
ABSTRACT
The conversion of a non-T cell-inflamed into a T cell-inflamed tumor microenvironment (TME) is a key to sensitizing tumors to T-cell-based immunotherapies. Recent data show that the extracellular superoxide dismutase (SOD3) alters endothelial basement membrane (EC-BM) composition, providing permissive signals that enhance tumor infiltration by effector T cells.
Abbreviations
AJ, adherens junction; EC, endothelial cell; EC-BM, endothelial basement membrane; HIF, hypoxia-inducible factor; ICAM-1, intercellular adhesion molecule-1; LAMA4, laminin-α4; SOD3, superoxide dismutase-3; TME, tumor microenvironment; VCAM-1, vascular cell adhesion molecule-1; VEGF, vascular-endothelial growth factor
The density of tumor-infiltrating T lymphocytes (TIL) determines patient prognosis and response to immunotherapy. Tumors nonetheless restrict T cell infiltration, and thus evade the immune system. T cell exclusion has been associated with activation of oncogenic pathways, inactivation of tumor suppressors, heightened production of certain inflammatory mediators, and accumulation of the products of cancer cell metabolism.Citation1 In addition to, or as a consequence of these cancer cell-intrinsic events, tumors actively alter the endothelium and that constitutes another factor that shapes the inflammatory infiltrate.
Leukocyte extravasation from the bloodstream into a tissue entails sequential adhesion steps regulated by adhesion receptors and chemokines that culminates in crossing of the endothelial cell (EC) layer. Adhesion receptors, common to all endothelia, are necessary for diapedesis of all leukocyte types. Downmodulation of the main EC adhesion molecules (ICAM-1 or VCAM-1) is a feature of tumors with “immune desertification”. Inflamed tumors can also fine-tune infiltration of effector and suppressor immune cells by modulating chemokine expression.Citation2 To reach the tumor parenchyma, leukocytes must also cross the EC-BM, the main extracellular matrix structure that lies beneath EC.Citation3 EC-BM constituents include type IV collagen, fibronectin, perlecan, nidogen, and laminins. Laminins are particularly interesting, since the laminin-α4 (LAMA4) subunit enhances, whereas laminin-α5 (LAMA5) inhibits T cell diapedesis.Citation3 How the TME influences EC-BM is not known precisely, although it can be predicted that tumor blood vessel abnormalities affect EC-BM composition and integrity.
Overexpression of angiogenic factors, such as the vascular-endothelial growth factor (VEGF), causes abnormal tumor vasculature, which is linked to T cell exclusion. Elevated permeability of the tumor vasculature impairs perfusion and increases interstitial pressure, both negative regulators of leukocyte trafficking. Clinical evidence suggests that anti-VEGF agents synergize with immune checkpoint inhibitors to improve disease control and overall survival in different cancer types, an effect associated with larger numbers of effector T cells in the TME.Citation4 Although an increase in TIL might be linked to transient normalization of the tumor vasculature, VEGF blockade can also create an immunodominant TME by impeding VEGF immunosuppressive activities.Citation4 We reported that perivascular SOD3 triggers a program that simultaneously induces vascular normalization and T cell infiltration, thus linking these two processes unequivocally.Citation5,Citation6
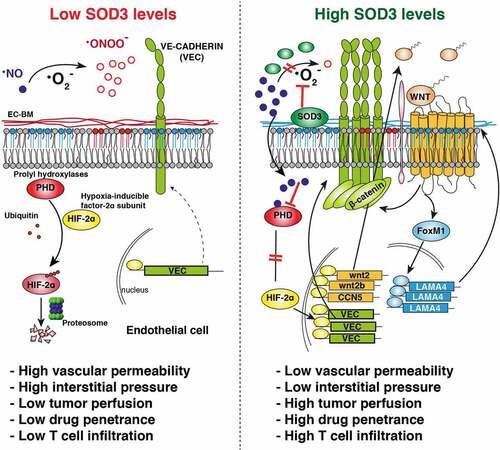
The anti-oxidant enzyme SOD3 is usually downregulated in tumors, which suggests that its loss is advantageous for cancer progression.Citation7 SOD3 catalyzes the dismutation of the superoxide radical (·O2−) in the extracellular space; this activity not only prevents oxidative damage of lipids or proteins but also preserves the bioavailability of nitric oxide (NO). NO is essential for SOD3-induced vascular normalization due to its inhibitory activity on the prolyl hydroxylase domain protein (PHD)-2. The PHD are central regulators of the hypoxia-inducible transcription factor (HIF)-α subunits, which they degrade in normoxia. High SOD3 levels, but not those of a catalytically inactive mutant, stabilize HIF-2α (but not HIF-1α) in EC in a NO-dependent manner.Citation5 This enhances HIF-2α-induced transcription of vascular-endothelial cadherin (VEC; cadherin 5, CD144), responsible for assembly of endothelial adherens junctions (AJ) and barrier architecture. SOD3 thus reduces EC monolayer permeability, prevents VEGF-induced destabilization of AJ, improves tumor perfusion, and increases delivery of chemotherapeutic drugs, all hallmarks of vessel normalization.Citation5 Specific HIF-2α ablation in EC prevents SOD3-induced vascular normalization in implanted tumors, which suggests that HIF-2α is a pivotal SOD3 mediator in vivo.Citation5
Whereas postnatal ablation of PHD2 induces hyperactive angiogenesis due to HIF-1α stabilization, hemizygous PHD2 deletion in EC stabilizes HIF-2α, induces VEC expression, and normalizes tumor vasculature.Citation8 NO also has antithetic, concentration-dependent effects on endothelial barrier function.Citation5 SOD3 does not affect NO synthesis in EC but appears to establish a delicate NO/·O2− balance in the TME that promotes specific HIF-2α (but not HIF-1α) stabilization and EC barrier tightening.
The SOD3/HIF-2α axis also renders the tumor endothelium more permissive to T cell infiltration. In vitro, SOD3 overexpression in EC increases transendothelial migration of naïve and activated CD4+ and CD8+ T cells, but not of myeloid (CD11b+) cells, in support of a specific EC-autonomous SOD3 effect on T cell transmigration. In vivo, perivascular SOD3 accumulation increases infiltration of adoptively transferred and endogenous tumor-specific T cells.Citation6 Specific HIF-2α deletion in the tumor endothelium causes a reduction in TIL numbers, which pinpoints HIF-2α as a necessary SOD3 mediator. SOD3 overexpression in the TME increases the CD8+/FoxP3+ ratio compared to controls,Citation6 which suggests distinct SOD3 effects on effector and regulatory T cell transmigration.
The mechanism by which the SOD3/HIF-2α pathway boosts T cell infiltration is independent of upregulation of endothelial adhesion receptors or T cell chemokines. HIF-2α instead upregulates transcription of specific WNT ligands in EC, triggering WNT pathway activation.Citation6 Pharmacological inhibition of the WNT pathway reverses the SOD3 effect on TIL infiltration in vitro and in vivo, which indicates its importance downstream of SOD3. A hallmark of WNT pathway activation is β-catenin stabilization. β-catenin levels are elevated in SOD3-overexpressing cells; however, β-catenin neither translocates to the nucleus nor induces transcription of canonical β-catenin-regulated genes in SOD3-overexpressing cells.Citation6 It accumulates massively in the juxtamembrane area, probably due to β-catenin sequestration by VEC at AJ.
SOD3 induces nuclear accumulation of another WNT-induced transcription factor, FoxM1, whose deficiency causes vascular abnormalities.Citation9 LAMA4, a permissive signal for T cell migration, is a transcriptional target of FoxM1.Citation3 LAMA4 silencing in EC abolishes SOD3-induced enhancement of T cell transmigration. In mouse tumor models, LAMA4 levels correlated positively with TIL density after SOD3 overexpression; in contrast, LAMA4 and TIL were reduced after endothelial HIF-2α deletion or WNT pathway inhibition.Citation6 These data support LAMA4 as a SOD3 mediator for TIL infiltration.
The SOD3/HIF-2α axis might operate in human malignancies. Indeed, SOD3 and VEC mRNA levels correlated positively in two colorectal cancer (CRC) cohorts, and most SOD3-stained tumor-associated EC showed HIF-2α staining.Citation5 SOD3 levels correlate positively with CD8+ T cell infiltration in a stage II CRC cohort, and with a T-cell inflamed signatureCitation10 in the COAD cohort from TCGA. SOD3 levels are also associated with lower recurrence rates in the stage II cohort (follow-up 120 months),Citation6 consistent with the prognostic value of CD8+ T cell infiltration in CRC.
These results identify a HIF-2α-dependent, SOD3-induced pathway that simultaneously triggers vascular normalization through VEC induction, and T cell infiltration through WNT pathway-induced LAMA4 expression (). The SOD3 downregulation observed in many human malignancies could be a mechanism for T cell desertification. Restoring SOD3 levels in the TME might thus transform immunologically “cold” into T cell-inflamed tumors, rendering them more susceptible to T cell-based immunotherapies.
Figure 1. SOD3 ties vascular normalization to tumor infiltration by T cells. Specific SOD3-induced signaling pathways in tumor-associated EC. SOD3 is downregulated in most tumors (left). In this situation, NO is oxidized to form ·ONOO− and PHD enzymes are active, which fosters proteasomal-induced HIF-2α degradation; VEC expression is low, tumor interstitial pressure is high, and LAMA4 is reduced in the EC-BM, which hampers T cell infiltration. SOD3 re-expression in the TME (right) prevents ·NO oxidation, which inhibits PHD enzymes and stabilizes HIF-2α. HIF-2α then initiates a transcription program that upregulates VEC, reducing interstitial pressure, and induces specific WNT ligands, which triggers autocrine/paracrine activation of the WNT pathway. By stabilizing β-catenin, WNT signaling then strengthens AJ and simultaneously triggers LAMA4 upregulation through FoxM1; this generates a LAMA4-enriched EC-BM permissive for T cell infiltration.
Disclosure of potential conflict of interest
No potential conflicts of interest were disclosed.
Additional information
Funding
References
- Trujillo JA, Sweis RF, Bao R, Luke JJ. T cell-inflamed versus non-T cell-inflamed tumors: A conceptual framework for cancer immunotherapy drug development and combination therapy selection. Cancer Immunol Res. 2018;6(9):990–3. doi:10.1158/2326-6066.CIR-18-0277.
- Lacalle RA, Blanco R, Carmona-Rodríguez L, Martín-Leal A, Mira E, Mañes S. Chemokine receptor signaling and the hallmarks of cancer. Int Rev Cell Mol Biol. 2017;331:181–244. doi:10.1016/bs.ircmb.2016.09.011.
- Di Russo J, Hannocks MJ, Luik AL, Song J, Zhang X, Yousif L, Aspite G, Hallmann R, Sorokin L. Vascular laminins in physiology and pathology. Matrix Biol. 2017;57-58:140–148. doi:10.1016/j.matbio.2016.06.008.
- Hilmi M, Neuzillet C, Calderaro J, Lafdil F, Pawlotsky JM, Rousseau B. Angiogenesis and immune checkpoint inhibitors as therapies for hepatocellular carcinoma: current knowledge and future research directions. J Immunother Cancer. 2019;7(1):333. doi:10.1186/s40425-019-0824-5.
- Mira E, Carmona-Rodríguez L, Pérez-Villamil B, Casas J, Fernández-Aceñero MJ, Martínez-Rey D, Martín-González P, Heras-Murillo I, Paz-Cabezas M, Tardáguila M, et al. SOD3 improves the tumor response to chemotherapy by stabilizing endothelial HIF-2α. Nat Commun. 2018;9(1):575. doi:10.1038/s41467-018-03079-1.
- Carmona-Rodríguez L, Martínez-Rey D, Fernández-Aceñero MJ, González-Martín A, Paz-Cabezas M, Rodríguez-Rodríguez N, Pérez-Villamil B, Sáez ME, Díaz-Rubio E, Mira E, et al. SOD3 induces a HIF-2α-dependent program in endothelial cells that provides a selective signal for tumor infiltration by T cells. J Immunother Cancer. 2020;8(1):e000432. doi:10.1136/jitc-2019-000432.
- Griess B, Tom E, Domann F, Teoh-Fitzgerald M. Extracellular superoxide dismutase and its role in cancer. Free Radic Biol Med. 2017;112:464–479. doi:10.1016/j.freeradbiomed.2017.08.013.
- Mazzone M, Dettori D, de Oliveira RL, Loges S, Schmidt T, Jonckx B, Tian YM, Lanahan AA, Pollard P, de Almodovar CR, et al. Heterozygous deficiency of PHD2 restores tumor oxygenation and inhibits metastasis via endothelial normalization. Cell. 2009;136(5):839–851. doi:10.1016/j.cell.2009.01.020.
- Kim IM, Ramakrishna S, Gusarova GA, Yoder HM, Costa RH, Kalinichenko VV. The forkhead box m1 transcription factor is essential for embryonic development of pulmonary vasculature. J Biol Chem. 2005;280(23):22278–22286. doi:10.1074/jbc.M500936200.
- Grasso CS, Giannakis M, Wells DK, Hamada T, Mu XJ, Quist M, Nowak JA, Nishihara R, Qian ZR, Inamura K, et al. Genetic mechanisms of immune evasion in colorectal cancer. Cancer Discov. 2018;8(6):730–749. doi:10.1158/2159-8290.CD-17-1327.