 3. (a) In a pilot screen of the Vβ repertoire of PBMC from IT20091 before and after IT, there was a suggestion that there were shifts in the repertoire that could be detected; 3b) In a subsequent study on IT200121, we asked what members of the Vβ repertoire in CD4 cells would respond to stimulation with MiaPaCa-2 by producing cytoplasmic IFN-γ before IT, in the middle of IT, and after IT. There were no changes seen in the CD4 Vβ repertoire; 3 c) With IT200121, we again asked what members of the Vβ repertoire, this time in CD8 cells, would respond to stimulation with MiaPaCa-2 by producing cytoplasmic IFN-γ before IT, in the middle of IT, and after IT. Enhanced cytoplasmic IFN-γ responses in CD8 cells were seen in Vβ1, 2, 4, 5.1, 13.6, 17, 22, with decreased responses seen in 13.2 after IT. 3d) Serum IgG antibody titers to EGFR, CEA, and HER2 doubled above baseline but were not significant.
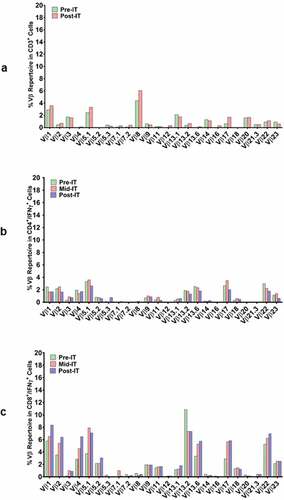
Figure 3. (Continued).
Discussion
This study provides encouraging clinical results in a heavily pretreated heterogeneous group of patients with disease ranging from no measurable disease to bulky multiple viscera lesions in the liver or surrounding vital structures, with most patients having bulky disease. There were no DLTs or cytokine storm. The most frequent side effects of chills, headache, hypertension, hypotension, and fatigue, were grades 1–3, with no persistent grade 3 (>72 hours), and no grade 4 or 5. The phase I dose schedule of one infusion per week for 3 weeks followed by a boost at 3 months, and phase II dose schedule of biweekly infusions for 4 weeks, were safe, and the targeted dose was technically feasible. The addition of low dose IL-2 and GM-CSF to the phase II regimen did not increase toxicities.
The remarkable median OS for 7 PC patients is 31 months with a median TTP of 7.0 months, with 2 patients alive at 54 and 38 months. Notably, everyone survived beyond a year, and one patient was stable at 25+ months and is alive at 38 months. The two patients (IT20091 and IT20104) who developed complete responses after restarting chemotherapy had robust specific IFN-γ EliSpots directed at MiaPaCa-2 and K562 cells (). The remarkable median OS for 7 PC patients is 31 months with a median TTP of 7.0 months, with 2 patients alive at 54 and 36 months. Notably, everyone survived beyond a year. IT20121 showed increased IFN-γ Elispots stimulated with MiaPaCa-2 from a mean of 267 (±38)/106 PBMC to a mean of 3203/106 PBMC post-therapy. It is not clear why IT20102 had a minor response with stable disease up to 6.5 months without making systemic immune responses. Since we evaluated the five patients in the phase I trial and the 2 patients in the phase II, the small numbers only permit a descriptive comment. Intriguingly, out of the total of seven patients, there were two CRs and one minor responder in the phase I trial and one responder in the phase II trial who developed long-term stable disease off all therapy for more than 48 months.
There was a higher proportion of CD8+ cells in ATC product in the PBMC of IT20104 and IT20121 that was associated with higher cytotoxicity and IFN-γ EliSpots after as few as 3 infusions of BATs. The PBMC from IT20104, IT20121, and IT20091, who were long-term survivors at 56, 38, and 31 months, showed elevated and persistent cytotoxicity and IFN-γ EliSpot responses with concomitant increases in NK cell activity post immunotherapy. The IFNγ responses in Vβ1, 2, 4, 5.1, 13.6, 17, 22 clones strongly suggest endogenous specific TCR clonal responses developed during therapy directed at multiple tumor antigens. Furthermore, changes in serum IP-10 and IL-8 suggest that the clinical responses may be associated with a systemic Th1 type shift in the patients that led to clinical responses and/or the maintenance of stable disease.
The MHC class I expression plays a critical role in T-cell mediated immune response.Citation27 In pancreatic tumor cells, HLA class I expression was associated with increased cytotoxic T-cells infiltration and a favorable prognosis in patients. Likewise, increased infiltration of CD3+, CD8+, and CD20+ tumor-infiltrating lymphocytes correlated with improved PFS and OS in human pancreatic carcinoma in the univariate analysis.Citation28 Although in this study EGFR expression, MHC class I expression and T cell infiltration in tumor biopsies are not known due to inaccessibility of tumor biopsies, but we have previously shownCitation23,Citation26 that BATs can target breast cancer cells even with low HER2 expression and that BATs mediated cytotoxicity is non-MHC mediated.Citation23,Citation26
The mechanism for the enhanced chemotherapy responses following the infusion of BATs in IT20091 and IT20104 is unclear; however, multiple infusions and larger doses of BATs may increase trafficking to and persistence of cytotoxic BATs in solid tumors. In an earlier report, multiple infusions of mesothelin-specific chimeric antigen receptor (CAR) mRNA-engineered T cells induced anti-pancreatic cancer tumor activity in 2 patients, with evidence of epitope spreading, resulting in stable disease in 1 patient for 6 months and for 77 days in the other patient.Citation29 The multiple infusion strategy may help overcome T cell exhaustion seen in CAR therapy for solid tumors.Citation30–Citation32 Multiple infusions of BATs not only lyse tumors releasing tumor-associated antigens, but also provide multiple local bursts of Th1 cytokines that overcome T cell exhausting, leading to in situ immunization of the endogenous immune system as seen in our phase I trial using anti-CD3 x anti-HER2 bispecific antibody armed ATC.Citation24 Furthermore, modulation of the tumor microenvironment may make it more effective for chemotherapy to penetrate the tumor. BATs “immunosensitization” may damage transporter pumps, alter cell membrane integrity, or modulate tumor stromal so therapeutic agents can penetrate and be effective. Secretion of Th1 cytokines and chemokines upon repeated BATs infusions may modify the apoptotic signaling pathways of the tumors or attenuate the expression of PD-1, PDL-1, or CTLA-4; the secretion of immunosuppressive cytokines such as IL-8, IL-10 or TGF-β; or other immunosuppressive circuits in the tumor microenvironment. Studies to dissect and understand these mechanisms to enhance anti-tumor effects are ongoing. Future approaches will combine BATs with “immunosensitization,” with the addition of checkpoint inhibitors.
Author contributions
VK, DS, AT, and LGL carried out the preclinical studies; LGL, MC, AD, GD, and AFS designed the clinical protocol; LGL, AT, AFS, MD, HA, and GD collected and analyzed the clinical results; AT and LGL collated and analyzed the immune evaluations; LGL, AT, AFS, and MC wrote the manuscript.
LGL, MC, AD, KF, MD, and AFS directed, coordinated, and managed patient care.
Originality disclosure
The data presented in this manuscript are original and has not been published elsewhere except in the form of abstracts and poster presentations at symposia and meetings.
Disclosure of potential conflicts of interest
LGL is co-founder of Transtarget Inc. and serves on the SAB for Rapa Therapeutics. AT is co-founder of Nova Immune Platform LLC. MC, AD, VK, DS, KF, MD, GD, HA, PP, and AFS have no conflicts of interest.
Acknowledgments
We acknowledge the nurse clinical coordinators Patricia Steele and Karen Meyers for their tireless efforts in serving the immunotherapy patients, the cGMP manufacturing staff for producing BATs, the immune evaluation staff for immune testing, and the outpatient infusion nurses in the Dresner Clinic at Karmanos Cancer Institute for their dedication to patient care.
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
References
- Burris HA III, Moore MJ, Andersen J, Green MR, Rothenberg ML, Modiano MR, Cripps MC, Portenoy RK, Storniolo AM, Tarassoff P, et al. Improvements in survival and clinical benefit with gemcitabine as first- line therapy for patients with advanced pancreas cancer: a randomized trial. JClinOncol. 1997;15:2403–11. doi:10.1200/JCO.1997.15.6.2403.
- Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, Seay T, Tjulandin SA, Ma WW, Saleh MN, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med. 2013;369(18):1691–1703. doi:10.1056/NEJMoa1304369.
- Conroy T, Desseigne F, Ychou M, Bouché O, Guimbaud R, Bécouarn Y, Adenis A, Raoul J-L, Gourgou-Bourgade S, de la Fouchardière C, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. New Eng J Medi. 2011;364(19):1817–1825. doi:10.1056/NEJMoa1011923.
- Louvet C, Labianca R, Hammel P, Lledo G, Zampino MG, André T, Zaniboni A, Ducreux M, Aitini E, Taïeb J, et al. Gemcitabine in combination with oxaliplatin compared with gemcitabine alone in locally advanced or metastatic pancreatic cancer: results of a GERCOR and GISCAD phase III trial. J Clin Oncol. 2005;23(15):3509–3516. doi:10.1200/JCO.2005.06.023.
- Rocha Lima CM, Green MR, Rotche R, Miller WH, Jeffrey GM, Cisar LA, Morganti A, Orlando N, Gruia G, Miller LL, et al. Irinotecan plus gemcitabine results in no survival advantage compared with gemcitabine monotherapy in patients with locally advanced or metastatic pancreatic cancer despite increased tumor response rate. J Clin Oncol. 2004;22(18):3776–3783. doi:10.1200/JCO.2004.12.082.
- Oettle H, Richards D, Ramanathan RK, van Laethem JL, Peeters M, Fuchs M, Zimmermann A, John W, Von Hoff D, Arning M, et al. A phase III trial of pemetrexed plus gemcitabine versus gemcitabine in patients with unresectable or metastatic pancreatic cancer. Ann Oncol. 2005;16(10):1639–1645. doi:10.1093/annonc/mdi309.
- Reni M, Cordio S, Milandri C, Passoni P, Bonetto E, Oliani C, Luppi G, Nicoletti R, Galli L, Bordonaro R, et al. Gemcitabine versus cisplatin, epirubicin, fluorouracil, and gemcitabine in advanced pancreatic cancer: a randomised controlled multicentre phase III trial. Lancet Oncol. 2005;6(6):369–376. doi:10.1016/S1470-2045(05)70175-3.
- Stathopoulos GP, Syrigos K, Aravantinos G, Polyzos A, Papakotoulas P, Fountzilas G, Potamianou A, Ziras N, Boukovinas J, Varthalitis J, et al. A multicenter phase III trial comparing irinotecan-gemcitabine (IG) with gemcitabine (G) monotherapy as first-line treatment in patients with locally advanced or metastatic pancreatic cancer. Br J Cancer. 2006;95(5):587–592. doi:10.1038/sj.bjc.6603301.
- Bernhard J, Dietrich D, Scheithauer W, Gerber D, Bodoky G, Ruhstaller T, Glimelius B, Bajetta E, Schüller J, Saletti P, et al. Clinical benefit and quality of life in patients with advanced pancreatic cancer receiving gemcitabine plus capecitabine versus gemcitabine alone: a randomized multicenter phase III clinical trial–SAKK 44/00-CECOG/PAN.1.3.001. J Clin Oncol. 2008;26:3695–3701. doi:10.1200/JCO.2007.15.6240.
- Lygidakis NJ, Spentzouris N, Theodoracopoulos M, Dedemadi G, Gemos K, Kyriakou A. Pancreatic resection for pancreatic carcinoma combined with neo- and adjuvant locoregional targeting immuno-chemotherapy–a prospective randomized study. Hepatogastroenterology. 1998;45:396–403.
- Colucci G, Giuliani F, Gebbia V, Biglietto M, Rabitti P, Uomo G, Cigolari S, Testa A, Maiello E, Lopez M, et al. Gemcitabine alone or with cisplatin for the treatment of patients with locally advanced and/or metastatic pancreatic carcinoma: a prospective, randomized phase III study of the Gruppo Oncologia dell’Italia Meridionale. Cancer. 2002;94(4):902–910. doi:10.1002/cncr.10323.
- Moore MJ, Hamm J, Dancey J, Eisenberg PD, Dagenais M, Fields A, Hagan K, Greenberg B, Colwell B, Zee B, et al. Comparison of gemcitabine versus the matrix metalloproteinase inhibitor BAY 12-9566 in patients with advanced or metastatic adenocarcinoma of the pancreas: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. 2003;21(17):3296–3302. doi:10.1200/JCO.2003.02.098.
- Heinemann V, Quietzsch D, Gieseler F, Gonnermann M, Schönekäs H, Rost A, Neuhaus H, Haag C, Clemens M, Heinrich B, et al. Randomized phase III trial of gemcitabine plus cisplatin compared with gemcitabine alone in advanced pancreatic cancer. J Clin Oncol. 2006;24(24):3946–3952. doi:10.1200/JCO.2005.05.1490.
- Berlin JD, Catalano P, Thomas JP, Kugler JW, Haller DG, Benson AB III. Phase III study of gemcitabine in combination with fluorouracil versus gemcitabine alone in patients with advanced pancreatic carcinoma: eastern cooperative oncology group trial E2297. J Clin Oncol. 2002;20:3270–3275. doi:10.1200/JCO.2002.11.149.
- Wang-Gillam A, Von Hoff D, Siveke J, Hubner R, Belanger B, Pipas JM, Chen L-T. nanoliposomal irinotecan in the clinical practice guideline for metastatic pancreatic cancer: applicability to clinical situations. J Clin Oncol. 2017;35(6):689–690. doi:10.1200/JCO.2016.70.1607.
- Rahma OE, Duffy A, Liewehr DJ, Steinberg SM, Greten TF. Second-line treatment in advanced pancreatic cancer: a comprehensive analysis of published clinical trials. Ann Oncol. 2013;24:1972–1979. doi:10.1093/annonc/mdt166.
- Scheithauer W, Ramanathan RK, Moore M, Macarulla T, Goldstein D, Hammel P, Kunzmann V, Liu H, McGovern D, Romano A. Dose modification and efficacy of nab-paclitaxel plus gemcitabine vs. gemcitabine for patients with metastatic pancreatic cancer: phase III MPACT trial. J Gastrointest Oncol. 2016;7(3):469–478. doi:10.21037/jgo.2016.01.03.
- Gill S, Ko YJ, Cripps C, Beaudoin A, Dhesy-Thind S, Zulfiqar M, Zalewski P, Do T, Cano P, Lam WYH, et al. PANCREOX: a randomized phase III study of 5-fluorouracil/leucovorin with or without oxaliplatin for second-line advanced pancreatic cancer in patients who have received gemcitabine-based chemotherapy. J Clin Oncol. 2016. doi:10.1200/JCO.2016.68.5776.
- Chiorean EG, Von Hoff DD, Tabernero J, El-Maraghi R, Ma WW, Reni M, Harris M, Whorf R, Liu H, Li JS, et al. Second-line therapy after nab-paclitaxel plus gemcitabine or after gemcitabine for patients with metastatic pancreatic cancer. Br J Cancer. 2016;115(2):188–194. doi:10.1038/bjc.2016.185.
- Zhang Y, Hochster H, Stein S, Lacy J. Gemcitabine plus nab-paclitaxel for advanced pancreatic cancer after first-line FOLFIRINOX: single institution retrospective review of efficacy and toxicity. Exp Hematol Oncol. 2015;4(1):29. doi:10.1186/s40164-015-0025-y.
- Cunningham D, Humblet Y, Siena S, Khayat D, Bleiberg H, Santoro A, Bets D, Mueser M, Harstrick A, Verslype C, et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med. 2004;351(4):337–345. doi:10.1056/NEJMoa033025.
- Reusch U, Sundaram M, Davol PA, Olson SD, Davis JB, Demel K, Nissim J, Rathore R, Liu PY, Lum LG. Anti-CD3 x anti-EGFR bispecific antibody redirects T cell cytolytic activity to EGFR-positive cancers in vitro and in an animal model. ClinCancer Res. 2006;12:183–190.
- Sen M, Wankowski DM, Garlie NK, Siebenlist RE, Van Epps D, LeFever AV, Lum LG. Use of anti-CD3 × anti-HER2/neu bispecific antibody for redirecting cytotoxicity of activated T cells toward HER2/neu + tumors. J Hematother Stem Cell Res. 2001;10(2):247–260. doi:10.1089/15258160151134944.
- Lum LG, Thakur A, Al-Kadhimi Z, Colvin GA, Cummings FJ, Legare RD, Dizon DS, Kouttab N, Maizel A, Colaiace W. Targeted T-cell therapy in stage IV breast cancer: a phase I clinical trial. Clin Cancer Res. 2015;21(10):2305–2314. doi:10.1158/1078-0432.CCR-14-2280.
- Grabert RC, Cousens LP, Smith JA, Olson S, Gall J, Young WB, Davol PA, Lum LG. Human T cells armed with Her2/neu bispecific antibodies divide, are cytotoxic, and secrete cytokines with repeated stimulation. Clin Cancer Res. 2006;12:569–576. doi:10.1158/1078-0432.CCR-05-2005.
- Thakur A, Rathore R, Kondadasula SV, Uberti JP, Ratanatharathorn V, Lum LG. Immune T cells can transfer and boost anti-breast cancer immunity. Oncoimmunology. 2018;7(12):e1500672. doi:10.1080/2162402X.2018.1500672.
- Ryschich E, Notzel T, Hinz U, Autschbach F, Ferguson J, Simon I, Weitz J, Frohlich B, Klar E, Buchler MW, et al. Control of T-cell-mediated immune response by HLA class I in human pancreatic carcinoma. Clin Cancer Res. 2005;11:498–504.
- Miksch RC, Schoenberg MB, Weniger M, Bosch F, Ormanns S, Mayer B, Werner J, Bazhin AV, D’Haese JG. Prognostic impact of tumor-infiltrating lymphocytes and neutrophils on survival of patients with upfront resection of pancreatic cancer. Cancers (Basel). 2019;11(1):39. doi:10.3390/cancers11010039.
- Beatty GL, Haas AR, Maus MV, Torigian DA, Soulen MC, Plesa G, Chew A, Zhao Y, Levine BL, Albelda SM, et al. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol Res. 2014;2:112–120. doi:10.1158/2326-6066.CIR-13-0170.
- Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. 2015;15:486–499. doi:10.1038/nri3862.
- Sen DR, Kaminski J, Barnitz RA, Kurachi M, Gerdemann U, Yates KB, Tsao H-W, Godec J, LaFleur MW, Brown FD, et al. The epigenetic landscape of T cell exhaustion. Science. 2016;354(6316):1165–1169. doi:10.1126/science.aae0491.
- Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, Smith JP, Walker AJ, Kohler ME, Venkateshwara VR, et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med. 2015;21(6):581–590. doi:10.1038/nm.3838.