
ABSTRACT
Development of T cell-directed immune checkpoint inhibitors (ICI) has revolutionized metastatic melanoma (MM) therapy, but <50% of treated patients experience durable responses. This phase I trial (NCT01946373) investigates the safety/feasibility of tumor-infiltrating lymphocyte (TIL) adoptive cell therapy (ACT) combined with dendritic cell (DC) vaccination in MM patients progressing on ICI.
An initial cohort (5 patients) received TIL therapy alone to evaluate safety and allow for optimization of TIL expansion protocols. A second cohort (first-in-man, 5 patients) received TIL combined with autologous tumor lysate-loaded DC vaccination.
All patients received cyclophosphamide/fludarabine preconditioning prior to, and intravenous (i.v.) IL-2 after, TIL transfer. The DC vaccine was given as five intradermal injections after TIL and IL-2 administration. [18F]-FDG PET/CT radiology was performed to evaluate clinical response, according to RECIST 1.1 (on the CT part). Immunological monitoring was performed by flow cytometry and T-cell receptor (TCR) sequencing.
In the safety/optimization cohort, all patients had a mixed response or stable disease, but none durable. In the combination cohort, two patients experienced complete responses (CR) that are still ongoing (>36 and >18 months, respectively). In addition, two patients had partial responses (PR), one still ongoing (>42 months) with only a small bone-lesion remaining, and one of short duration (<4 months). One patient died early during treatment and did not receive DC. Long-lasting persistency of the injected TILs was demonstrated in blood.
In summary, we report clinical responses by TIL therapy combined with DC vaccination in 4 out of 4 treated MM patients who previously failed ICI.
Introduction
Treatment of metastatic melanoma (MM) has been revolutionized by the development of immune checkpoint inhibitors (ICI) and, for the ~50% of patients that have BRAF-mutated tumors, targeted therapy with BRAF/MEK inhibitors. However, less than half of ICI-treated MM patients experience durable responses and most BRAF/MEK inhibitor-treated patients progress because of acquired resistance. Since there is no standard treatment for MM patients who have progressed on or are not eligible for these new therapies, there is a need for other therapies.Citation1
Adoptive cell therapy (ACT) with the transfer of ex vivo-expanded tumor-infiltrating lymphocytes (TILs) to lymphodepleted MM patients, together with high-dose IL-2 to support TIL persistence, has resulted in high rates of clinical responses.Citation2 However, there are currently no approved TIL ACT regimens and, in addition, the procedure to produce clinical-grade TIL is technically demanding and co-administration of IL-2 is associated with considerable toxicity requiring supervision and management by highly trained personnel. Thus, TIL ACT is practiced in clinical trials, in few medical centers, as last-line therapy.
The underlying mechanism for primary resistance to ICI or secondary resistance after initial response remains elusive. Failure to re-activate sufficient numbers of tumor-specific T cells by ICI, due to the presence of MDSCs, lack of an IFN-γ gene signature or insufficient number of neoantigens can contribute to resistance.Citation3 However, ACT with autologous TILs from patients who have failed on ICI can mediate tumor regressions in a low but significant proportion of treated patients, Citation4–Citation6 arguing against inherent functional defects in TILs from ICI resistant patients.
Dendritic cells (DC) are professional antigen-presenting cells that can be used as potent inducers of tumor-specific immune responses in a vaccine setting. Dendritic cells can be generated in vitro from monocytes and pulsed with proteins, peptides or whole-tumor lysates or transfected with RNA coding for tumor-specific epitopes.Citation7 Dendritic cell vaccine trials have generally not resulted in strong clinical responses, but induction of T cell and delayed-type hypersensitivity (DTH) responses are often observed.Citation7 To enhance their therapeutic potential, DC vaccine-combinations with other cancer regimens including other immunotherapeutic therapies have been tested. For example, combining a whole-tumor lysate-loaded DC vaccine with anti-VEGF mAb in patients with ovarian cancer resulted in significantly prolonged survival compared to the single agents.Citation8 Similarly, combining DC vaccination with CTLA4 blockade in melanoma showed encouraging results with response rates higher than expected with each therapy alone.Citation9 There are also several ongoing trials combining DC vaccination and PD-1 blockade, Citation10 and results from mouse models indicate enhanced therapeutic efficacy with this combinatorial approach.Citation7
There is a rationale for combining DC vaccines with ACT. In a pilot phase I clinical trial combining whole-tumor lysate DC vaccine with TILs in eight enrolled MM patients, we demonstrated the feasibility and safety of this therapeutic approach.Citation11 Patients received low-dose conditioning of orally administered cyclophosphamide that had no apparent lymphodepleting effect. Although the small study size precluded analysis of clinical responses, one patient had complete remission and two had stable disease. We also observed long-term, although low frequency, persistency of dominant T cell clones derived from the infusion product in the blood of treated patients.
The results from the previous trial have spurred a continuation trial. In the new and revised protocol, one important change is the temporal order of TIL versus DC vaccine, starting with TIL therapy followed by DC tumor vaccination instead of the reverse. The intention is to re-stimulate injected TILs with the DC vaccine, which is loaded with antigens derived from the tumor from which TILs were expanded, thereby extending their lifespan. The TILs were expanded from tumors using an optimized protocol allowing treatment with much higher doses of TIL than in the previous trial. In addition, the DC vaccine was produced by an improved protocol resulting in stronger IL-12 production and T cell activation.Citation12 Stronger preconditioning chemotherapy consisting of intravenous (i.v.) cyclophosphamide and fludarabine, as well as low doses of i.v. IL-2 after TIL ACT was also added. Stronger preconditioning has been shown to increase response rates in adoptive T cell trials, Citation2 likely by creating a niche for the infused cells via the removal of both competing T cells and suppressive immune cells. The in vivo TIL expansion and persistence would thereafter be supported by the IL-2 treatment, which is broadly employed in adoptive T cell trials. We chose a lower dose to avoid the well know toxicity of IL-2 and to thereby possibly avoid having to halt this supportive treatment prematurely. The trial was divided into two cohorts, a first cohort that received TIL ACT alone to evaluate safety and optimize TIL production before commencing the second cohort that received TIL ACT combined with DC vaccination. We report promising results with clinical responses in 4/4 patients subjected to the combined treatment, and observed long-lasting persistency of T cell clones derived from infused TILs in the blood of treated patients (NCT01946373).
Materials and methods
Patients
The trial (NCT01946373) protocol was approved by the Regional Ethical Review Board (2013/516-31/1), the Institutional Review Board and the Swedish Medical Products Agency (EudraCT number 2012–000450-63). It was conducted in accordance with the Helsinki declaration ethical principles and the International Conference on Harmonization Good Clinical Practice (ICH GCP) guidelines, and was monitored by the Center for Clinical Cancer Studies, Theme Cancer, Karolinska University Hospital Solna, Stockholm, Sweden. Upon enrollment, all patients were informed about the study, including the routines for the handling of patient material and the full anonymization of published data, and signed an informed consent to be included. During preconditioning, TIL infusion and IL-2 supportive therapy, patients were hospitalized and treated at CAST (Center for Allogeneic Stem-cell Transplantation), Karolinska University Hospital.
Eligible patients () were 18–74 years old, had progressive inoperable stage III or stage IV (according to AJCC) malignant melanoma, measurable disease (according to RECIST 1.1), were ambulatory in performance status 0–2 (according to ECOG) with a life expectancy of at least 3 months and with a tumor lesion available for surgery or for core biopsy. Main exclusion criteria were active CNS metastases, severe comorbidity or active autoimmune disease.
Table 1. Patient characteristics at treatment initiation.
Study design
The trial was a single-center, open-label, phase I trial to evaluate the safety, feasibility and immunologic response of adoptive T cell transfer with or without dendritic cell (DC) vaccination in patients with metastatic melanoma (). Primary endpoints were safety and feasibility and secondary were immunologic response and objective response according to RECIST 1.1, although irRC was also evaluated.
Figure 1. Protocol time line. A schematic view of the treatment protocol for the safety/optimization cohort (top) and for the combinatorial treatment cohort (bottom). ACT, adoptive cell transfer. DCV, dendritic cell vaccine.
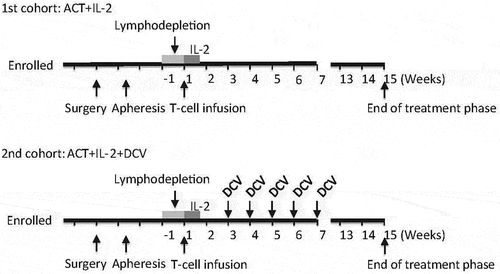
All patients received lymphodepleting chemotherapy with cyclophosphamide 60 mg/kg/day for 2 days followed by fludarabine 25 mg/m2/day for 5 days. The next day, TIL-infusion was administered and after 24 hours 100 000 IU/kg IL-2 was administered intravenously every 8 hours for up to 14 doses if tolerated. During lymphodepleting treatment, TIL-infusion and IL-2 treatment, patients were hospitalized at the CAST unit at Karolinska university hospital and monitored daily.
As per recommendation by the Swedish MPA, treatment of patients in a first cohort with lymphodepletion, TIL therapy and IL-2 supportive was completed first to determine safety and feasibility before commencing treating patients in the second cohort that were given the same treatment and, if clinically stable and with neutrophil count >0.5E9, 5 weekly doses of intradermal DC-vaccination.
Toxicity was assessed using National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE version 4.0). Clinical efficacy was evaluated using [18 F]-2-fluoro-2-deoxy-d-glucose (FDG) PET/CT, according to RECIST 1.1 (evaluated on the CT scans) before the start of treatment, approximately 10 weeks after TIL treatment for patients in the safety/optimization cohort and approximately 4 weeks after the last DC-vaccination for patients in the combinatorial treatment cohort. Responding patients were monitored with [18F]-FDG PET/CT approximately every 3–6 months according to local clinical routine to follow response duration.
The DC vaccine efficacy was tested clinically by DTH-testing prior to treatment and after the last DC vaccine using tumor lysate (10 µg) and NY-ESO-1 peptide (10 µg) injections or tetanus toxoid tuberculin and saline injections as controls.
Expansion of TILs from autologous tumors
Autologous young TILs were expanded from one or several excised melanoma tumors or, when no resectable tumor was available, from 6 to 8 16 G core-needle tumor biopsies (), as described previouslyCitation11,Citation13(Supplemental material and methods). TILs could be expanded from all patients, but for three patients in the first cohort, REP expansion was less successful. Patient #3 TILs expanded slowly and the patient received fewer TILs while patient #6 had a completely failed REP and received pre-REP TILs. For patient #7, the REP in the bioreactor failed and the patient received two backup REP cultures, a GRex-100 M flask and a VueLife cell culture bag.
Table 2. TIL characteristics and objective response.
Production of DC vaccine
Dendritic cells were generated from monocytes enriched from a leukapheresis product and tumor lysate was generated by freeze thawing of tumor fragments from the same tumor that was used for TIL generation, as described previouslyCitation11(Supplemental material and methods). Mature DCs (mDCs) were generated as describedCitation12 and after harvest 1/10 of the cells were incubated with 20 µg/ml of the NY-ESO-1157–165 (C165 V) peptide (SLLMWITQV; GMP-grade, GIN-laboratories, Uppsala University) for 30 min at 37°C before mixing with the rest of the mDCs. The mDCs were frozen in 17.5E6 aliquots using a computerized gradient freezer (PLANER®). On the day of DC-vaccination, one vial of frozen DCs was thawed, washed and resuspended in 2 × 0.2 ml 0.15 M NaCl for intradermal injection at two sites (upper arms) using insulin syringes (Micro-Fine, BD Medical, San Diego).
Immunomonitoring
Blood samples were collected before and at several time-points after TIL therapy. Isolation of PBMCs was performed by density gradient centrifugation over Ficoll-Hypaque (GE, Uppsala, Sweden) and samples were frozen. PBMCs and TILs were analyzed for T cell maturation and exhaustion by flow cytometry (Supplemental material and methods). In addition, TIL samples were analyzed for T cell-specificity using MHC-multimers for known shared melanoma tumor-associated antigens (TAA); NY-ESO-1, MART-1, MAGE-A1 and -A3, gp100 and tyrosinase (Melanoma collection 1 dextramers, Immunodex; Copenhagen, Denmark), accordingly to manufacturer’s recommendations. For PBMC samples, only NY-ESO-1 was analyzed.
Analysis of TCR repertoire
Deep sequencing of the TCRB gene complementarity-determining region 3 (CDR3) was performed on the final TIL product as well as on PBMC samples collected before and after therapy to identify high-frequency T cell clones. Frequencies of clones were analyzed and exported (Survey level ImmunoSeq; Adaptive Biotechnologies).
Results
Administered treatments and patient characteristics
Of 14 patients with progressive MM enrolled in the study between October 2013 and March 2018, 10 were treated (). All enrolled patients except one (#7) had progressed on ICI therapy (given for stage IV disease). The patients had either primary resistance or developed acquired resistance to ICI, with varying ICI-treatment periods, spanning from 3 to 12 months. Progression on ICI had been verified with radiology before the patient was assessed for suitability for enrollment into the trial. Upon enrollment, we conducted a baseline PET-CT on all patients, which again verified the progression. At least 3 months had passed from the last ICI administration before TIL ACT treatment was given. Patients were consecutively enrolled into the first and the second cohort, with an intention to treat the first cohort with TIL therapy alone, and the second cohort with a combination of TILs and an autologous tumor lysate-loaded DC vaccine (). This was achieved with the exception of one patient (#14) in the second cohort, who died rapidly after TIL therapy before DC vaccination could be started.
Characteristics of TILs and DC vaccine
Production of TILs was possible for all patients. However, one patient (#2) had to be excluded due to both bacterial contamination in TILs pre-REP cultures and too few CD34+ cells in the second leukapheresis, making lymphodepleting chemotherapy impossible. Three more patients had to be excluded due to rapid disease progression (#4, #5 and #12).
Median production time for TILs was 35 days (range 28–40, ) including both expansion in cell culture plates and REP (static flasks plus bioreactor). The average yield given back to patients was 10.2E9 for the safety/optimization cohort (range 26E6-35E9) and 41.1E9 for the combinatorial therapy cohort (range 29E9-65.5E9). The difference in yields between the two cohorts is a result of the optimization of the REP protocol during the treatment of patients in the first cohort. It is notable that TILs could be expanded from all patients, even though a majority of patients were progressing on ICI and that in some cases only core biopsies from liver- and lymph node metastases were available. Production was, however, less successful for three patients in the first cohort (#3, #6 and #7), in all three cases due to failed REP.
The viability of the T cell product at final harvest was high (mean 97%, range 87–100%), and cultures contained predominantly T cells (mean 98%, range 93–100%), for most patients with strong skewing toward the CD8+ subpopulation (mean 62.7%, range 12–92%). The TIL product from two patients (#13 and #14) did however contain a large proportion of CD4 + T cells, 54 and 87%, respectively.
Patients in the combination treatment cohort received a tumor lysate-loaded DC-vaccine. A proportion of the DC-vaccine was pulsed with a NY-ESO-1157–165 (C165 V) peptide with the aim to be able to follow the elicited T cell response against the vaccine. The enriched product contained 93% monocytes (range 92–94%). After DC differentiation and tumor lysate-loading, mDCs showed a mature phenotype (Average: 95% CD80+, 47% CD83+, 95% CD86+, 97% HLA-DR+, 79% CCR7). Four patients in the second cohort received five doses of intradermal (i.d.) DC vaccine of high viability (Average: 92%, range 87%-95%). One patient (#14) from the second cohort died early during treatment course and could not receive DC vaccinations.
Clinical efficacy
The number of administered TILs was lower, and more variable in the safety/optimization cohort than in the combinatorial therapy cohort, as commented above. This, as well as the fact that in the first cohort, three patients (#6, #7, #8) had uveal melanomas, known to be more resistant to immune therapy, Citation14 and one patient had had CNS metastases (#1, treated with gamma knife radiosurgery and stable at time of inclusion), while four patients in the second cohort (#9, #10, #11, #14) had cutaneous melanoma and one (#13) had melanoma of unknown origin, makes any comparison of clinical outcome between the two cohorts difficult.
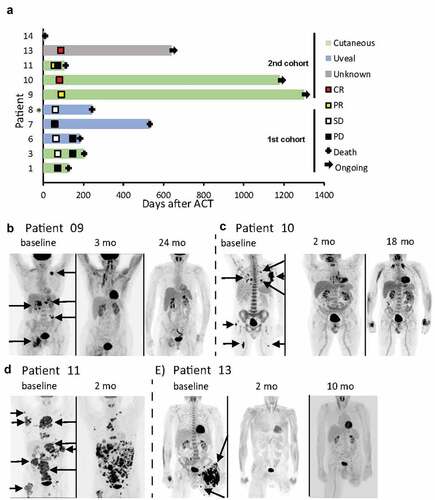
In the safety/optimization cohort, all treated patients experienced mixed response or stable disease that were, however, not durable (, ). In the combinatorial treatment cohort, five patients were enrolled and commenced therapy as per protocol. One patient (#14) died before receiving the DC vaccine and was not evaluable for response. Of the four patients receiving the complete combined treatment, all had objective response according to RECIST 1.1 (). These consisted of two durable complete responses (CR) (#10 and #13, duration of >36 months and >18 months, respectively, -e, ), one durable high-quality partial response (PR) with only one small bone-lesion remaining (#9, duration >42 months, ) and one short-term PR (#11, duration <4 months, ). The short-term response in #11 was only considered a mixed response by irRC. Responses were also visualized by PET/CT (-e, at baseline and different time-points after TIL transfusion). These images clearly showed decreased isotope-labeled glucose [18F] uptake corresponding to the degree of response, demonstrating that shrinking tumors also had decreased metabolic activity.
Figure 2. Clinical responses. Swimmers plot (a, * Date of progressive disease unknown) and PET/CT scans for patients 9 (b), 10 (c), 11 (d) and 13 (e) are shown. Arrows indicating several FDG-avid lesions. Patient 11 had previously undergone amputation of the left leg because of progressing melanoma lesions.
Adverse events
All 10 patients received lymphodepleting chemotherapy, but two patients (#1 and #14) received the chemotherapy at reduced dose due to mildly impaired kidney function. All patients reacted with expected hematological toxicity, but one patient (#3) experienced prolonged toxicity and transfusion of autologous stem cells derived from the second leukapheresis had to be performed.
The majority of patients reacted to TIL transfusions with mild to moderate toxicity (), such as fever and chills. One patient (#13) had a more long-lasting reaction to TIL transfusion with fever, chills and peripheral edema, resembling capillary leakage syndrome and decision was taken not to administer IL-2.
The most severe adverse events in the trial occurred in response to IL-2. In general, IL-2 treatment was accompanied by well-known side effects such as fever, chills, hypotension, fluid retention and electrolyte derangement. All patients were administered paracetamol to manage fever and chills. To master more severe effects such as durable high temperature, elevated heart rate, decreasing blood pressure as signs of threatening CRS of IL-2 treatment, most patients after thorough consideration were administered low doses of corticosteroids (hydrocortisone 100 mg iv, which has short duration). In the safety/optimization cohort, four of five treated patients received all 14 doses of IL-2, whereas in the combinatorial treatment cohort, none of five patients received all doses. The main reason for stopping IL-2 infusions was capillary leakage, a known side effect of IL-2 therapy.
One patient (#14) died during treatment. Baseline [18F]-FDG PET/CT before the start of the lymphodepleting regimen revealed tumor-related hydronephrosis without a major influence on creatinine value. A nephrostomy was placed and chemotherapy was reduced. Chemotherapy and TIL transfusion were administered without major toxicity. IL-2 support was stopped after 10 doses, because of symptoms of capillary leakage and metabolic acidosis and the patient improved. However, 2 days after the last IL-2 injection, the patient suddenly de-saturated. Pulmonary embolism, pulmonary edema or septic shock were suspected. The patient could not keep oxygen saturation, had to be intubated and moved to the intensive care unit where he died due to therapy refractory metabolic acidosis. Autopsy did not offer any macroscopic explanation to the fatal outcome.
None of the four patients receiving DC-vaccinations experienced any particular toxicity to these injections. Interestingly, both patients with CR (#10 and #13) and the high-quality partial responder (#9) developed autoimmune skin reactions, vitiligo, after the combined therapy.
Immunomonitoring and functional ex vivo analysis
Monitoring of T cell differentiation stages in blood and TILs of patients in the combinatorial treatment cohort showed that pre-ACT blood contained a mix of all T cell differentiation stages, including a substantial proportion of naïve T cells (Supplementary Fig 1). TILs on the other hand contained mainly effector memory (EM) T cells and in some patients central memory (CM) T cells. Blood taken early after TIL ACT contained mostly EM T cells and the even more differentiated effector T cells (EMRA) while CM T cells were usually low. In later blood samples, T cell composition started to normalize with populations of all differentiation stages, including naïve T cells, present again.
The activation/exhaustion marker PD-1 (CD3+ CD8+: , CD3+ CD4+: ) was absent in pre-ACT blood samples from all patients except one (#9) where PD-1 was slightly elevated. Levels of PD-1 on TILs were variable, but for most patients, it was lower than on blood T cells post-ACT. For the two PR patients (#9 and #11), PD-1 was higher in blood than in TILs already at the first time point after TIL therapy and levels then increased until reaching a plateau. For one CR patient (#10), PD-1 was very low in TILs and remained low in blood samples after ACT, but suddenly raised to very high levels after 78 weeks. For the second CR patient (#13), TILs had high PD-1 levels but the first sample after ACT showed virtually no PD-1 on T cells while subsequent samples had high PD-1 levels.
Figure 3. Monitoring of immune cells during treatment. T cell activation/exhaustion were monitored in the TIL product and in patient blood before or at different time points after TIL ACT by staining with antibodies for CD3, CD8, CD4 and PD-1 and analysis by FACS (CD8:A, CD4:B). Staining with dextramers to detect common tumor-associated antigen-specific T cells was performed in TIL and in blood samples before and after TIL ACT. Only one patient (#13) had NY-ESO-1 specific T cells both in TIL and in blood samples (c).
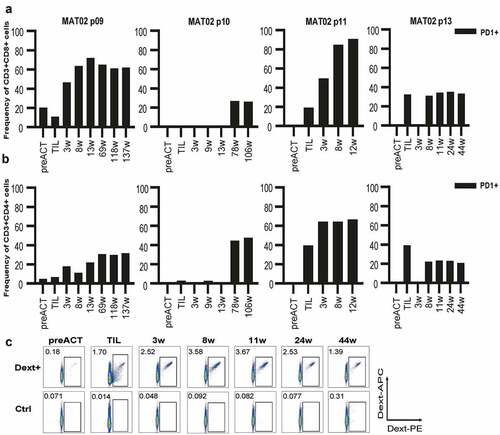
For the combinatorial treatment cohort, T cell-specificity in TILs was analyzed with a panel of dextramers for HLA-A*02:01 restricted epitopes from common melanoma antigens (MART-1, NY-ESO-1, MAGE-A3, tyrosinase, gp100 and MAGE-A1) while PBMC samples were analyzed for NY-ESO-1 reactivity only. TILs from patient #9 were significantly stained with dextramer against MART-1 and patient #11 displayed reactivity against MART-1, gp100, tyrosinase and NY-ESO-1 while patient #10 did not recognize any of the common antigens (supplementary fig 2 and data not shown). Only one responding patient (#13, ) demonstrated significant positivity to NY-ESO-1 dextramer in both blood and TILs, with 0.18% positive cells in PBMCs before treatment, 1.7% in TILs and 2.5% in PBMC at the first time-point (week 3) after TIL ACT. At the second time-point (week 8) after TIL ACT, NY-ESO-1 specific T cells had increased to 3.6%. The tumor-lysate pulsed DC vaccine was pulsed with the same NY-ESO-1157–165 (C165 V) peptide as the epitope detected by the NY-ESO-1 dextramer. It is, therefore, possible that the increase in NY-ESO-1 positive T cells in the blood at the second time-point was boosted by the DC vaccination, which was started on week 4.
The effect of DC vaccination was clinically assessed by DTH-testing for tumor cell lysate and NY-ESO-1 peptide. Although all patients responded with skin reactions to the tetanus toxoid positive control, none reacted to tumor cell lysate. Only one patient (#11) responded with skin reaction against the NY-ESO-1 peptide and this reaction was found only before but not after vaccination (results not shown).
Analysis of T cell clones in TILs and PBMCs
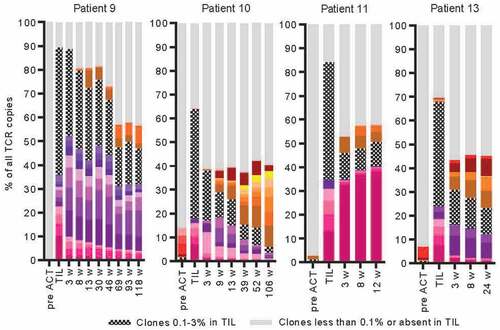
For the four responding patients in the combinatorial treatment cohort (#9, #10, #11 and #13), TIL products and PBMCs from before and at several time points after treatment were analyzed for clonality by deep-sequencing of the TCRB chain locus (). The TIL product from all four patients contained dominant clones accounting for up to 10%, 7%, 13% and 7% of T cells in each of the patients. For all four patients, there was very little overlap between TCR repertoires found in TIL products and in PBMCs before TIL therapy. In contrast, after therapy, patient PBMCs contained a substantial proportion of T cell clones originating from the TIL product. This proportion decreased over time, but TIL-originating clones were found in patient PBMCs as late as 118 (#9) and 106 (#10) weeks after T cell therapy, which were the latest time points analyzed. Interestingly, for the two CR patients (#10 and #13), levels of TIL-originating T cell clones dropped quicker than for the long-term PR patient (#9) that after an initial decrease had stable levels of TIL-originating clones constituting around half of the T cells present in blood at 69, 93 and 118 weeks after TIL therapy. However, not all clones from TILs persisted. Some decreased quickly while others expanded in vivo. For the short-term PR patient (#11) the last sample was taken at 12 weeks and there was a stable level of around 50% TIL-originating T cell clones in all samples from this patient. These consisted mainly of one single clone (>35% of all T cells) that had expanded even further in vivo compared to levels found in TILs (13%).
Figure 4. Monitoring of T cell clones during treatment. T cell clone frequencies were detected by deep sequencing of the TCRB locus in DNA from the TIL product and in patient blood before or at different time points after TIL ACT. Clones that constituted 3% or more of all T cells in at least one of the samples (TIL or blood) is depicted with a separate color. For the rest of the detected T cell clones, those that constituted 0.1%-1% in TIL are depicted as hashed bars and those that constituted less than 0.1% or were not at all detected in TILs are depicted as gray.
Discussion
The 3-year overall survival (OS) of patients with stage IV melanoma can reach >50% as a result of ICI therapy.Citation15,Citation16 However, for patients that fail to respond or progress on ICI therapy, there is a need for the development of alternative treatment strategies, and ACT with TILs is an attractive strategy. In a previous phase I study including eight patients with stage IV MM, we established that combination therapy with TILs and a tumor lysate-loaded DC vaccine is feasible and safe.Citation11 One patient experienced CR that is ongoing more than 100 months after treatment. In the current study, a safety/optimization cohort with five patients receiving TIL ACT only, and a combinatorial treatment cohort, with 4/5 patients receiving the combination of TIL ACT and DC vaccination, were included. To support multiplication, engraftment and longevity of infused TILs, low-dose IL-2 was administered to both cohorts after TIL infusion. The DC vaccine administered to the second cohort was a whole-tumor lysate pulsed DC vaccine, matured with a cocktail of Toll receptor agonists and IFN-γ.Citation12 All included patients had MM and all except one had progressed after ICI therapy. We report promising results with clinical responses (2 CR, 1 long-term PR and 1 short-term PR) in all four evaluable patients receiving combined treatment, while patients receiving only TILs experienced mixed response or stable disease that were not durable. One patient in the second cohort died soon after TIL transfer and could not be evaluated.
Although it would have been preferable to compare clinical outcomes of the two cohorts, they differed in the type of patients recruited, with three uveal melanoma patients (#6, #7, #8) and one patient (#1) with treated and stable CNS metastasis in the first cohort, and four cutaneous melanomas (#9, #10, #11, #14) plus one patient with unknown primary tumor (#13) in the second cohort. Cutaneous and uveal melanomas are both of neural crest origin, but are in many aspects different types of cancers. Uveal melanoma is, for example, known to have a lower mutational burden and to be more resistant to immune therapy.Citation14,Citation17 Furthermore, the two cohorts also differed in the amount of TILs that were administered due to the optimization of the expansion protocol during the treatment of patients in the first cohort. Taken together, this makes a direct comparison between the cohorts invalid.
Although the generation of two autologous cellular products is challenging, there is an advantage of having two products in a protocol since it minimizes the risk of leaving included patients without treatment. In addition, mouse models have demonstrated that a combination of ACT and DC vaccination is superior to single-agent treatments.Citation18–Citation22 We were able to generate TILs for all patients and DCs for all patients in the second cohort. For three patients that lacked metastases suitable for resection (#3, #7, #8), TILs could be expanded from core biopsies in sufficient numbers to start full-scale REP.
Blood of all four patients with clinical responses contained dominant T cell clones originating from TILs after ACT and there was little overlap with T cell repertoires in blood before therapy. Using a panel of dextramers specific for HLA-A2 restricted tumor antigens, we could conclude that patients #9, #10 and #13 had T cell clones in their TIL product that could recognize one or several of the known shared tumor antigen. However, only in one patient (#13) NY-ESO-1 specific T cells were found already in pre-ACT blood, but in even higher levels in TILs and in post-ACT blood samples. The increase of NY-ESO-1 specific T cells may have been due to contact with tumor but also to boosting by the DC vaccine, which was loaded with NY-ESO-1 peptide. That only one responder had NY-ESO-1 reactivity in blood after DC vaccinations have made it difficult to demonstrate antigen-specific-boosting induced by our DC vaccination. It is important to note is that the anti-tumor effect of the TIL is likely not mainly due to the recognition of shared TAA but to the recognition of neoepitopes generated by the high mutational load in melanomas. These neoepitopes would in most cases be unique for each patient. Thus, the lack of reactivity to most of the shared antigens is in no way surprising, and further studies on the detailed specificity of the TIL products are ongoing.
Important to note is that all but one of the enrolled patients were refractory to ICI. Efficacy of TIL ACT in patients progressing on ICI has previously been shown, Citation5,Citation6,Citation23 and the high proportion of responders that were noted in this trial shows that the TIL therapy/DC vaccine combination is strikingly successful in these patients. Why ICI does not activate TILs that efficiently target the tumor in these refractory/relapsing patients, even though the same TILs apparently are able to recognize and eliminate tumor cells after ex vivo expansion and, in our trial, DC vaccination, remains to be established. However, expansion ex vivo generates a huge amount of TILs, likely more massive than ICI can achieve in vivo and this expansion is, in our trial, also further boosted in vivo by DC vaccination. In addition, ICI will act on all T cells in the body while in TIL ACT therapy preconditioning removes other T cells as well as immunosuppressive cells and creates a niche where TILs will have increased possibilities to act on the tumor. TILs are thought to be enriched for tumor-specific T cells compared to blood and would thereby be more efficient at eliminating tumor cells. Still, response to TIL ACT may have been aided by previous ICI therapy, e.g., by recruiting more tumor-specific T cells to the tumor prior to excision and ex vivo TIL expansion, as well as by decreasing regulatory T cells and MDSCs in the tumor.Citation24
Toxicity related to TIL infusion was seen in both cohorts, in most cases as expectedCitation25 mild or moderate, such as transient fever and chills directly after infusion. In contrast, considerable toxicity was observed even with the low-dose IL-2 regimen used, perhaps due to the advanced stage of disease of the patients. Mild toxicities were treated with paracetamol and more severe toxicities were treated with hydrocortisone, which could have a negative impact on T cells. However, both responders and non-responders did receive hydrocortisone showing that any inhibitory effect on TIL must have been limited. IL-2 related toxicity was resolved when infusions were discontinued. Only four patients (all in the safety/optimization cohort) tolerated all 14 doses of IL-2, possibly due to the low TIL doses given. One patient (#14) in the combinatorial treatment cohort developed metabolic acidosis, which was not resolved despite IL-2 was halted and resulted in a fatal outcome.
Interestingly, all patients reaching PR or CR did so despite IL-2 therapy being halted prematurely. One might speculate whether the DC vaccine aided TIL persistence in those patients where IL-2 therapy had to be halted. Persistence of infused TILs has been shown to correlate with improved clinical response.Citation2 In addition, one CR patient (#13) was not even started on IL-2 due to strong reactions to TIL infusion alone. This patient had a high frequency of CD4 + T cells in infused TILs, which may have produced IL-2 themselves, increasing treatment efficacy and causing toxicity. In our previous TIL ACT and DC vaccine trial, Citation11 we argued that the combination with a DC vaccine would circumvent the need for IL-2 therapy and therefore did not treat patients with IL-2. There have been similar efforts to perform TIL ACT without or with low-dose IL-2, administered subcutaneouslyCitation13 or intravenously in a decrescendo regime.Citation23 As in our previous trial, these trials have reported clinical responses but patient groups treated were too small to evaluate efficacy compared to trials using high-dose IL-2.
One important factor for achieving clinical responses with TIL ACT is the number of infused cells and their persistence. We performed both immunomonitoring and TCRB sequencing with the aim to follow infused TILs. TCRB sequencing clearly shows that infused TILs persisted for long time and persistence was most impressive in the long-term PR patient, possibly due to continuous proliferation in response to the small remaining tumor lesion. In the short-term PR patient, the blood samples consisted of a large proportion of one single clone that originated from TILs. This patient had a large tumor mass, and after ACT, he had shrinkage of some tumor masses but the progression of others. One may speculate that this mixed response occurred since TILs consisted of few tumor-specific clones and that the big tumor mass contained cells heterogeneous in their expression of TAA, resulting in antigen-escape.
Immunomonitoring data showed that TILs contained both CM and EM T cells. However, after infusion, they continued to differentiate with an increase of the EMRA population and a decrease of the CM population, probably due encounter with tumor cells and later the DC vaccine. In line with this, expression of the activation and exhaustion marker PD-1 increased in blood T cells after ACT for most patients. Prolonged expression of PD-1 may be a sign of exhaustion, which could be negative for treatment. However, so far 3/4 responding patients have no sign of progression, despite expressing high levels of PD-1 on their T cells.
Side effects related to DC vaccination were mild and consisted mainly of irritation at the vaccination site. An interesting observation is the widespread patchy vitiligo developed in all three long-term responder patients (2 CR and 1 PR). In two patients (#9 and #13), this was noted after the first DC vaccination, and for one patient (#10) 3 months after the last vaccination. Thus, it is plausible that administered DCs attract TILs to enter into the dermis, destroying melanocytes that express differentiation antigens, such as gp100 and MART-1, that are also found in tumor cells. However, autoimmune-like manifestations, such as vitiligo and uveitis, have also been observed after TIL monotherapy, Citation26 and more frequently following ACT with gp100 or MART-1-specific TCR-transduced T cells.Citation27
In summary, we found that therapy of MM with a combination of TIL ACT and tumor lysate-loaded DC vaccination is feasible and safe and we observed impressive clinical responses in all patients treated with the combination. The promising clinical data in this limited number of patients support the continued evaluation of the TIL ACT/DC vaccine combination in a larger patient cohort to confirm therapeutic benefit. Thereby the exact contribution of each of the two cellular products could also be established.
Disclosure of potential conflicts of interest
R. Kiessling is a board member/consultant for Clinical Laser Thermia Systems and is a Scientific Advisor for Anocca AB and Phion Pharmaceutics and receives research grants from these companies. R. Tell is an employee of Isofol Medical. J. Hansson has been PI on trials sponsored by Astra-Zeneca, Bristol Myers-Squibb, Merck, Novartis, Roche and Clinical Laser Thermia Systems and has received research grants from Bristol Myers-Squibb and Merck. T. Lövgren has received honoraria for lecturing from Bristol Myers-Squibb.
Supplemental Material
Download ()Acknowledgments
We would like to thank patients and medical staff involved in this trial. We would also like to thank A.M. Salazar (Oncovir Inc.) for providing Hiltonol, used for the generation of the DC vaccine, and K. Heimersson and B. Näsman-Glaser for their expert technical assistance.
Additional information
Funding
References
- Eggermont AMM, Crittenden M, Wargo J. Combination immunotherapy development in melanoma. Am Soc Clin Oncol Educ Book. 2018;38:197–11. doi:10.1200/EDBK_201131.
- Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science. 2015;348(6230):62–68. doi:10.1126/science.aaa4967.
- Jenkins RW, Barbie DA, Flaherty KT. Mechanisms of resistance to immune checkpoint inhibitors. Br J Cancer. 2018;118(1):9–16. doi:10.1038/bjc.2017.434.
- Andersen R, Borch TH, Draghi A, Gokuldass A, Rana MAH, Pedersen M, Nielsen M, Kongsted P, Kjeldsen JW, Westergaard MCW, et al. T cells isolated from patients with checkpoint inhibitor-resistant melanoma are functional and can mediate tumor regression. Ann Oncol. 2018;29(7):1575–1581. doi:10.1093/annonc/mdy139.
- Besser MJ, Shapira-Frommer R, Itzhaki O, Treves AJ, Zippel DB, Levy D, Kubi A, Shoshani N, Zikich D, Ohayon Y, et al. Adoptive transfer of tumor-infiltrating lymphocytes in patients with metastatic melanoma: intent-to-treat analysis and efficacy after failure to prior immunotherapies. Clin Cancer Res. 2013;19(17):4792–4800. doi:10.1158/1078-0432.CCR-13-0380.
- Forget MA, Haymaker C, Hess KR, Meng YJ, Creasy C, Karpinets T, Fulbright OJ, Roszik J, Woodman SE, Kim YU, et al. Prospective analysis of adoptive TIL therapy in patients with metastatic melanoma: response, impact of anti-CTLA4, and biomarkers to predict clinical outcome. Clin Cancer Res. 2018;24(18):4416–4428. doi:10.1158/1078-0432.CCR-17-3649.
- Garg AD, Coulie PG, Van den Eynde BJ, Agostinis P. Integrating next-generation dendritic cell vaccines into the current cancer immunotherapy landscape. Trends Immunol. 2017;38(8):577–593. doi:10.1016/j.it.2017.05.006.
- Tanyi JL, Bobisse S, Ophir E, Tuyaerts S, Roberti A, Genolet R, Baumgartner P, Stevenson BJ, Iseli C, Dangaj D, et al. Personalized cancer vaccine effectively mobilizes antitumor T cell immunity in ovarian cancer. Sci Transl Med. 2018;10(436). doi:10.1126/scitranslmed.aao5931.
- Wilgenhof S, Corthals J, Heirman C, van Baren N, Lucas S, Kvistborg P, Thielemans K, Neyns B. Phase II study of autologous monocyte-derived mRNA electroporated dendritic cells (TriMixDC-MEL) plus ipilimumab in patients with pretreated advanced melanoma. J Clin Oncol. 2016;34(12):1330–1338. doi:10.1200/JCO.2015.63.4121.
- Nesselhut J, Marx D, Lange H, Regalo G, Cillien N, Chang RY, Nesselhut T. Systemic treatment with anti-PD-1 antibody nivolumab in combination with vaccine therapy in advanced pancreatic cancer. J Clin Oncol. 2016;34(15_suppl):3092. doi:10.1200/JCO.2016.34.15_suppl.3092.
- Poschke I, Lovgren T, Adamson L, Nystrom M, Andersson E, Hansson J, Tell R, Masucci GV, Kiessling R. A phase I clinical trial combining dendritic cell vaccination with adoptive T cell transfer in patients with stage IV melanoma. Cancer Immunol Immunother. 2014;63(10):1061–1071. doi:10.1007/s00262-014-1575-2.
- Lovgren T, Sarhan D, Truxova I, Choudhary B, Maas R, Melief J, Nyström M, Edbäck U, Vermeij R, Scurti G. Enhanced stimulation of human tumor-specific T cells by dendritic cells matured in the presence of interferon-gamma and multiple toll-like receptor agonists. Cancer Immunol Immunother. 2017;66(10):1333–1344. doi:10.1007/s00262-017-2029-4.
- Ellebaek E, Iversen TZ, Junker N, Donia M, Engell-Noerregaard L, Met O, Hölmich L, Andersen R, Hadrup S, Andersen M, et al. Adoptive cell therapy with autologous tumor infiltrating lymphocytes and low-dose Interleukin-2 in metastatic melanoma patients. J Transl Med. 2012;10:169. doi:10.1186/1479-5876-10-169.
- Yang J, Manson DK, Marr BP, Carvajal RD. Treatment of uveal melanoma: where are we now? Ther Adv Med Oncol. 2018;10:1758834018757175. doi:10.1177/1758834018757175.
- Zappasodi R, Merghoub T, Wolchok JD. Emerging concepts for immune checkpoint blockade-based combination therapies. Cancer Cell. 2018;33(4):581–598. doi:10.1016/j.ccell.2018.03.005.
- Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Rutkowski P, Lao CD, Cowey CL, Schadendorf D, Wagstaff J, Dummer R, et al. Five-year survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med. 2019;381(16):1535–1546. doi:10.1056/NEJMoa1910836.
- Jager MJ, Shields CL, Cebulla CM, Abdel-Rahman MH, Grossniklaus HE, Stern MH. Uveal melanoma. Nat Rev Dis Primers. 2020;6(1):24. doi:10.1038/s41572-020-0158-0.
- Koike N, Pilon-Thomas S, Mule JJ. Nonmyeloablative chemotherapy followed by T-cell adoptive transfer and dendritic cell-based vaccination results in rejection of established melanoma. J Immunother. 2008;31(4):402–412. doi:10.1097/CJI.0b013e31816cabbb.
- Lutz-Nicoladoni C, Wallner S, Stoitzner P, Pircher M, Gruber T, Wolf AM, Gastl G, Penninger JM, Baier G, Wolf D, et al. Reinforcement of cancer immunotherapy by adoptive transfer of cblb -deficient CD8 + T cells combined with a DC vaccine. Immunol Cell Biol. 2012;90(1):130–134. doi:10.1038/icb.2011.11.
- Park MY, Kim CH, Sohn HJ, Oh ST, Kim SG, Kim TG. The optimal interval for dendritic cell vaccination following adoptive T cell transfer is important for boosting potent anti-tumor immunity. Vaccine. 2007;25(42):7322–7330. doi:10.1016/j.vaccine.2007.08.037.
- Song S, Zhang K, You H, Wang J, Wang Z, Yan C, Liu F. Significant anti-tumour activity of adoptively transferred T cells elicited by intratumoral dendritic cell vaccine injection through enhancing the ratio of CD8(+) T cell/regulatory T cells in tumour. Clin Exp Immunol. 2010;162(1):75–83. doi:10.1111/j.1365-2249.2010.04226.x.
- Tamai H, Watanabe S, Zheng R, Deguchi K, Cohen PA, Koski GK, Shu S. Effective treatment of spontaneous metastases derived from a poorly immunogenic murine mammary carcinoma by combined dendritic-tumor hybrid vaccination and adoptive transfer of sensitized T cells. Clin Immunol. 2008;127(1):66–77. doi:10.1016/j.clim.2007.12.001.
- Andersen R, Donia M, Ellebaek E, Borch TH, Kongsted P, Iversen TZ, Hölmich LR, Hendel HW, Met Ö, Andersen MH. Long-lasting complete responses in patients with metastatic melanoma after adoptive cell therapy with tumor-infiltrating lymphocytes and an attenuated IL2 regimen. Clin Cancer Res. 2016;22(15):3734–3745. doi:10.1158/1078-0432.CCR-15-1879.
- de Coana YP, Wolodarski M, Poschke I, Yoshimoto Y, Yang Y, Nystrom M, Edbäck U, Brage SE, Lundqvist A, Masucci GV, et al. Ipilimumab treatment decreases monocytic MDSCs and increases CD8 effector memory T cells in long-term survivors with advanced melanoma. Oncotarget. 2017;8(13):21539–21553. doi:10.18632/oncotarget.15368.
- Rohaan MW, van den Berg JH, Kvistborg P, Haanen J. Adoptive transfer of tumor-infiltrating lymphocytes in melanoma: a viable treatment option. J Immunother Cancer. 2018;6(1):102. doi:10.1186/s40425-018-0391-1.
- Dudley ME, Wunderlich JR, Yang JC, Sherry RM, Topalian SL, Restifo NP, Royal RE, Kammula U, White DE, Mavroukakis SA, et al. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J Clin Oncol. 2005;23(10):2346–2357. doi:10.1200/JCO.2005.00.240.
- Johnson LA, Morgan RA, Dudley ME, Cassard L, Yang JC, Hughes MS, Kammula US, Royal RE, Sherry RM, Wunderlich JR, et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood. 2009;114(3):535–546. doi:10.1182/blood-2009-03-211714.