
ABSTRACT
Background
High tumor mutational burden (TMB) predicts checkpoint blockade responsiveness, although the association with outcomes may be nuanced in certain tissue contexts. The correlation between TMB and cytotoxic chemotherapy sensitivity is unknown. This study evaluated the relationship between TMB and outcome in patients with solid tumors receiving cytotoxic chemotherapy.
Methods
University of California San Diego patients who received cytotoxic chemotherapy within one year after biopsy for TMB evaluation were included in a retrospective analysis. Physician notes and imaging reports in the electronic medical record were reviewed to determine clinical benefit and progression-free survival (PFS).
Results
Among 1526 patients with TMB availability, there were 294 eligible patients who received chemotherapy. There were no significant differences in TMB between those with stable disease ≥6 months/partial response/complete response versus others (t-test, p = .22). There were no significant differences in PFS for patients with TMB <10 vs. TMB ≥10 mutations/Mb (log-rank test, median and 95% CI: 6.0 (4.8–7.4) vs. 5.4 (4.3–6.6) months; p = .21). Nor were there significant differences in PFS for patients with a TMB <10 vs. TMB ≥10 mutations/mb for breast (p = .07), lung (p = .47), or gastrointestinal cancer (p = .53).
Conclusions
In summary, TMB was not predictive of stable disease ≥6 months/partial response/complete response or PFS in patients receiving cytotoxic chemotherapy.
Trials Registration
NCT02478931
Introduction
Tumor mutational burden (TMB) is the total number of mutations identified per megabase (1,000,000 basepairs) of coding DNA. Tumors with higher TMB likely harbor more neo-antigens and, hence, a high TMB should increase the chances of immune recognition and eradicaton of cancer cells after immunotherapy.Citation1,Citation2 Indeed, prior studies have shown that high TMB is predictive of better outcome after anti-PD-1/PD-L1 directed immunotherapies .Citation3–Citation9
Cytotoxic chemotherapy has been the mainstay of treatment for metastatic solid tumors. Chemotherapy regimens have traditionally been selected based on tumor type. Protein markers have been explored as predictive biomarkers for response to chemotherapy ,Citation10 however these markers may not be predictive for all tumor types .Citation11 There is evidence that somatic mutations in genes such as ATM and BRCA can predict responsiveness to platinum chemotherapy .Citation12 Additional biomarkers predictive of response to cytotoxic chemotherapy would be of benefit for a personalized approach to cancer treatment. The relationship between TMB and chemotherapy response is unknown. Many cytotoxic therapies affect DNA replication, and specific mutations affecting DNA repair pathways have been associated with chemotherapy responsiveness .Citation12 However, mutations can also occur in sites that are not relevant to chemotherapy response. Thus, a high TMB could theoretically make a tumor more sensitive to DNA-damaging chemotherapy; furthermore, high TMB might decrease tumor cell viability in a manner analogous to aneuploidic tumor suppression, making it vulnerable to cytotoxics .Citation13,Citation14 Alternatively, a high TMB might be associated with the presence of resistance mutations and, hence, attenuate chemotherapy sensitivity.
The current study evaluated the relationship between TMB and clinical benefit (stable disease (SD) ≥6 months/partial or complete remission (PR or CR)) or progression-free survival (PFS) in patients with solid tumors receiving cytotoxic chemotherapy, with the goal of evaluating the potential of TMB to serve as biomarker for outcome after chemotherapy.
Methods
This is a retrospective cohort study of patients enrolled in the University of California San Diego Study (UCSD) of Personalized Cancer Therapy to Determine Response and Toxicity (UCSD-PREDICT), which encompasses an institutional review board (IRB)-approved observational cohort study at UCSD designed to learn more about personalized cancer therapy, including dosing, response to treatment, and side effects. This study was performed in accordance with the UCSD IRB guidelines for data analysis and for any investigational treatments for which patients gave consent.
A database of 1526 patients was generated from consecutive patients with tumor mutational burden (TMB) on FoundationOne molecular testing results from November 2012 through December 2016. For the current study, eligible patients received cytotoxic chemotherapy, which was initiated within one year after obtaining the tissue biopsy for TMB (providing that they had not had chemotherapy within one year prior to the biopsy); the rate of SD ≥6 months/PR/CR and PFS was evaluated for the chemotherapy regimen after the biopsy. Patients who received chemotherapy concurrent with radiation, had a hematologic malignancy diagnosis, or who were on adjuvant chemotherapy without measurable disease following surgery where excluded. Patients receiving anti-PD-1/PD-L1-directed immunotherapies were excluded from the analysis. The chemotherapy regimen closest to the date of the biopsy for TMB was used. Start date for PFS was the start date of the chemotherapy. Physician notes, medication lists, and radiology reports of scans were reviewed in the electronic medical record to determine rate of SD ≥6 months/PR/CR, start and end data of therapy, and date of progression. When discrepancies arose between radiology reports and physician notes, the documented physician assessment was prioritized for clinical response and progression. Patients who had treatment interrupted by surgical resection, those who died prior to progression, or patients who were lost to follow-up were censored at the date of last evaluation. Patients treated outside of UCSD who lacked detailed information about treatments and response information in the electronic medical record were excluded from the analysis.
TMB was extracted from previously generated FoundationOne reports available in the electronic medical record. Formalin-fixed paraffin embedded tumor samples were previously sent to Foundation Medicine a clinical laboratory improvement amendments certified laboratory and analyzed using the FoundationOne next generation sequencing assay as previously described .Citation15 Average sequencing depth of coverage was greater than 250×, with >100× at >99% of exons. The number of somatic mutations detected on next generation sequencing after interrogating up to 1.2 megabases of the genome was quantified and that value extrapolated to the whole exome using a validated algorithm .Citation16 Alterations likely or known to be bona fide oncogenic drivers and germline polymorphisms are excluded and TMB was measured in mutations per megabase.
Clinical benefit was stratified by SD <6 months and progressive disease versus SD ≥6 months/PR/CR and were compared with ANOVA. Patients with SD who were censored prior to 6 months of therapy were excluded from the clinical benefit analysis. PFS was compared between patients with TMB ≥10 and <10 mutations/megabase using the log-rank test (Kaplan Meier analysis). All statistical analyses were verified by our biostatistician (DAB). SAS v. 9.4 was used and p-values ≤0.05 were considered significant.
Results
The PREDICT database was screened for patients who had TMB reported and received chemotherapy within one year after the tissue biopsy for TMB. SD ≥6 months/PR/CR and PFS were ascertained. Of the 1526 patients with reported TMB in the database, 294 individuals were eligible for the study and included in the analysis. The consort diagram for the study is shown in . Demographics are shown in . TMB ranged from 1 to 347 with a median of 5 mutations/megabase. Seventeen percent of patients had a TMB of 10 mutations/megabase or greater.
Table 1. Demographics for 294 subjects who initiated chemotherapy within 1 year of TMB.
Figure 1. Consort diagram for study.

SD ≥6 months/PR/CR was evaluable in 284 of the 294 total patients and was as follows: 11 patients (4%) achieved CR; 137 patients (48%), PR; 36 patients (13%), SD ≥6 months; 24 patients (8%) with SD<6 months; and 76 patients (27%) with progressive disease. Patients with stable disease who were censored prior to 6 months of evaluation were excluded from the analysis.
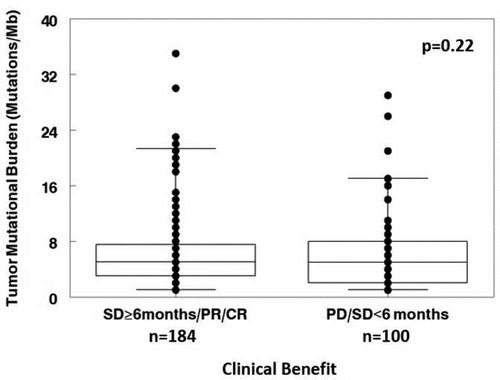
There were no significant differences in TMB between patients with SD≥6 months/PR/CR and those with SD <6 months/progressive disease (p = .22). Median and 25–75% IQR were 5 (3–7.5) and 5 (2–8) mutations/megabase, respectively (). Subset analysis did not find significant differences between TMB in patients with SD ≥6 months/PR/CR and those with SD <6 months/progressive disease for lung (median (25–75% IQR): 6.5 (5–15) vs. 6 (3–9) mutations/megabase; p = .69; N = 45 patients), breast (median (25–75% IQR): 4 (1–5) vs. 6 (4–9) mutations/megabase; p = .22; N = 40 patients), or gastrointestinal cancers (median (25–75% IQR): 4 (2–7) vs. 5 (3–6); p = .68; N = 94 patients). Of 284 evaluable patients, 12 (71%) with TMB high (≥20 mutations/megabase) achieved SD ≥6 months/PR/CR versus 172 (64%) with low/intermediate TMB (p = .79). We also stratified by PD, SD, and PR/CR and still did not see an association between TMB and outcome (ANOVA, p = .66).
Figure 2. Tumor mutational burden vs. SD≥6 months/PR/CR. Data represent the median and 25–75% interquartile range for tumor mutational burden. Patients with SD≥6 months/PR/CR after chemotherapy had similar tumor mutational burden to those with progression disease (PD) or SD<6 months (median (25–75%): 5 (3–7.5) vs. 5 (2–8) mutations/megabase; p = .22). Patients with stable disease who were censored prior to 6 months of evaluation were excluded from the analysis.
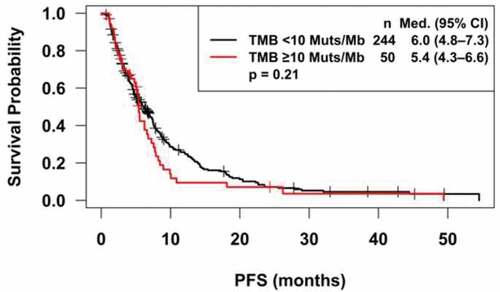
Of 294 evaluable patients, there were no differences in PFS for patients with TMB <10 vs. TMB ≥10 mutations/megabase (p = .21) (); median (95% CI) PFS was 6.0 (4.8–7.3) vs. 5.4 (4.3–6.6) months, respectively. Subset analysis found no significant differences in PFS for patients with a TMB <10 vs. TMB ≥10 mutations/megabase for breast (median (95% CI): 6.9 (4.8–9.3) vs. 2.9 (1.1–7.6) months; p = .07), lung (median (95% CI): 4.8 (3.9–7.3) vs. 5.3 (2.0–6.6) months; p = .47), or gastrointestinal cancer (median (95% CI): 7.4 (5.0–9.0) vs 5.4 (1.9–10.9) months; p = .53).
Figure 3. Kaplan Meier survival analysis. Progression-free survival for patients with tumor mutational burden of 10 or greater is similar to those with TMB less than 10 (p = .21).
Discussion
Despite the advent of novel targeted therapies and immunotherapeutic approaches, cytotoxic chemotherapy remains a standard first-line therapeutic approach for many metastatic solid tumors. Many cytotoxic regimens are associated with significant side effects, thus identifying which patients would derive the greatest benefit from therapy is important. Somatic mutations such as ATM and BRCA can predict responsiveness to platinum chemotherapy .Citation12 Protein markers such as TUBB3 ,Citation17–Citation19 TOPO1 ,Citation20 TOP2A ,Citation21 and thymidylate synthaseCitation22,Citation23 have previously been described as predictive of response to commonly administered chemotherapies for certain disease types; however, they have not been predictive of response for all tumor types and their use is still controversial. Thus, novel biomarkers for chemotherapy response are of interest for a precision medicine approach. Several studies indicate that TMB is a predictive marker for immunotherapy response, at least in some settings .Citation3–Citation9 However, relationships between TMB and cytotoxic chemotherapy have not been characterized.
Our study found no correlation between clinical benefit (defined as SD≥6 months/PR/CR) and TMB. PFS was also not related to TMB. The analysis combined all chemotherapy and tumor types; thus, the results are likely generalizable. We performed subset analyses in three cancer types and were unable to determine differences between PFS and TMB or clinical benefit and TMB. It is unclear if, however, for specific chemotherapies and/or other tumor types, TMB might be predictive of chemotherapy outcome.
A prior study compared outcomes for 46 patients with HER2-positive metastatic breast cancer receiving HER-2 directed therapies with chemotherapy and found that high TMB was associated with a significantly improved overall survival, but not PFS ;Citation24 it is unclear if patients receiving anti-PD-1/PD-L1 directed immunotherapies were excluded from the overall survival analysis. However, we recently noted that high TMB is associated with longer survival in immunotherapy-naïve patients regardless of treatment, suggesting that a high TMB is a favorable prognostic factor (Riviere and Kurzrock R, data submitted for publication). Thus, a high TMB may have a prognostic role in cancer outcomes distinct from predicting response to immunotherapy. A study of 74 patients with colorectal cancer found an improved PFS for irinotecan-based chemotherapy as compared to oxaliplatin based chemotherapy in TMB low patients (<6 mutations/megabase), but no differences in PFS between TMB low and intermediate/high patients .Citation25 A recent study explored the relationships between markers of response to chemotherapy with TMB-high status, but did not have clinical outcome data .Citation26 To our knowledge, no additional studies have explored relationships between TMB and chemotherapy.
The current study is limited in that it is retrospective; thus response was determined from interpretation of physician notes and radiology reports, which may be biased compared to central review of a prospective study. Since the TMB was obtained retrospectively from the electronic medical record, information on the differences between the allele frequency, passenger mutations, and non-clonal mutational status of the analyzed samples between the high and low TMB groups was not available. The lack of relationship between high TMB and response to chemotherapy may also be due to mutations arising in sites of DNA unrelated to DNA responsiveness despite a high TMB.
Conclusions
The current retrospective study evaluated the relationship between TMB, a potentially important predictor of immunotherapy response ,Citation3 and outcome after cytotoxic chemotherapy based on TMB. There were no significant differences in rates of SD≥6 months/PR/CR or in PFS associated with higher TMB in patients treated with chemotherapy. The findings of our retrospective analysis suggest that higher TMB is unlikely to be a biomarker for chemotherapy response. These results will need to be validated in prospective randomized studies.
Abbreviations
Authors’ contributions
PR assembled the database and reviewed the manuscript. DAB performed the statistical analysis and reviewed the manuscript. MN performed the chart review, analyzed the data, and wrote the manuscript. RK conceptualized the study and wrote the manuscript. AG and GF reviewed the manuscript.
Availability of data and material
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Disclosure of potential conflicts of interest
Dr. Kurzrock receives consultant fees from Actuate Therapeutics, X-Biotech and NeoMed, as well as research funds from Incyte, Genentech, Pfizer, Foundation Medicine, Guardant, Sequenom, and Merck Serono, speaker fees from Roche, and has an ownership interest in Curematch Inc. Paul Riviere discloses prior salary support from Peptide Logic, LLC.
Ethics approval and consent to participate
This study was performed in accordance with the UCSD IRB guidelines for data analysis and for any investigational treatments for which patients gave consent.
Additional information
Funding
References
- Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–5. doi:10.1126/science.aaa1348.
- Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, Walsh LA, Postow MA, Wong P, Ho TS. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014;371(23):2189–2199. doi:10.1056/NEJMoa1406498.
- Goodman AM, Kato S, Bazhenova L, Patel SP, Frampton GM, Miller V, Stephens PJ, Daniels GA, Kurzrock R. Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol Cancer Ther. 2017;16(11):2598–2608. doi:10.1158/1535-7163.MCT-17-0386.
- Khagi Y, Kurzrock R, Patel SP. Next generation predictive biomarkers for immune checkpoint inhibition. Cancer Metastasis Rev. 2017;36:179–190. doi:10.1007/s10555-016-9652-y.
- Chan TA, Yarchoan M, Jaffee E, Swanton C, Quezada SA, Stenzinger A, Peters S. Development of tumor mutation burden as an immunotherapy biomarker: utility for the oncology clinic. Ann Oncol. 2019;30:44–56. doi:10.1093/annonc/mdy495.
- Fancello L, Gandini S, Pelicci PG, Mazzarella L. Tumor mutational burden quantification from targeted gene panels: major advancements and challenges. J Immunother Cancer. 2019;7:183. doi:10.1186/s40425-019-0647-4.
- Samstein RM, Lee CH, Shoushtari AN, Hellmann MD, Shen R, Janjigian YY, Barron DA, Zehir A, Jordan EJ, Omuro A. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet. 2019;51:202–206. doi:10.1038/s41588-018-0312-8.
- Zhu J, Zhang T, Li J, Lin J, Liang W, Huang W, Wan N, Jiang J. Association Between Tumor Mutation Burden (TMB) and outcomes of cancer patients treated with PD-1/PD-L1 inhibitions: a meta-analysis. Front Pharmacol. 2019;10:673. doi:10.3389/fphar.2019.00673.
- Goodman AM, Sokol ES, Frampton GM, Lippman SM, Kurzrock R. Microsatellite-stable tumors with high mutational burden benefit from immunotherapy. Cancer Immunol Res. 2019;7:1570–1573. doi:10.1158/2326-6066.CIR-19-0149.
- Nikanjam M, Arguello D, Gatalica Z, Swensen J, Barkauskas DA, Kurzrock R. Relationship between protein biomarkers of chemotherapy response and microsatellite status, tumor mutational burden and PD-L1 expression in cancer patients. Int J Cancer. 2020;146(11):3087–3097.
- Friboulet L, Olaussen KA, Pignon JP, Shepherd FA, Tsao MS, Graziano S, Kratzke R, Douillard J-Y, Seymour L, Pirker R. ERCC1 isoform expression and DNA repair in non-small-cell lung cancer. N Engl J Med. 2013;368:1101–1110. doi:10.1056/NEJMoa1214271.
- Choi M, Kipps T, Kurzrock R. ATM mutations in cancer: therapeutic implications. Mol Cancer Ther. 2016;15:1781–1791. doi:10.1158/1535-7163.MCT-15-0945.
- Weaver BA, Silk AD, Montagna C, Verdier-Pinard P, Cleveland DW. Aneuploidy acts both oncogenically and as a tumor suppressor. Cancer Cell. 2007;11:25–36. doi:10.1016/j.ccr.2006.12.003.
- Holland AJ, Cleveland DW. Boveri revisited: chromosomal instability, aneuploidy and tumorigenesis. Nat Rev Mol Cell Biol. 2009;10:478–487. doi:10.1038/nrm2718.
- Frampton GM, Fichtenholtz A, Otto GA, Wang K, Downing SR, He J, Schnall-Levin M, White J, Sanford EM, An P. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol. 2013;31(11):1023–1031. doi:10.1038/nbt.2696.
- Johnson DB, Frampton GM, Rioth MJ, Yusko E, Xu Y, Guo X, Ennis RC, Fabrizio D, Chalmers ZR, Greenbowe J, et al. Targeted next generation sequencing identifies markers of response to PD-1 blockade. Cancer Immunol Res. 2016;4(11):959–967. doi:10.1158/2326-6066.CIR-16-0143.
- Hetland TE, Hellesylt E, Florenes VA, Trope C, Davidson B, Kaern J. Class III beta-tubulin expression in advanced-stage serous ovarian carcinoma effusions is associated with poor survival and primary chemoresistance. Hum Pathol. 2011;42:1019–1026. doi:10.1016/j.humpath.2010.10.025.
- Mozzetti S, Ferlini C, Concolino P, Filippetti F, Raspaglio G, Prislei S, Gallo D, Martinelli E, Ranelletti FO, Ferrandina G. Class III beta-tubulin overexpression is a prominent mechanism of paclitaxel resistance in ovarian cancer patients. Clin Cancer Res. 2005;11:298–305.
- Ploussard G, Terry S, Maille P, Allory Y, Sirab N, Kheuang L, Soyeux P, Nicolaiew N, Coppolani E, Paule B. Class III beta-tubulin expression predicts prostate tumor aggressiveness and patient response to docetaxel-based chemotherapy. Cancer Res. 2010;70:9253–9264. doi:10.1158/0008-5472.CAN-10-1447.
- Braun MS, Richman SD, Quirke P, Daly C, Adlard JW, Elliott F, Barrett JH, Selby P, Meade AM, Stephens RJ. Predictive biomarkers of chemotherapy efficacy in colorectal cancer: results from the UK MRC FOCUS trial. J Clin Oncol. 2008;26(16):2690–2698. doi:10.1200/JCO.2007.15.5580.
- Rodrigo RS, Nathalie A, Elodie T, Gonzalo GA, Philippe T, Francoise D, Julien D, Angela C, Bérénice B, Jean-Yves B. Topoisomerase II-alpha protein expression and histological response following doxorubicin-based induction chemotherapy predict survival of locally advanced soft tissues sarcomas. Eur J Cancer. 2011;47:1319–1327. doi:10.1016/j.ejca.2011.02.010.
- Sun JM, Ahn JS, Jung SH, Sun J, Ha SY, Han J, Park K, Ahn M-J. Pemetrexed plus cisplatin versus gemcitabine plus cisplatin according to thymidylate synthase expression in nonsquamous non-small-cell lung cancer: a biomarker-stratified randomized phase II trial. J Clin Oncol. 2015;33:2450–2456. doi:10.1200/JCO.2014.59.9324.
- Qiu LX, Tang QY, Bai JL, Qian XP, Li RT, Liu BR, Zheng M-H. Predictive value of thymidylate synthase expression in advanced colorectal cancer patients receiving fluoropyrimidine-based chemotherapy: evidence from 24 studies. Int J Cancer. 2008;123:2384–2389. doi:10.1002/ijc.23822.
- Park SE, Park K, Lee E, Kim J-Y, Ahn JS, Im Y-H, Lee C, Jung H, Cho SY, Park W-Y. Clinical implication of tumor mutational burden in patients with HER2-positive refractory metastatic breast cancer. Oncoimmunology. 2018;7:e1466768. doi:10.1080/2162402X.2018.1466768.
- Pai S, Carneiro B, Kalyan A, Costa R, Helenowski I, Radmeaker A, et al. Correlation of tumor mutation burden and chemotherapy outcomes in colorectal cancer. American Association of Cancer Research. Proceedings: AACR Annual Meeting 2017; April 1-5, 2017; Washington, DC.
- Nikanjam M, Arguello D, Gatalica Z, Swensen J, Barkauskas DA, Kurzrock R. Relationship between protein biomarkers of chemotherapy response and microsatellite status, tumor mutational burden and PD-L1 expression in cancer patients. Int J Cancer. 2020;146:3087–3097. doi:10.1002/ijc.32661.