
ABSTRACT
Vaccinia viruses (VACV) are a novel class of immune-oncolytic therapeutics and their mechanism of action is based both on their capacity to replicate selectively in cancer cells and to elicit danger signals that can boost anti-tumor immunity. We recently reported that the intratumor expression of MLKL, a necroptosis inducing factor, generates a protective anti-tumor immunity. Here, we combined both approaches to test the use of VACV to deliver MLKL into the tumor. We generated VACV vectors expressing MLKL and evaluated the effects of MLKL on antitumor efficacy. In vitro infection of cancer cells with MLKL-expressing vectors led to cell death with necroptotic hallmarks. In syngeneic mouse tumor models, VACV expressing MLKL induced an outstanding antitumor activity, which was associated with a robust immunity directed against neo-epitopes. In conclusion, delivery of MLKL by VACV vectors boosts the intrinsic anti-tumor properties of these viral vectors by promoting in situ immunogenic cell death of infected cancer cells.
Introduction
The principle that it is possible to fight cancer by activating a patient’s own immune system is a very appealing paradigm. In the last decade, cancer immunotherapies have emerged as new powerful weapons in the anticancer arsenal.Citation1 However, many patients do not respond to these therapiesCitation2 and combinations with novel immunotherapies are being evaluated for achieving clinical responses in resistant tumors.
In the last decade, new insights into the working mechanisms of some conventional anticancer treatments showed that cancer cells can die in an immunogenic way.Citation3 Consequently, the capacity of anticancer therapeutics to induce immunogenic cell death (ICD) has emerged as a critical factor in eliciting antitumor responses.Citation4 Necroptosis is a genetically regulated form of necrotic cell death with ICD features.Citation5 The necroptotic pathway culminates with phosphorylation of mixed lineage kinase domain-like (MLKL) protein, which is subsequently recruited to the plasma membrane, which becomes destabilized and permeable.Citation6 Recently, we demonstrated that intratumoral delivery of mRNA coding for MLKL induces potent antitumor immunity and antitumor activity in mouse tumor models.Citation7
Oncolytic vaccinia viruses (VACV) showed initial promise for the treatment of tumors and their capacity to act as immunotherapeutic agents demonstrated as key in clinical trials.Citation8 Replication of VACV in tumors serves as a potent danger signal and results in lysis of tumor cells and release of cancer antigens.Citation9 Combined, such mechanisms overcome the immune-suppressive tumor environment and can result in T cell responses targeting tumor antigens. However, candidate viruses tested to date demonstrated a suboptimal capacity to establish antitumor immunity in patients.Citation10 In this work, we aimed to improve oncolytic VACV treatments by combining these vectors with the delivery of the necroptosis executioner MLKL. We demonstrated that intratumoral administration of VACV expressing MLKL activates potent antitumor T cell responses directed against tumor neo-antigens, and that such immunity mediates an outstanding improvement in antitumor efficacy.
Results and discussion
MLKL expression controlled by the P7.5late VACV promoter is compatible with unimpaired viral life cycle
In order to activate necroptosis-like tumor cell death after infection with oncolytic viruses, we generated a set of VACV vectors expressing mouse MLKL. As MLKL induces cell death, we hypothesized that MLKL expression at late stages of the viral life cycle would be necessary to allow for generation of progeny virus. Therefore, we evaluated the strong late VACV promoter P1111 and the moderate-strength late VACV promoter P7.5lateCitation11,Citation12 for the control of MLKL transcription. In addition, in order to test the influence of virus replication on antitumor activity, both replicating and non-replicating MLKL-expressing VACV vectors were constructed: the replication-competent recombinant VACV was based on VACV strain Western Reserve with inactivated viral thymidine kinase (WR/TK-),Citation13 and the non-replicating vector virus was based on the highly attenuated vaccine strain Modified Vaccinia virus Ankara (MVA).Citation14
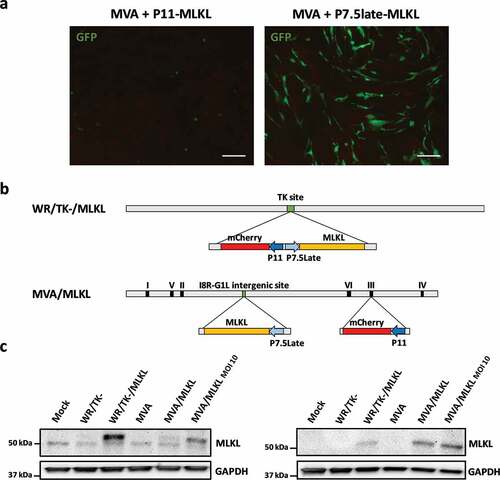
We observed that cells infected with VACV (WR/TK- or MVA) and transfected at the same time with the P11-MLKL DNA died at early time points and no foci of VACV replication could be detected. This toxic effect of MLKL can be observed in (left panel), where CEF (Chicken Embryo Fibroblasts) cells infected with MVA and transfected with the P11-MLKL plasmid expressed low levels of GFP (encoded by the P11-MLKL plasmid) and rapidly lost their fibroblast-like morphology to a typical dead cell rounded morphology. In contrast, this effect was milder when transfecting the P7.5late-MLKL construct (, right panel); such transfection readily allowed for the formation of recombinant viruses and the MLKL-expressing VACVs WR/TK-/MLKL and MVA/MLKL could be clonally isolated and amplified. depicts a schematic diagram of such vectors. Expression of mouse MLKL from the novel viruses was evaluated by Western-blot assay in non-murine cell lines to avoid interference of endogenous MLKL (). Although MLKL-expression cassettes are inserted in different loci within VACV genome () and this can affect MLKL expression, similar amounts of MLKL expression were detected in CEF cells. This suggests that the lower amounts of MLKL produced following infection of HeLa cells with MVA/MLKL is likely the result of the abortive infection of MVA in these cells.
Figure 1. Construction of VACVs expressing MLKL. (a) Fluorescent images of CEF cells 24 hours after infection with Vaccinia virus (VACV) MVA strain and transfection with a plasmid codifying for mouse MLKL under the control of P7.5Late or P11 promoters. Cells were infected with a MOI of 5 to allow 100% infection. GFP is codified by the transfected plasmid and is used to observe toxicity due to overexpression of MLKL by the P11 promoter (scale bar = 100 μm). (b) Schematic diagram of WR/TK-/MLKL and MVA/MLKL recombinant viruses. (c) Confirmation of mouse MLKL expression. HeLa and CEF cells were infected with a MOI of 5 and, 24 hours post-infection, a Western-blot analysis was performed. For the non-replicating MVA/MLKL vector infections were increased to MOI 10 in order to readily detect MLKL expression in HeLa cells.
MLKL-expressing VACV vectors led to cell death with necroptotic hallmarks
We next compared the outcome of parental and MLKL-expressing VACV on cancer cells. Expression of MLKL by the late VACV promoter P7.5late did not interfere with the capacity of VACV to replicate in cancer cell lines () or with the plaque size phenotype (). Very importantly, VACV expressing MLKL demonstrated a superior capacity to kill tumor cells compared with their parental counterpart (-d).
Figure 2. VACV vectors expressing MLKL activate necroptosis-related phenotype in cancer cells. (a) Replication capacity in a panel of cancer cells. Different tumor cell lines were infected with viruses at a MOI of 1 and virus production was measured by plaque-assay 48 hours post-infection. Means +SD are plotted. (b) Plaque size in MA104 cells. MA104 cells were infected at a MOI of 0.05 and, 3 days after infection, the diameter of plaques was measured after dying with crystal violet. MVA vectors were not included in this experiment due to their inability to form plaques. The size of 12 representative plaques and mean ±SD are depicted. (c-d) Cytotoxicity in mouse tumor cell lines. Cells were infected with a MOI of 1 and, 3 days (c) or at different time points after infection (d), % of cells killed was evaluated. (e) Detection of necroptosis-related HMGB1 release. HMGB1 concentration detected by ELISA assay on supernatants of CT26 cells infected with a MOI of 5 with indicated viruses. (f) Images of CT26 cells 24 hours after infection with indicated viruses (MOI of 5). Necroptosis-related swelling of WR/TK-/MLKL-infected cells could be observed (black arrows, scale bar = 25 μm). *, p < .05 vs non-MLKL expressing counterpart.
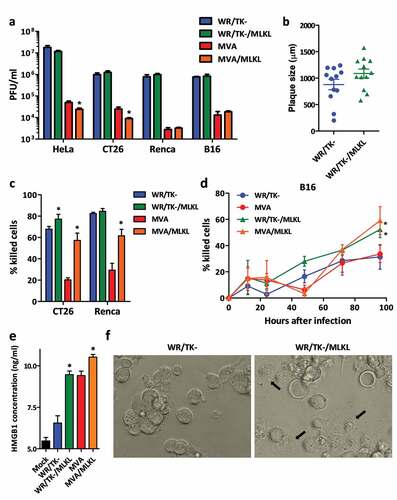
Next, we tested whether infection of cancer cells with MLKL-expressing VACV results in necroptotic hallmarks. Higher levels of HMGB1 protein were detected in the supernatant of cells infected with MLKL-expressing VACV vectors (). In addition, cancer cells infected with VACV expressing MLKL resulted in a visually different cytopathic effect (, black arrows), with increased cell swelling (bigger cells with a different phase contrast under the microscope). Both the release of intracellular HMGB1 and swelling of cells are associated with necroptosis activation.Citation7,Citation15
MLKL-expressing VACV activates systemic antitumor immune responses and improves antitumor activity
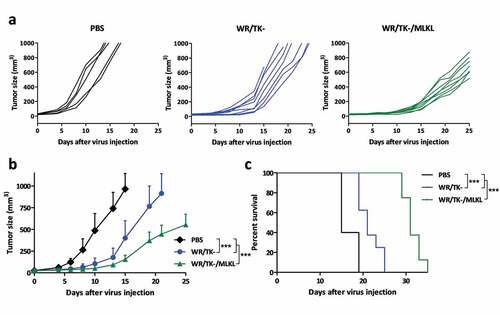
We eventually evaluated the impact on tumor growth of intratumoral injection of MLKL-expressing VACV in syngeneic B16 melanoma tumor model. WR/TK- expressing mouse MLKL could significantly delay tumor growth (-b) and highly increased the median survival time of mice () compared to the control virus.
Figure 3. Intratumoral administration of MLKL-expressing vectors protects against primary tumor growth. C57BL/6 mice harboring subcutaneous B16 tumors were randomized and injected twice (days 0 and 4) with an intratumoral dose of 1 × 107 plaque-forming units (pfu) of indicated viruses. Injection of PBS was used as a control. Tumor volume of individual animals (a), mean of treatments (b), and overall survival (c) are plotted for 5–8 mice/group +SD. *, p < .05; **, p < .01; ***, p < .001.
Moreover, we evaluated if the delivery of VACV expressing MLKL into a primary tumor could result in a systemic anti-tumor response. Therefore, we inoculated B16 cells in the left hand flank of mice and, 3 days later, in the right flank. After tumors were established in both sides, only the tumor in the left flank was treated with the indicated viruses and growth of the treated and untreated tumors were monitored. Same way as in , VACV-mediated expression of MLKL, either via MVA or WR/TK-, greatly reduced the volume of directly injected tumors compared to their control counterpart (MVA or WR/TK-)(). Importantly, a pronounced delay in the growth of the untreated distal tumor was observed in mice treated with VACV expressing MLKL, whereas treatment with the control MVA and WR/TK- strain did not affect the untreated tumor growth (-b). In addition, mice treated with the replicative WR/TK-/MLKL were better protected against growth of a distal tumor than mice treated with MVA/MLKL, and overall survival was also significantly increased when treated with the replication-competent strain (). This indicates that replication of the vector does play an important role in the antitumor activity of oncolytic viruses. This finding is somewhat in contradiction with a recent report showing that intratumoral inoculation of inactivated replication-deficient VACV MVA is sufficient for achieving an optimal antitumor activity.Citation16
Figure 4. VACV-mediated delivery of the necroptosis inducer MLKL activates antitumor immunity. (a-b) Intratumoral administration of MLKL-expressing vectors protects against distal tumors. C57BL/6 mice harboring a primary and a secondary (on the counterflank) B16 tumors were injected twice (days 0 and 4) with an intratumoral dose into the primary tumor of 1 × 107 plaque-forming units (pfu) of indicated viruses. Tumor growth of the untreated secondary tumor of individual animals (a) and mean of treatments (b), tumor growth of the directly injected primary tumor (c), and overall survival (d) are plotted +SD for 5–8 mice/group. (e) Intratumoral administration of MLKL-expressing vectors induces antitumor T cell responses directed against tumor neo-antigens. Mice harboring B16 tumors were treated twice (days 0 and 4) with an intratumoral dose of indicated viruses and, at day 8 after virus-administration, splenocytes were analyzed for their reactivity to indicated peptides by IFN-γ ELISPOT. Individual values of 5 mice/group and mean ±SD are plotted. *, p < .05; **, p < .01; ***, p < .001.
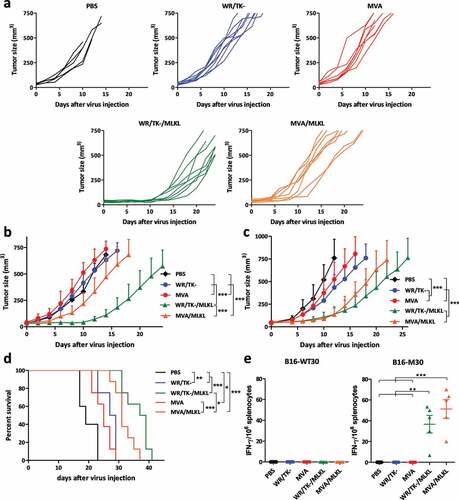
Importantly, the B16 tumor model allows evaluation of neo-epitope specific T cell responses. By ELISPOT, we determined the T cell response directed against the major histocompatibility complex (MHC) class II-restricted neo-epitope B16-M30 or its wild-type counterpart B16-WT30.Citation17 VACV expressing MLKL performed better than their control counterparts, showing pronounced reactivity against the neo-epitope but not responding against the wild type peptide ().
These results reinforce the idea that oncolytic vectors currently in use need to be further improved in order to elicit robust antitumor immunity. Different oncolytic vectors show certain levels of immunogenic cell death (ICD) activation, but most of them activate forms of ICD that are different from necroptosis; For example, Newcastle disease virus and Adenoviruses predominantly activate autophagy,Citation18,Citation19 and Semliki Forest virus activates immunogenic apoptosis.Citation20 VACV has previously been shown to activate events of necroptosis in certain human cell lines, but such activation does not result in subsequent activation of tumor-associated antigen-specific T-cells.Citation20 In this work, control VACV vectors elicited a poor antitumor immunity (), confirming this hindrance. However, our results also highlight that modifying such vectors to efficiently activate necroptosis is feasible. For such activation, we previously demonstrated the advantages of delivering the necroptosis mediator MLKL7; such delivery was via a mRNA and here we demonstrated that such delivery is also efficient via a viral vector. In the same way as with mRNA delivery, intratumoral injection of VACV vectors expressing MLKL demonstrated to successfully induce neo-epitope-specific T cell responses, which are able to control the growth of primary and distal tumors. Further combinations with immunotherapies are under study; both oncolytic VACVCitation21 and delivery of mRNA coding for MLKL7 have demonstrated to have synergistic anti-tumor effects with immune check point blockades, and combination with MLKL-expressing VACV is likely to result in outstanding antitumor efficacies.
In summary, delivery of MLKL into the tumor through a viral vector is feasible and results in improved anti-tumor responses. Further development of such agents and their combination with other immunotherapies are an interesting prospect for developing a therapy able to induce responses in wide range of patients.
Materials and methods
Mouse models
C57BL/6 mice were injected subcutaneously with 5 × 105 B16 cells into the left flank. At day 6–10 after implantation, indicated viruses were injected intratumorally at a dose of 1 × 107 pfu/mice. In a different experimental set-up, a second tumor was implanted on the right flank of mice at day 3 after primary implantation. Tumor sizes were measured every 2 days and tumor volume was calculated as the length x width x height (in mm3). Mice were euthanized when tumor volume reached 1000 mm3.
IFN-γ ELISPOT
For ELISPOT assay, splenocytes were prepared from C57BL/6 mice bearing a B16 tumor and treated as indicated, and 2.5 × 105 cells were cultured for 24 hours in anti-IFN-γ (Diaclone) pre-coated 96-well plates in the presence of 10 μg/ml of peptide. The synthetic peptides used for restimulation were: B16-M30 mut (PSKPSFQEFVDWENVSPELNSTDQPFL) and B16-M30 WT (PSKPSFQEFVDWEKVSPELNSTDQPFL) originated from gene Kif18b.Citation17
Cells and viruses
MA104, B16, CT26, HeLa, and Renca cell lines were purchased from the American Type Culture Collection (ATCC). Primary chicken embryo fibroblasts (CEF) were prepared from 10-day-old chicken embryos (SPF eggs, VALO). All cell lines were maintained in recommended culture media containing 5–10% fetal bovine serum and antibiotics at 37°C, 5% CO2.
All vector viruses used in this work (WR/TK-, WR/TK-/MLKL, MVA, MVA/MLKL) express the red fluorescent marker gene mCherry using the VACV-specific late promoter P11. Replication-competent VACV (WR/TK- and WR/TK-/MLKL) are based on the VACV strain Western Reserve (WR) with a truncated viral thymidine kinase (TK) gene. For the construction of WR/TK-/MLKL, heterologous gene sequences encoding the murine MLKL protein were inserted by guided homologous recombination into the TK gene locus to be expressed under transcriptional con trol of the VACV-specific modified late promoter P7.5Late (). For generating MVA/MLKL, the P7.5Late-MLKL expression cassette was introduced by homologous recombination into the intergenic site between the viral genes MVA069 R and MVA070 L.Citation22 Viruses were purified as previously describedCitation13 and titrated by plaque assay in CEF cells which allow for productive replication of MVA recombinant viruses.
Protein analysis
For evaluating MLKL expression, HeLa or CEF cells were infected at an MOI of 5 (PFU/cell) and, 24 hours after infection, whole-cell protein extracts were obtained. Samples were separated by 10% SDS-PAGE gel, transferred to a nitrocellulose membrane, and mouse MLKL protein was detected using a monoclonal anti-MLKL primary antibody (Rabbit, Cell Signaling) and a polyclonal anti-Rabbit conjugated with HRP (Goat, Cell Signaling). A monoclonal anti-GADPH antibody (Rabbit, Cell Signaling) was used for the loading control.
Enzyme-linked immunosorbent assay
A HMGB1 ELISA kit (Tecan) was used to determine HMGB1 released after 24 hours of infection with indicated viruses (MOI of 5).
Virus growth and cytotoxicity assays
2x105 cells were seeded in 24-well plates and infected at an MOI of 1 (PFU/cell) with indicated viruses. Four hours after infection, cultures were washed twice with PBS and incubated in fresh virus-free medium. Forty-eight h after infection cultures were harvested, frozen-thawed thrice, and viral titers were determined by plaque assay in CEF cells.
Cytotoxicity assays were performed by seeding 5 × 104 cells per well in 96-well plates and infecting at an MOI of 1; at indicated time points, a nonradioactive cell proliferation assay kit (Promega) was used to stain the cultures and determine percentage of cells killed.
Statistical analyses
Standard Student’s t test (two-tailed) was used for analyzing results in . For comparison of antitumor efficacies in , , and , a Two-Way ANOVA and Bonferroni posttest was chosen. A One-Way ANOVA and Tukey’s Multiple Comparison test were used in . For comparison of survival curves, a log rank test was used. In all cases, significance was achieved if p < .05.
Declaration of interest statement
LVH and XS are named as inventors on patent application WO2019/048639, which claims IP rights on the use of MLKL for anti-tumor treatment.
Acknowledgments
This work was supported by Deutsche Krebs Hilfe project number 70112543 (JJR and GS), by a VIB Technology Transfer Fund project (XS), and by Spanish Ministry of Science I+D+i project number PID2019-109102RA-I00 (JJR). JJR is funded by the Spanish Ministry of Science through a Ramon y Cajal program (RYC2018-025425-I).
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
References
- Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. 2015 Apr 13;27(4):450–5. doi:10.1016/j.ccell.2015.03.001.
- O’Donnell JS, Long GV, Scolyer RA, Teng MW, Smyth MJ. Resistance to PD1/PDL1 checkpoint inhibition. Cancer Treat Rev. 2017 Jan;52:71–81. doi:10.1016/j.ctrv.2016.11.007.
- Krysko DV, Garg AD, Kaczmarek A, Krysko O, Agostinis P, Vandenabeele P. Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer. 2012 Dec;12(12):860–875. doi:10.1038/nrc3380.
- Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol. 2017 Feb;17(2):97–111. doi:10.1038/nri.2016.107.
- Aaes TL, Kaczmarek A, Delvaeye T, De Craene B, De Koker S, Heyndrickx L, Delrue I, Taminau J, Wiernicki B, De Groote P et al. Vaccination with necroptotic cancer cells induces efficient anti-tumor immunity. Cell Rep. 2016 Apr 12;15(2):274–287. doi:10.1016/j.celrep.2016.03.037.
- Dondelinger Y, Declercq W, Montessuit S, Roelandt R, Goncalves A, Bruggeman I, Hulpiau P, Weber K, Sehon C, Marquis R et al. MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep. 2014 May 22;7(4):971–981. doi:10.1016/j.celrep.2014.04.026.
- Van Hoecke L, Van Lint S, Roose K, Van Parys A, Vandenabeele P, Grooten J, Tavernier J, De Koker S, Saelens X. Treatment with mRNA coding for the necroptosis mediator MLKL induces antitumor immunity directed against neo-epitopes. Nat Commun. 2018 Aug 24;9(1):3417. doi:10.1038/s41467-018-05979-8.
- Moehler M, Heo J, Lee HC, Tak WY, Chao Y, Paik SW, Yim HJ, Byun KS, Baron A, Ungerechts G, et al. Vaccinia-based oncolytic immunotherapy pexastimogene devacirepvec in patients with advanced hepatocellular carcinoma after sorafenib failure: a randomized multicenter phase IIb trial (TRAVERSE). Oncoimmunology. 2019;8(8):1615817. doi:10.1080/2162402X.2019.1615817.
- Thorne SH. Immunotherapeutic potential of oncolytic vaccinia virus. Front Oncol. 2014;4:155. doi:10.3389/fonc.2014.00155.
- Harrington K, Freeman DJ, Kelly B, Harper J, Soria JC. Optimizing oncolytic virotherapy in cancer treatment. Nat Rev Drug Discov. 2019 Sep;18(9):689–706. doi:10.1038/s41573-019-0029-0.
- Chakrabarti S, Brechling K, Moss B. Vaccinia virus expression vector: coexpression of beta-galactosidase provides visual screening of recombinant virus plaques. Mol Cell Biol. 1985 Dec;5(12):3403–3409. doi:10.1128/MCB.5.12.3403.
- Masternak K, Wittek R. cis- and trans-acting elements involved in reactivation of vaccinia virus early transcription. J Virol. 1996 Dec;70(12):8737–8746. doi:10.1128/JVI.70.12.8737-8746.1996.
- Rojas JJ, Sampath P, Bonilla B, Ashley A, Hou W, Byrd D, Thorne S. Manipulating TLR signaling increases the anti-tumor T cell response induced by viral cancer therapies. Cell Rep. 2016 Apr 12;15(2):264–273. doi:10.1016/j.celrep.2016.03.017.
- Sutter G, Moss B. Nonreplicating vaccinia vector efficiently expresses recombinant genes. Proc Natl Acad Sci U S A. 1992 Nov 15;89(22):10847–10851. doi:10.1073/pnas.89.22.10847.
- Zhu K, Liang W, Ma Z, Xu D, Cao S, Lu X, Liu N, Shan B, Qian L, Yuan J et al. Necroptosis promotes cell-autonomous activation of proinflammatory cytokine gene expression. Cell Death Dis. 2018 May 1;9(5):500. doi:10.1038/s41419-018-0524-y.
- Dai P, Wang W, Yang N, Serna-Tamayo C, Ricca JM, Zamarin D, Shuman S, Merghoub T, Wolchok JD, Deng L et al. Intratumoral delivery of inactivated modified vaccinia virus Ankara (iMVA) induces systemic antitumor immunity via STING and Batf3-dependent dendritic cells. Sci Immunol. 2017 May 19;2(11):eaal1713. doi:10.1126/sciimmunol.aal1713.
- Kreiter S, Vormehr M, van de Roemer N, Diken M, Löwer M, Diekmann J, Boegel S, Schrörs B, Vascotto F, Castle JC et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature. 2015 Apr 30;520(7549):692–696. doi:10.1038/nature14426.
- Tazawa H, Kuroda S, Hasei J, Kagawa S, Fujiwara T. Impact of autophagy in oncolytic adenoviral therapy for cancer. Int J Mol Sci. 2017 Jul 10;18(7):1479. doi:10.3390/ijms18071479.
- Ye T, Jiang K, Wei L, Barr MP, Xu Q, Zhang G, Ding C, Meng S, Piao H. Oncolytic newcastle disease virus induces autophagy-dependent immunogenic cell death in lung cancer cells. Am J Cancer Res. 2018;8:1514–1527.
- Ma J, Ramachandran M, Jin C, Quijano-Rubio C, Martikainen M, Yu D, Essand M. Characterization of virus-mediated immunogenic cancer cell death and the consequences for oncolytic virus-based immunotherapy of cancer. Cell Death Dis. 2020 Jan 22;11(1):48. doi:10.1038/s41419-020-2236-3.
- Rojas JJ, Sampath P, Hou W, Thorne SH. Defining effective combinations of immune checkpoint blockade and oncolytic virotherapy. Clin Cancer Res. 2015 Dec 15;21(24):5543–5551. doi:10.1158/1078-0432.CCR-14-2009.
- Wyatt LS, Earl PL, Xiao W, Americo JL, Cotter CA, Vogt J, Moss B. Elucidating and minimizing the loss by recombinant vaccinia virus of human immunodeficiency virus gene expression resulting from spontaneous mutations and positive selection. J Virol. 2009 Jul;83(14):7176–7184. doi:10.1128/JVI.00687-09.