
ABSTRACT
Macroautophagy (autophagy) is an essential cellular catabolic process required for survival under conditions of starvation. The role of autophagy in cancer is complex, context-dependent and at times contradictory, as it has been shown to inhibit, promote or be dispensable for tumor progression. In this study, we evaluated the contribution of the immune system to the reliance of tumors on autophagy by depleting autophagy-related 7 (ATG7) in murine tumor cells and grafting into immunocompetent versus immunodeficient hosts. Although loss of ATG7 did not affect tumor growth in vitro or in immunodeficient mice, our studies revealed that cancer cell reliance on autophagy was influenced by anti-tumor immune responses, including those mediated by CD8+ T cells. Furthermore, we provide insights into possible mechanisms by which autophagy disruption can enhance anti-tumor immune responses and suggest that autophagy disruption may further benefit patients with immunoreactive tumors.
Introduction
Macroautophagy (herein referred to as autophagy) is a cellular maintenance and survival mechanism that traffics cytosolic components to lysosomes for degradation and recycling via double-membraned vesicles termed autophagosomes. The ATG7 protein is essential for autophagy,Citation1 as its ubiquitin E1-like activity facilitates the conjugation of ATG5 to ATG12 and of phosphatidylethanolamine (PE) to LC3-I (MAP1LC3B cleaved by ATG4 enzymes), generating the lipidated form LC3-II. This event leads to the loading of autophagic cargo and cargo receptors, such as p62/SQSTM1, to autophagosomes and subsequent degradation in lysosomes.Citation2 Basal levels of autophagy maintain cellular homeostasis by limiting the accumulation of damaged proteins and organelles, and autophagy can be upregulated upon nutrient deprivation in order to maintain essential intracellular metabolite levels.Citation3
These pro-survival functions of autophagy can be hijacked by cancer cells to enable rapid proliferation under conditions of metabolic stress, which are typically observed in the tumor microenvironment. The majority of studies examining the role of autophagy in cancer have provided evidence that autophagy promotes tumor growth.Citation4-Citation17 However, work from other groups, including our own, have demonstrated that autophagy loss does not impact tumor growth.Citation18,Citation19 Other studies have found that autophagy loss can promote tumor growth.Citation20,Citation21 Thus, the role of autophagy in cancer is complex and may depend on numerous factors unique to individual tumors and the tumor microenvironment.Citation22
We hypothesized that the presence or absence of an intact immune response as a component of the tumor models may contribute to differing outcomes of autophagy loss in published studies. Xenografts of human tumors in immunodeficient mice continue to play important roles in cancer research and drug development due to the abundance of cell lines that exemplify the genetic landscape of primary human tumors. However, the recent clinical success of immune-checkpoint antibodies blocking CTLA-4Citation23 or PD-1/PD-L1Citation24,Citation25 have highlighted the importance of immune system function, particularly tumor-reactive T cells, in pre-clinical models of cancer. Therefore, we evaluated the role of autophagy in several syngeneic models of cancer with varying degrees of tumor immune reactivity by deleting the essential autophagy gene Atg7. We found that cancer cell dependence on ATG7 was most pronounced in the context of an underlying immune response against the tumors, and partially driven by CD8+ T cells. Additionally, we provide insight into how disruption of cancer cell autophagy may further augment the immune response against tumors.
Results
ATG7 is dispensable for murine cancer cell growth in vitro or in immunocompromised mice
To examine the role of autophagy in murine cancer cell proliferation, Atg7 was deleted in B16F10 (melanoma), MC38, and CT26 (colorectal carcinoma) cell lines using CRISPR/Cas9. These three cell lines were selected for their collective breadth of anti-tumor immune activity ranging from non-responsive to highly responsive.Citation26-Citation28 The initial population of edited B16F10 cells had undetectable levels of ATG7 protein (ATG7KO). For MC38 and CT26, gene editing resulted in incomplete ATG7 loss, thus single cells that lacked ATG7 protein were isolated and expanded (MC38 ATG7KO#1, CT26 ATG7KO#40, and CT26 ATG7KO#89) for use in subsequent studies. To validate functional loss of ATG7 and autophagy, lysates of edited cells were probed for ATG5, LC3 and p62. As ATG7 is essential for the formation of ATG5-ATG12 and LC3-PE conjugates,Citation2 ATG7-deficient cells completely lacked ATG5-ATG12 conjugates as well as lipidated LC3 (LC3-II) (). An accumulation of the autophagy cargo receptor SQSTM1/p62Citation29 was also observed in ATG7KO cell lines, indicative of decreased autophagic flux (). Finally, since autophagy supports cell survival when nutrients are scarce,Citation1,Citation30 Control (Ctrl) and ATG7KO cell lines were subjected to serum/amino acid starvation followed by a brief recovery in complete medium. All ATG7-deficient cell lines had a significant survival disadvantage when nutrients were removed (), demonstrating that autophagy is dysfunctional in these cell lines.
Figure 1. Loss of ATG7 blocks autophagy and sensitizes cells to nutrient deprivation.
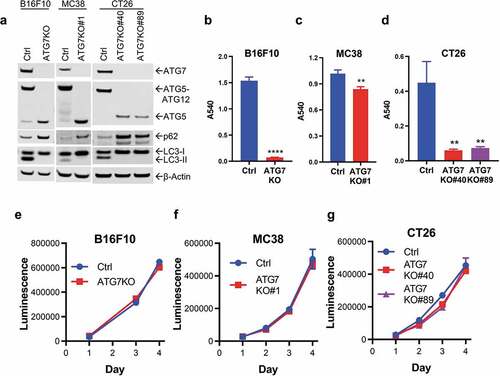
Reports have been conflicting as to whether autophagy supports cancer cell growth in vitro and in vivo.Citation4-Citation19 Therefore, we first evaluated the proliferation of ATG7KO cell lines under nutrient rich conditions in vitro. Compared to Ctrl cells, loss of ATG7 did not impact the proliferation of B16F10, MC38 or CT26 cells in vitro (), suggesting that autophagy is dispensable for the proliferation of murine cancer cell lines under standard culture conditions. These data are consistent with our previous studies showing autophagy-independent proliferation in nutrient-rich conditions in a panel of human cancer cell lines.Citation18 The metabolic composition of blood plasma is vastly different than that of standard nutrient-rich culture media,Citation31,Citation32 and the actual nutrient composition of the tumor microenvironment likely falls between complete culture media and the severe starvation conditions utilized in . Consequently, in vitro proliferation assays may not be predictive of cancer cell dependence on the metabolite-sensitive cellular process of autophagy in a physiologic setting.
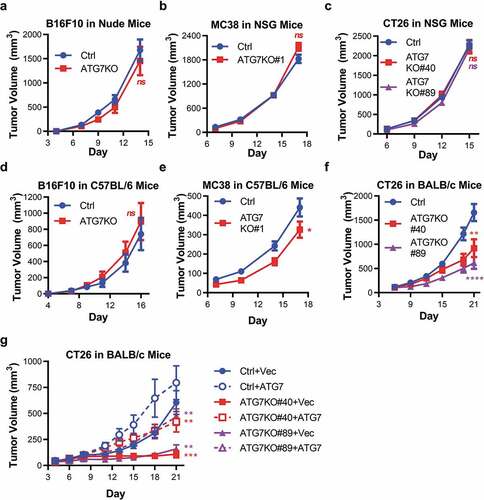
To determine if autophagy supports murine cancer cell tumorigenicity in vivo, Ctrl and ATG7KO cell lines were engrafted into immunodeficient mice. Loss of ATG7 did not impact the growth of B16F10 tumors (in nude mice), MC38, or CT26 tumors (in NSG mice) (). To ensure that the ATG7 knockout was maintained throughout the course of the in vivo studies, tumor lysates were probed for ATG7 and ATG5. ATG7 protein levels were markedly reduced and a prominent free ATG5 band was observed in all the ATG7KO tumors (Supplemental Figure 1a-c), indicating that ATG7 loss of function is largely maintained in the knockout tumors. Faint ATG7 and ATG5-12 bands could be observed in the ATG7KO tumors, which is likely a result of contaminating autophagy-competent host tissue that infiltrated the tumors. Taken together, these results suggest that autophagy is dispensable for murine tumor growth in vitro and in immunodeficient in vivo tumor models.
Figure 2. Murine tumors have differential reliance upon ATG7 when grown in immuno-competent hosts.
Tumors have differential reliance on autophagy when grown in immunocompetent hosts
We next assessed if the presence of an intact immune system impacted the requirement of cancer cell-intrinsic autophagy in tumor growth by evaluating tumorigenicity of the ATG7-deficient cell lines in immunocompetent (C57BL/6 or BALB/c) hosts. Compared to Ctrl tumors, loss of ATG7 did not affect the growth of B16F10 tumors in C57BL/6 mice (). Loss of ATG7 modestly, but significantly, impaired the growth of MC38 tumors in C57BL/6 mice (). Loss of ATG7 had the most pronounced effect on CT26 tumors, as both ATG7KO clones (#40 and #89) on average grew significantly slower than Ctrl tumors when engrafted into BALB/c mice (). Similar to the studies performed with immunocompromised mice, the decrease in ATG7 protein and increase in free ATG5 was maintained in ATG7KO B16F10, MC38, and CT26 tumors harvested from immunocompetent hosts (Supplemental Figure 1a-c), confirming that the autophagy defect was preserved throughout the course of the studies.
To ensure that the observed tumor growth inhibition of the CT26 ATG7KO tumors was due specifically to ATG7 loss and not due to clonal variation, rescue studies were performed to determine whether ATG7 re-expression would reverse tumorigenicity defects observed with ATG7 loss. Vector only (+Vec) or ATG7 with V5 tag (+ATG7) were stably expressed in CT26 Ctrl, ATG7KO#40 and ATG7KO#89 cell lines. ATG7 re-expression resulted in restored ATG5-12 conjugate formation, LC3 lipidation, reduced p62 protein levels and enhanced cellular capacity to survive nutrient deprivation (Supplemental Figure 2a-b). Thus, re-expression of ATG7 restored autophagic capacity to the level observed in Ctrl cells. Additionally, ATG7 expression did not alter proliferation of the CT26 cell lines under standard culture conditions (Supplemental Figure 2c).
The ATG7-deficient and rescue cell lines were next engrafted into immunocompetent BALB/c mice. Similar to the results in , a pronounced inhibition of tumor growth was observed in the ATG7KO+Vec cell lines compared to the Ctrl+Vec and Ctrl+ATG7 cell lines. Re-expression of ATG7 significantly enhanced the growth of both ATG7KO clones similar to the growth rate of the Ctrl+Vec tumors (). The re-introduced ATG7 protein was observed in tumor lysates of the appropriate samples, as well as a decrease in free ATG5 and a decrease in p62 in ATG7-expressing vs –deficient tumors (Supplemental Figure 2d). The ability of exogenous ATG7 to restore both functional autophagy and in vivo tumor growth in the ATG7KO cell lines demonstrates that autophagy supports the growth of CT26 tumors when engrafted into immunocompetent hosts.
CD8+ T cells promote cancer cell dependence on autophagy
Given that the dependence of tumor growth on ATG7 in immunocompetent hosts ranged from non-existent (B16F10), to modest (MC38), to substantial (CT26), we investigated which factors contribute to the impact of autophagy disruption on tumor growth. The immune responses of mouse cancer cell lines implanted into immunocompetent hosts are well-characterized.Citation26-Citation28 For example, B16F10 cells are defined as immunologically “cold” because the tumors have sparse immune infiltrate, do not respond to immune checkpoint blockade (ICB) immunotherapy and have low cytolytic activity score (defined by log-average of granzyme A (Gzma) and perforin (Prf1) expression).Citation33 On the opposite end of the spectrum, CT26 tumors are classified as immunologically “hot” because they have an abundance of immune infiltrate, respond well to immunotherapy and have high cytolytic activity score. MC38 are intermediate, as they have substantial immune infiltrate but have limited response to the ICB agents anti-CTLA-4 and anti-PD-L1 and evoke a relatively low T cell-mediated cytolytic activity score.Citation26,Citation28,Citation34 Given the correlation between immunogenicity and in vivo sensitivity to ATG7 loss, we reasoned that anti-tumor immunity may drive cancer cell reliance upon autophagy.
To analyze levels of immune infiltrates in our syngeneic models, we used immunohistochemistry (IHC) to detect and quantify CD3+ and CD8+ cells within B16F10, MC38, and CT26 tumors isolated from immunocompetent hosts. Compared to B16F10 tumors, both MC38 and CT26 tumors were significantly enriched for CD3+ and CD8+ cells, and there was no significant difference between MC38 and CT26 tumors (). These data confirm that the immunogenicity profile of the cell lines used in this study correlates with previously published results.Citation26-Citation28
Figure 3. CD8+ and CD4+ T cell contribution to cancer-cell dependence on ATG7 in vivo.
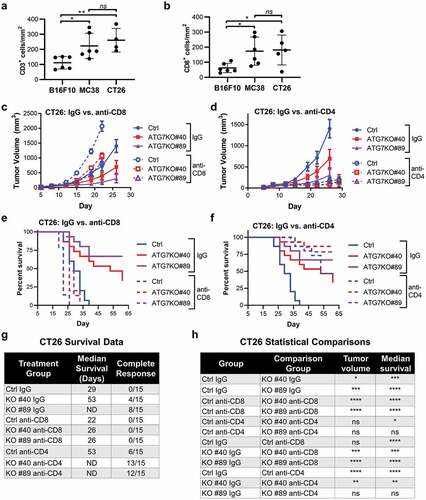
Since CT26 tumors have a combination of relatively high CD8+ T cell infiltrate, elevated cytolytic activity score and enhanced response to ICB,Citation26–Citation28,Citation34 we investigated whether CD8+ T cell mediated anti-tumor immunity rendered these tumors reliant upon autophagy for survival. To assess the contribution of CD8+ T cells, CT26 Ctrl or ATG7KO tumors were engrafted into BALB/c mice that were treated with IgG control or anti-CD8 antibodies, the latter of which significantly decreased CD8+ T cells to a nearly undetectable level in mice at the time of tumor implant (Supplemental Figure 3a). The depletion of CD8+ T cells significantly restored growth of both ATG7KO tumor lines to levels comparable with Ctrl tumors grown in mice treated with the control IgG (). CD8+ T cell depletion non-significantly enhanced the growth of Ctrl tumors (), suggesting that while CD8+ T cells contribute to the reduced growth of autophagy-deficient tumors, they are not the sole determinant.
CD4+ helper T cells can also impede tumor growth,Citation35 thus we assessed the contribution of CD4+ T cells to the tumorigenicity of ATG7KO tumors. Treatment with anti-CD4 antibodies depleted CD4+ T cells (Supplemental Figure 3b) and resulted in a marked growth reduction of both Ctrl and ATG7KO tumors (). The decreased tumor burden observed upon CD4 depletion is likely a result of the elimination of CD4+ pro-tumorigenic regulatory T cells (Tregs), as deletion of CD25, a marker of Tregs, suppresses growth of CT26 tumors.Citation36
We further interrogated tumor response in this study by allowing tumors to progress until euthanasia criteria were met in order to evaluate the impact of autophagy loss on disease-mediated survival. In IgG treated mice, loss of ATG7 dramatically prolonged median survival compared to Ctrl tumor bearing mice and generated complete regressions in 12/30 mice (). Treatment with anti-CD8 decreased survival in all tumor bearing mice and diminished the difference in survival between Ctrl and ATG7KO mice. Loss of ATG7 only yielded a 4-day extension of median survival compared to Ctrl tumors in CD8+ T cell depleted mice, while ATG7KO prolonged median survival by ≥24 days in IgG treated mice. Additionally, none of the ATG7KO tumor-bearing mice had complete responses when CD8+ T cells were depleted (). In contrast to the impact of CD8 depletion on tumor growth, depletion of CD4 + T cells significantly prolonged the survival of both Ctrl and ATG7KO tumor-bearing mice (). Due to the strong suppression of tumor growth by treatment with anti-CD4 antibodies, median survival was not reached in the ATG7KO groups and the study lacked statistical power to conclude whether CD4+ T cells play a significant role in the survival advantage conferred by ATG7 deletion.
To further interrogate why autophagy disruption has more profound effects in the immunocompetent setting, RNA sequencing (RNAseq) was performed on ATG7-competent or ATG7-deficient CT26 cells grown in immune-competent (BALB/c) or immune-deficient (NSG) mice. In NSG mice, loss of ATG7 in tumors resulted in the significant upregulation of 12 genes, and the significant downregulation of 3 genes when compared to autophagy-proficient tumors ( and Supplemental Table 1). Of these 15 gene expression changes, 14 were conserved in BALB/c mice. However, in immunocompetent BALB/c mice, disruption of tumor-intrinsic autophagy had a substantially greater impact on the transcriptional profile of tumors, with upregulation of 816 genes and downregulation of 114 genes upon ATG7 loss ( and Supplemental Table 2). These data demonstrate that the presence of a host immune system triggers far more changes in the tumor transcriptome upon autophagy loss in the CT26 model. Due to the use of bulk RNAseq and not single-cell RNAseq, we are unable to distinguish whether the gene expression changes originate in the tumor cells or infiltrating host tissue.
Figure 4. Gene expression and pathway alterations upon Atg7 loss.
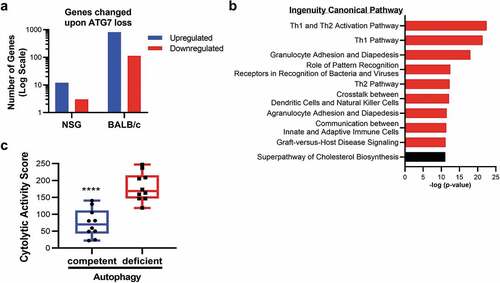
To decipher pathways impacted by autophagy disruption, Ingenuity Pathway Analysis (IPA) was performed on the genes modulated by ATG7 loss in immune-competent mice. This analysis revealed that nine of the top ten canonical pathways significantly enriched in autophagy-deficient tumors were related to immunity (), with the top 2 being Th1 and Th1/Th2 pathways. Consistent with its role in the Th1 response, one of the most highly and significantly upregulated genes in autophagy-deficient tumors from BALB/c mice was Ifng (Log2 fold-change 1.94, p-value <0.0001). We also noted enhanced expression of Cd8a (Log2 fold-change 1.2, p-value <0.0001) (Supplemental Table 2), indicative of increased infiltration of CD8+ T cells. Furthermore, autophagy-deficient tumors had an enhanced cytolytic activity score, as defined by log-average of Gzma and Prf1 expression (), which revealed that autophagy-deficient tumors had an abundance of cells with potential to mediate tumor killing, such as cytotoxic CD8+ T cells or NK cells. The enrichment of these immune-related genes indicates that disruption of cancer cell autophagy can enhance the recruitment, and possibly the activity, of effector cells to the tumor microenvironment, resulting in decreased tumor burden.
Discussion
To probe the consequence of tumor-intrinsic autophagy disruption, we evaluated autophagy loss in tumors implanted in both immunocompetent and immunodeficient mice. Although we found that cancer cell intrinsic autophagy is dispensable for tumor growth in immunodeficient hosts, consistent with our previous results,Citation18 we discovered that cancer cell reliance on autophagy varies dramatically when different tumor models are engrafted in immunocompetent hosts and is likely driven by the underlying immune response to the tumors. Our findings highlight the importance of testing cancer cell growth mechanisms in immunocompetent models in order to capture immune-mediated responses that may impact seemingly tumor-intrinsic effects. These findings, along with other recent studies demonstrating a role for autophagy in suppressing anti-tumor immune responses,Citation37,Citation38 help define factors which contribute to tumor reliance on autophagy, and shed light as to why autophagy inhibition may have different outcomes in different circumstances.
Previous studies have explored the contribution of host factors to dependence on tumor-intrinsic autophagy, specifically immune cell infiltrates.Citation4,Citation13 Our study further characterizes adaptive immune responses that may be enhanced by autophagy disruption. In support of the concept that preexisting immunity is required, the “cold” B16F10 cancer cell line is not responsive to ATG7 loss, the “warm” MC38 model displayed modest sensitivity, and the “hot” CT26 cancer cell line yielded the most robust responses to autophagy disruption when implanted into immunocompetent mice. These findings are consistent with studies demonstrating that autophagy loss through ATG7 or ATG12 knockdown does not impact growth of B16F10 or 4T1 tumors,Citation19 another syngeneic model that lacks response to anti-CTLA-4 and anti-PD-L1, has lower levels of immune infiltrate, and decreased cytolytic activity compared to CT26 tumors.Citation26,Citation28 However, autophagy blockade through inhibition of Vps34, an enzyme critical to both autophagic and endocytic pathways, was efficacious in the “cold” B16F10 model.Citation37 Thus, despite the correlation between preexisting immunity and sensitivity to ATG7 loss in our study, it is possible that other factors contribute to the differential sensitivity of these tumor models, and that different methods of autophagy disruption may warrant different outcomes.
To further understand which immune cell types contribute to the decreased growth of ATG7KO tumors, we depleted CD8+ T cells and found that reducing adaptive immune responses against CT26 tumors diminished the survival advantage conferred by ATG7 deletion, similar to work published in pancreatic cancer models.Citation38 Although our results with CD4 depletion were inconclusive, pathway analysis revealed upregulation of TH1 and TH2 pathways, supporting a potential role for CD4+ T helper cells in the control of tumor growth upon autophagy loss. Autophagy in cancer cells can limit immune-mediated cell death by autophagolysosome-mediated sequestration and degradation of granzyme B, a protease secreted by cytotoxic T cells and natural killer (NK) cells.Citation14,Citation15 Given that depletion of CD8+ T cells did not completely rescue the growth of ATG7KO cancer cells, it is possible that NK cells also contribute to the killing of autophagy-deficient CT26 tumors. Recent studies have demonstrated roles of NK cells and CD8+ T cells (driven by CD103+ dendritic cells)Citation37,Citation38 in promoting immune responses after tumor-intrinsic autophagy loss. Thus, it is likely that in our studies, cooperation between multiple immune cell types influence tumor growth inhibition in response to ATG7KO.
By interrogating pathways altered upon ATG7 loss, we found that tumors lacking ATG7 had enhanced expression of immune-related genes and increased cytolytic activity scores. These findings are consistent with others who have described that loss of ATG7 induces a CD8+/Th1 gene signature in an APC-driven spontaneous mouse model of colorectal cancer.Citation4 It has also been reported that autophagy disruption enhances expression of cytokines such as CCL5,Citation16,Citation39 CXCL10Citation17 and CXCL5,Citation5 and CCL5 blockade reversed tumor growth suppression by Vps34 inhibitors.Citation37 Indeed, we observed increased expression of Ccl5, Cxcl10 and Cxcl5 in our autophagy-deficient tumors (Supplemental Table 2), indicating that these corresponding proteins could contribute to the enhanced infiltration and/or activation of effector immune cells such as T or NK cells. Finally, recent work in pancreatic cancer found that blockade of tumor-intrinsic autophagy can activate T cells through enhanced MHC-I presentation.Citation38 The mechanism(s) by which autophagy disruption alters the tumor infiltrating lymphocyte repertoire is likely multifaceted, and additional studies will be required to reveal and validate the roles of cytokines and additional immune cell types in the response to tumor-intrinsic autophagy loss.
Our findings relied on deletion of ATG7, and while ATG7 is a core component of autophagic machinery due to its role in ubiquitin-like reactions, ATG7 has roles beyond macroautophagy.Citation40 Thus, use of orthogonal genetic approaches would confirm whether enhanced anti-tumor immunity upon ATG7 loss are autophagy dependent. Studies using the B16F10 and 4T1 models yielded similar results with both ATG7 and ATG12 knockdown.Citation19 Recent work in pancreatic cancer similarly illustrates enhanced T cell responses upon autophagy deficiency with either ATG7 knockdown or expression of dominant negative ATG4B, and that the enhanced immunogenicity is due to autophagic degradation of MHC-I.Citation38 Another recent study demonstrated that genetic or pharmacological inhibition of VPS34 enhanced anti-tumor immunity.Citation37 In sum, these results suggest that tumor-intrinsic autophagy alters immune visibility of tumors, although these data do not rule out a contribution for non-autophagic pathways.
Our data might suggest that, in immunogenic tumors, autophagy inhibition in cancer would be therapeutically advantageous. However, we deleted ATG7 only in cancer cells, whereas therapeutic autophagy inhibitors would have systemic effects. Similar to the context-dependent nature of autophagy in cancer, the roles of autophagy across different tissues are equally complex and could influence the anti-tumor effects of autophagy loss in cancer cells. For example, whole-body ablation of ATG7 drastically decreases lifespan in adult animals.Citation11 Similar to its multifaceted roles in cancer, autophagy has context-dependent roles in the immune system that could be either deleterious or advantageous in the response to tumors.Citation41 Autophagy in T cells can promote T cell expansionCitation42 and autophagy in antigen-presenting cells can promote both MHC class II and MHC class I antigen presentation.Citation43 However, disruption of autophagy in CD8+ T cells increases glucose metabolism resulting in enhanced control of syngeneic breast and prostate tumors.Citation44 Beyond effects on the immune system, stromal autophagy promotes tumor growth through alanine secretion,Citation45 and acute, whole-body deletion of ATG7 retards tumor growth via arginine starvation.Citation46 The effects of small molecule inhibitors would likely be less pronounced than those observed with complete genetic loss, as suggested by the tolerability of systemic Vps34 inhibition at efficacious doses.Citation37 In conclusion, we add further definition to the context-dependent nature of autophagy-driven tumor growth by describing that an underlying immune response promotes cancer cell sensitivity to ATG7 loss, which in turn can further enhance anti-tumor immunity.
Materials and methods
Cell lines
B16F10 and CT26 cells were obtained from the American Type Culture Collection and MC38 cells were kindly provided by the laboratory of Antoni Ribas at UCLA. CT26 and MC38 were cultured in RPMI and B16F10 in DMEM, all supplemented with 10% FBS and penicillin/streptomycin (complete medium). Cells were maintained in a 37°C humidified incubator with 5% CO2 and tested negative for mycoplasma throughout the course the study.
ATG7-deficient cell lines were generated by CRISPR-Cas9- mediated deletion of the Atg7 gene. An sgRNA vector containing the guide sequence 5ʹ ACGTCCAGGGCACTATTAAA was purchased from PNA Bio, as well as a Cas9 expression vector (pRGEN-Cas9-CMV) and a reporter vector containing the same guide sequence, constitutively expressed RFP, and expressed EGFP when the guide sequence is excised. Cell lines were transiently transfected with sgRNA vector, Cas9 vector, and reporter vector using Lipofectamine 2000 (Invitrogen), according to PNA Bio instructions. Cells were harvested and sorted for RFP+EGFP+ on a FACS Aria II (BD Biosciences). Control (Ctrl) cells were transfected with the Cas9 and reporter vectors and sorted for RFP+. For cell lines that did not have complete loss of ATG7, as indicated by western blot (CT26 and MC38), RFP+EGFP+ cells were subsequently expanded from single-cell clones to isolate cells that lacked ATG7.
In order to re-express ATG7, CT26 cells were stably transduced with lentivirus containing murine Atg7 (NM_001253717.1) in the pLenti6.3/V5-DEST vector (ThermoFisher) and selected with 5 ug/ml blasticidin (ThermoFisher). The pLenti6.3/V5-GW/lacZ vector (ThermoFisher) was used as the negative vector control.
In vitro growth assays
To assess cell proliferation, 1,000 cells/well were plated in 96-well plates in complete medium and cell abundance was measured by CellTiter-Glo (Promega) at indicated time points, per manufacturer’s instructions.
Sensitivity to nutrient deprivation was evaluated by first plating 2 × 105 cells/well in 12-well plates in complete medium. Cells were allowed to adhere overnight and then washed and starved in Hank’s balanced salt solution (HBSS) (HyClone SH30030.02) for 2 days (MC38) or 3 days (B16F10, CT26) and allowed to recover for 2 days (B16F10, CT26) or 4 days (MC38) in complete medium. Cell abundance was measured by staining with SRB, dissolving in 10 mM Tris Base and reading absorbance at 540 nm (A540), as previously described.Citation47
Immunoblotting
Cells cultured in vitro were lysed in NuPAGE LDS buffer with reducing agent (ThermoFisher), and tumor xenograft fragments were lysed in RIPA buffer (50 mM Tris-HCL (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.5% Sodium Deoxycholate, 0.1% SDS, protease inhibitors, and phosphatase inhibitors), followed by sonication. Immunoblotting was performed as previously described,Citation48 with antibodies against ATG7 (Cell Signaling Technology #8558), ATG5 (Abcam #ab108327), p62 (Cell Signaling Technology #5114), LC3B (Sigma #L7543) and β-Actin (Sigma #A1978). Primary antibodies were diluted to 1:1000, and secondary antibodies were diluted to 1:15000 in TBS Odyssey Blocking Buffer with 0.2% Tween. ATG7 blots were blocked in Blocking Buffer for Fluorescent Western Blotting (Rockland #MB-070) and all other blots were blocked in TBS Odyssey Blocking Buffer (LI-COR #927-50000). Blots were imaged on an Odyssey CLx scanner using Image Studio software (Version 5.2) (Li-Cor).
In vivo studies
All procedures performed on animals were in accordance with regulations and established guidelines and were reviewed and approved by Pfizer’s Institutional Animal Care and Use Committee. All mice were female between the ages of 8–12 weeks. For tumor growth studies, 5 × 105 (B16F10), 1 × 106 (MC38) or 2 × 106 (CT26) cells were suspended in complete media without antibiotics and subcutaneously implanted into the hind flanks of athymic nude mice (Charles River), NSG, C57BL/6 or BALB/c mice (Jackson Laboratory). For RNAseq studies, 2 × 106 CT26 ATG7KO#40+ ATG7 (autophagy-competent) or ATG7KO#40+ Vec (autophagy-deficient) cells were implanted as described above and harvested 8 days post-implant. Samples were processed for RNAseq as described below. For T cell infiltration studies, B16F10, MC38, or CT26 cells were implanted as described above, harvested when tumors reached 400 mm3, and processed for IHC as described below. For T cell depletion studies, BALB/c mice were dosed intraperitoneally with 100 µg IgG2b clone LTF-2 isotype control (BioXCell #BP0090), anti-CD8α clone 2.43 (BioXCell #BP0061) or anti-mouse CD4 Clone GK1.5 (BioXCell #BP0003-1) suspended in PBS for two consecutive days prior to tumor implant. 1 × 106 CT26 cells were implanted as described above, and mice were dosed with antibodies twice-a-week until day 26 post-implant. CD8+ T cell depletion was measured by flow cytometry on spleens and blood harvested the same day as tumor implant, as described below. Mice were euthanized when tumors reached 1500 mm3. Tumor volumes were determined by caliper measurements obtained in two dimensions and was calculated as (width2x length)/2.
Flow cytometry
Mouse tissue was harvested, dissociated and stained with antibodies as previously described.Citation48 Dissociated cells were stained with Fixable Viability Dye eFluor™ 780 (ThermoFisher #65-0865-14) diluted 1:1000 in Dulbecco’s PBS (DPBS) for 15 minutes at 4°C, washed in FACS buffer (0.5% FBS, 2 mM EDTA in DPBS), blocked with UltraLEAF™ Purified anti-mouse CD16/32 (Biolegend #101330) in FACS buffer for 10 minutes at 4°C, and stained with antibodies against CD45 (BD #553080), CD3 (Biolegend #100312), CD4 (Biolegend #116006) and CD8b (BD #740761) diluted 1:100 in FACS buffer for 15 minutes at 4°C, followed by analysis on a Fortessa X-20 flow cytometer.
RNAseq
Processing of snap-frozen tumor samples and RNA-Seq profiling was conducted by Novogene, USA. RNA from autophagy-competent and autophagy-deficient tumor tissue samples (five samples per group from NSG mice and 10 samples per group from BALB/c mice) were pair-end sequenced with read length of 2 x 150bps. Raw reads were first adapter trimmed with Trimmomatic-0.36Citation49 and then mapped to the UCSC mm10 reference genome using Bowtie 2 (v2.2.5).Citation50 Expected counts and normalized expression levels of genes in transcripts per million (TPM) were generated by RSEM (v1.2.20).Citation51 Genes specifically differentially regulated in autophagy-deficient tumor samples compared to autophagy-competent tumor samples were identified using the DESeq2Citation52 package with criteria of adjusted p-value (FDR) ≤ 0.01, absolute fold change ≥1.5 and the maximum of group mean ≥10. Hierarchical clustering of the significantly regulated genes was performed in R using Euclidean distance method and ward.D clustering method. Cytolytic activity (CYT) was defined as the log-average (geometric mean) of Gzma and Prf1 expression value (TPM) as described by Rooney et al.Citation33 All pathway enrichment analysis was performed using Ingenuity® Pathway Analysis (IPA®).
Immunohistochemistry and digital image analysis
Immunohistochemistry and digital image analysis were performed as previously described.Citation48 Excised tumors were fixed in 10% neutral buffered formalin for 48 hours prior to being paraffin embedded. Five micron sections were deparaffinized in xylene and rehydrated through a graded series of alcohols to deionized water. Sections underwent heat-induced epitope retrieval in Borg Decloaker (Biocare Medical) for 30 minutes, endogenous peroxidase block with Peroxidazed 1 (Biocare Medical) for 10 min, and protein block with Background Punisher (Biocare Medical) for 10 min. Anti-CD3 (Clone SP162, Abcam #ab135372) or anti-CD8 (Clone D4W2Z, Cell Signaling #98941) was applied at 1:300 for 60 minutes followed by MACH2 Rabbit HRP-Polymer (Biocare Medical) for 30 minutes, and Vina Green chromogen (Biocare Medical) for 12 minutes. After chromogen staining, slides were rinsed in dH2O, counterstained for 10 seconds in Tacha’s hematoxylin (Biocare Medical), dehydrated in 100% alcohol, cleared in xylene, and coverslipped with Permount medium (Fisher). When dry, slides were scanned on a Leica/Aperio AT2 whole slide digital scanner and analyzed using custom algorithms created in Visiopharm software. Visiopharm IHC marker applications with threshold parameters were applied uniformly to identify CD3 and CD8 chromogen-positive area in viable regions of tumor sections.
Statistics
Statistical analyses were performed as previously described.Citation48 Unpaired two-tailed t-tests and log-rank tests of Kaplan-Meier survival curves were performed in GraphPad Prism software version 7.04. Analysis of variance (ANOVA) was used in most in vivo tumor growth studies on log transformed tumor volume. Where indicated, two-sided nonparametric Wilcoxon rank-sum tests were performed when some animals in the study had unmeasurable tumors (tumor volume = 0 mm3). No multiple comparison adjustment was applied to the P values. ANOVA and Wilcoxon rank-sum tests were performed in R v3.3.3.
Abbreviations
| ATG12 | = | autophagy related 12 |
| ATG5 | = | autophagy related 5 |
| ATG7 | = | autophagy related 7 |
| CRISPR | = | clustered regularly interspaced short palindromic repeats |
| CTLA-4 | = | cytotoxic T lymphocyte protein 4 |
| CTRL | = | control |
| FDR | = | false discovery rate |
| GZMA | = | granzyme A |
| IHC | = | immunohistochemistry |
| KO | = | knockout |
| MAP1LC3B/LC3 | = | microtubule associated protein 1 light chain 3 beta |
| NSG | = | NOD scid gamma |
| PD-1 | = | programmed cell death 1 |
| PD-L1 | = | programmed cell death ligand 1 |
| PE | = | phosphatidylethanolamine |
| PRF1 | = | perforin |
| SQSTM/p62 | = | sequestosome 1 |
Disclosure of potential conflicts of interest
All authors were employees of Pfizer at the time the work was performed.
Supplemental Material
Download ()Acknowledgments
We thank the Zhang laboratory from the Broad Institute and McGovern Institute of Brain Research at the Massachusetts Institute of Technology for providing CRISPR reagents, Alan Opsahl for digital image analysis, and Vlad Buklan and Roger Conant for harvesting mouse tissue.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol. 2005;169(3):425–10. doi:10.1083/jcb.200412022.
- Mizushima N, Ohsumi Y, Yoshimori T. Autophagosome formation in mammalian cells. Cell Struct Funct. 2002;27(6):421–429. doi:10.1247/csf.27.421.
- Mortimore GE, Poso AR. Intracellular protein catabolism and its control during nutrient deprivation and supply. Annu Rev Nutr. 1987;7(1):539–564. doi:10.1146/annurev.nu.07.070187.002543.
- Levy J, Cacheux W, Bara MA, L’Hermitte A, Lepage P, Fraudeau M, Trentesaux C, Lemarchand J, Durand A, Crain A-M, et al. Intestinal inhibition of Atg7 prevents tumour initiation through a microbiome-influenced immune response and suppresses tumour growth. Nat Cell Biol. 2015;17(8):1062–1073. doi:10.1038/ncb3206.
- Rao S, Tortola L, Perlot T, Wirnsberger G, Novatchkova M, Nitsch R, Sykacek P, Frank L, Schramek D, Komnenovic V, et al. A dual role for autophagy in a murine model of lung cancer. Nat Commun. 2014;5:3056. doi:10.1038/ncomms4056.
- Yang A, Rajeshkumar NV, Wang X, Yabuuchi S, Alexander BM, Chu GC, Von Hoff DD, Maitra A, Kimmelman AC. Autophagy is critical for pancreatic tumor growth and progression in tumors with p53 alterations. Cancer Discov. 2014;4(8):905–913. doi:10.1158/2159-8290.CD-14-0362.
- Guo JY, Karsli-Uzunbas G, Mathew R, Aisner SC, Kamphorst JJ, Strohecker AM, Chen G, Price S, Lu W, Teng X. Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev. 2013;27(13):1447–1461. doi:10.1101/gad.219642.113.
- Guo JY, Chen HY, Mathew R, Fan J, Strohecker AM, Karsli-Uzunbas G, Kamphorst JJ, Chen G, Lemons JMS, Karantza V, et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011;25(5):460–470. doi:10.1101/gad.2016311.
- Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, Bause A, Li Y, Stommel J, Dell'Antonio G, et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011;25:717–729. doi:10.1101/gad.2016111.
- Kim MJ, Woo SJ, Yoon CH, Lee J-S, An S, Choi Y-H, Hwang S-G, Yoon G, Lee S-J. Involvement of autophagy in oncogenic K-Ras-induced malignant cell transformation. J Biol Chem. 2011;286(15):12924–12932. doi:10.1074/jbc.M110.138958.
- Karsli-Uzunbas G, Guo JY, Price S, Teng X, Laddha SV, Khor S, Kalaany NY, Jacks T, Chan CS, Rabinowitz JD. Autophagy is required for glucose homeostasis and lung tumor maintenance. Cancer Discov. 2014;4(8):914–927. doi:10.1158/2159-8290.CD-14-0363.
- Xie X, Koh JY, Price S, White E, Mehnert JM. Atg7 overcomes senescence and promotes growth of BrafV600E-driven melanoma. Cancer Discov. 2015;5(4):410–423. doi:10.1158/2159-8290.CD-14-1473.
- Yang A, Herter-Sprie G, Zhang H, Lin EY, Biancur D, Wang X, Deng J, Hai J, Yang S, Wong -K-K, et al. Autophagy sustains pancreatic cancer growth through both cell-autonomous and nonautonomous mechanisms. Cancer Discov. 2018;8(3):276–287. doi:10.1158/2159-8290.CD-17-0952.
- Noman MZ, Janji B, Kaminska B, Van Moer K, Pierson S, Przanowski P, Buart S, Berchem G, Romero P, Mami-Chouaib F, et al. Blocking hypoxia-induced autophagy in tumors restores cytotoxic T-cell activity and promotes regression. Cancer Res. 2011;71(18):5976–5986. doi:10.1158/0008-5472.CAN-11-1094.
- Baginska J, Viry E, Berchem G, Poli A, Noman MZ, van Moer K, Medves S, Zimmer J, Oudin A, Niclou SP, et al. Granzyme B degradation by autophagy decreases tumor cell susceptibility to natural killer-mediated lysis under hypoxia. Proc Natl Acad Sci U S A. 2013;110(43):17450–17455. doi:10.1073/pnas.1304790110.
- Mgrditchian T, Arakelian T, Paggetti J, Noman MZ, Viry E, Moussay E, Van Moer K, Kreis S, Guerin C, Buart S, et al. Targeting autophagy inhibits melanoma growth by enhancing NK cells infiltration in a CCL5-dependent manner. Proc Natl Acad Sci U S A. 2017;114(44):E9271–E9. doi:10.1073/pnas.1703921114.
- Wei H, Wei S, Gan B, Peng X, Zou W, Guan JL. Suppression of autophagy by FIP200 deletion inhibits mammary tumorigenesis. Genes Dev. 2011;25(14):1510–1527. doi:10.1101/gad.2051011.
- Eng CH, Wang Z, Tkach D, Toral-Barza L, Ugwonali S, Liu S, Fitzgerald SL, George E, Frias E, Cochran N, et al. Macroautophagy is dispensable for growth of KRAS mutant tumors and chloroquine efficacy. Proc Natl Acad Sci U S A. 2016;113(1):182–187. doi:10.1073/pnas.1515617113.
- Starobinets H, Ye J, Broz M, Barry K, Goldsmith J, Marsh T, Rostker F, Krummel M, Debnath J. Antitumor adaptive immunity remains intact following inhibition of autophagy and antimalarial treatment. J Clin Invest. 2016;126(12):4417–4429. doi:10.1172/JCI85705.
- Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402(6762):672–676. doi:10.1038/45257.
- Takamura A, Komatsu M, Hara T, Sakamoto A, Kishi C, Waguri S, Eishi Y, Hino O, Tanaka K, Mizushima N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011;25(8):795–800. doi:10.1101/gad.2016211.
- White E. The role for autophagy in cancer. J Clin Invest. 2015;125(1):42–46. doi:10.1172/JCI73941.
- Maio M, Grob JJ, Aamdal S, Bondarenko I, Robert C, Thomas L, Garbe C, Chiarion-Sileni V, Testori A, Chen -T-T, et al. Five-year survival rates for treatment-naive patients with advanced melanoma who received ipilimumab plus dacarbazine in a phase III trial. J Clin Oncol. 2015;33(10):1191–1196. doi:10.1200/JCO.2014.56.6018.
- Brahmer JR, Tykodi SS, Chow LQ, Hwu W-J, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366(26):2455–2465. doi:10.1056/NEJMoa1200694.
- Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443–2454. doi:10.1056/NEJMoa1200690.
- Mosely SI, Prime JE, Sainson RC, Koopmann J-O, Wang DYQ, Greenawalt DM, Ahdesmaki MJ, Leyland R, Mullins S, Pacelli L, et al. Rational selection of syngeneic preclinical tumor models for immunotherapeutic drug discovery. Cancer Immunol Res. 2017;5(1):29–41. doi:10.1158/2326-6066.CIR-16-0114.
- Yu JW, Bhattacharya S, Yanamandra N, Kilian D, Shi H, Yadavilli S, Katlinskaya Y, Kaczynski H, Conner M, Benson W, et al. Tumor-immune profiling of murine syngeneic tumor models as a framework to guide mechanistic studies and predict therapy response in distinct tumor microenvironments. PLoS One. 2018;13(11):e0206223. doi:10.1371/journal.pone.0206223.
- Zhong W, Myers JS, Wang F, Wang K, Lucas J, Rosfjord E, Lucas J, Hooper AT, Yang S, Lemon LA, et al. Comparison of the molecular and cellular phenotypes of common mouse syngeneic models with human tumors. BMC Genomics. 2020;21(1):2. doi:10.1186/s12864-019-6344-3.
- Johansen T, Lamark T, Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N. Selective autophagy mediated by autophagic adapter proteins. Autophagy. 2011;7(3):279–296. doi:10.4161/auto.7.3.14487.
- Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, Mukherjee C, Shi Y, Gélinas C, Fan Y, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell. 2006;10(1):51–64. doi:10.1016/j.ccr.2006.06.001.
- Psychogios N, Hau DD, Peng J, Guo AC, Mandal R, Bouatra S, Sinelnikov I, Krishnamurthy R, Eisner R, Gautam B, et al. The human serum metabolome. PLoS One. 2011;6(2):e16957. doi:10.1371/journal.pone.0016957.
- Cantor JR, Abu-Remaileh M, Kanarek N, Freinkman E, Gao X, Louissaint A, Lewis CA, Sabatini DM. Physiologic medium rewires cellular metabolism and reveals uric acid as an endogenous inhibitor of UMP synthase. Cell. 2017;169(2):258–72 e17. doi:10.1016/j.cell.2017.03.023.
- Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. 2015;160(1–2):48–61. doi:10.1016/j.cell.2014.12.033.
- Selby MJ, Engelhardt JJ, Johnston RJ, Lu L-S, Han M, Thudium K, Yao D, Quigley M, Valle J, Wang C. Preclinical development of ipilimumab and nivolumab combination immunotherapy: mouse tumor models, in vitro functional studies, and cynomolgus macaque toxicology. PLoS One. 2016;11(9):e0161779. doi:10.1371/journal.pone.0161779.
- Borst J, Ahrends T, Babala N, Melief CJM, Kastenmuller W. CD4+ T cell help in cancer immunology and immunotherapy. Nat Rev Immunol. 2018;18(10):635–647. doi:10.1038/s41577-018-0044-0.
- O’Konek JJ, Ambrosino E, Bloom AC, Pasquet L, Massilamany C, Xia Z, Terabe M, Berzofsky J. Differential Regulation of T-cell mediated anti-tumor memory and cross-protection against the same tumor in lungs versus skin. Oncoimmunology. 2018;7:e1439305. doi:10.1080/2162402X.2018.1439305.
- Noman MZ, Parpal S, Van Moer K, Xiao M, Yu Y, Viklund J, De Milito A, Hasmim M, Andersson M, Amaravadi RK, et al. Inhibition of Vps34 reprograms cold into hot inflamed tumors and improves anti-PD-1/PD-L1 immunotherapy. Sci Adv. 2020;6(18):eaax7881. doi:10.1126/sciadv.aax7881.
- Yamamoto K, Venida A, Yano J, Biancur DE, Kakiuchi M, Gupta S, Sohn ASW, Mukhopadhyay S, Lin EY, Parker SJ. Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature. 2020;581(7806):100–105. doi:10.1038/s41586-020-2229-5.
- Yang S, Imamura Y, Jenkins R, Canadas I, Kitajima S, Aref A, Brannon A, Oki E, Castoreno A, Zhu Z. Autophagy inhibition dysregulates TBK1 signaling and promotes pancreatic inflammation. Cancer Immunol Res. 2016;4(6):520–530. doi:10.1158/2326-6066.CIR-15-0235.
- Galluzzi L, Green DR. Autophagy-independent functions of the autophagy machinery. Cell. 2019;177(7):1682–1699. doi:10.1016/j.cell.2019.05.026.
- Amaravadi RK, Kimmelman AC, Debnath J. Targeting autophagy in cancer: recent advances and future directions. Cancer Discov. 2019;9(9):1167–1181. doi:10.1158/2159-8290.CD-19-0292.
- Pua HH, Dzhagalov I, Chuck M, Mizushima N, He YW. A critical role for the autophagy gene Atg5 in T cell survival and proliferation. J Exp Med. 2007;204(1):25–31. doi:10.1084/jem.20061303.
- Munz C. Autophagy beyond intracellular MHC class II antigen presentation. Trends Immunol. 2016;37:755–763. doi:10.1016/j.it.2016.08.017.
- DeVorkin L, Pavey N, Carleton G. Autophagy regulation of metabolism is required for CD8(+) T cell anti-tumor immunity. Cell Rep. 2019;27:502–13 e5.
- Sousa CM, Biancur DE, Wang X, Halbrook CJ, Sherman MH, Zhang Li, Kremer D, Hwang RF, Witkiewicz AK, Ying H, et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature. 2016;536(7617):479–483. doi:10.1038/nature19084.
- Poillet-Perez L, Xie X, Zhan L, Yang Y, Sharp DW, Hu ZS, Su X, Maganti A, Jiang C, Lu W. Autophagy maintains tumour growth through circulating arginine. Nature. 2018;563(7732):569–573. doi:10.1038/s41586-018-0697-7.
- Vichai V, Kirtikara K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat Protoc. 2006;1(3):1112–1116. doi:10.1038/nprot.2006.179.
- Arensman MD, Yang XS, Leahy DM, Toral-Barza L, Mileski M, Rosfjord EC, Wang F, Deng S, Myers JS, Abraham RT, et al. Cystine-glutamate antiporter xCT deficiency suppresses tumor growth while preserving antitumor immunity. Proc Natl Acad Sci U S A. 2019;116(19):9533–9542. doi:10.1073/pnas.1814932116.
- Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–2120. doi:10.1093/bioinformatics/btu170.
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi:10.1038/nmeth.1923.
- Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011;12(1):323. doi:10.1186/1471-2105-12-323.
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. doi:10.1186/s13059-014-0550-8.