
ABSTRACT
The presence of a tumor can alter host immunity systematically. The immune-tumor interaction in one site may impact the local immune microenvironment in distal tissues through the circulation, and therefore influence the efficacy of immunotherapies to distant metastases. Improved understanding of the immune-tumor interactions during immunotherapy treatment in a metastatic setting may enhance the efficacy of current immunotherapies. Here we investigate the response to αPD-1/αCTLA4 and trimAb (αDR5, α4-1BB, αCD40) of 67NR murine breast tumors grown simultaneously in the mammary fat pad (MFP) and lung, a common site of breast cancer metastasis, and compared to tumors grown in isolation. Lung tumors present in isolation were resistant to both therapies. However, in MFP and lung tumor-bearing mice, the presence of a MFP tumor could increase lung tumor response to immunotherapy and decrease the number of lung metastases, leading to complete eradication of lung tumors in a proportion of mice. The MFP tumor influence on lung metastases was mediated by CD8+ T cells, as CD8+ T cell depletion abolished the difference in lung metastases. Furthermore, mice with concomitant MFP and lung tumors had increased tumor specific, effector CD8+ T cells infiltration in the lungs. Thus, we propose a model where tumors in an immunogenic location can give rise to systemic anti-tumor CD8+ T cell responses that could be utilized to target metastatic tumors. These results highlight the requirement for clinical consideration of cross-talk between primary and metastatic tumors for effective immunotherapy for cancers otherwise resistant to immunotherapy.
Introduction
Tumors often metastasize to multiple sites throughout the body, and cancer metastasis is the leading cause of death among cancer patients.Citation1 Systemic immune responses against tumors have been shown to impact distal tissues and tumors through the lymphatics and vascular systems.Citation2–Citation5 Tumor cells and the surrounding tumor microenvironment (TME) in one metastatic lesion have the potential to influence the TME composition in distant tumors. These changes are mediated by circulating factors including growth factors, tumor-derived extracellular vesicles, and immune cell populations that are propagated by the tumor cells and TME.Citation5–Citation9
Systemic and distant influences of localized tumors are evidenced by studies of the pre-metastatic niche and abscopal effect of local radiation.Citation2,Citation3 Secreted and cellular factors from the primary tumor influence the pre-metastatic tissue to create an environment conducive for tumor establishment.Citation2 The abscopal effect describes a phenomenon where local radiation directed toward one tumor leads to regression of tumors elsewhere in an immune dependent mechanism.Citation3 Therefore, the changes mediated by tumor growth are not limited to the tumor site and can impose widespread changes to both healthy and diseased tissue throughout the host.
A reasonable explanation for these effects is that metastatic tumors in various anatomical sites are not completely separate systems and that communication between tumors could impact therapeutic responses. Studies investigating this hypothesis in murine models are limited, and to date only focus on simultaneous tumor growth in subcutaneous models.Citation10–Citation12 Experimental investigation into this area is especially warranted since there is emerging evidence that certain metastases can reduce primary tumor responses or the overall response of patients to immunotherapy.Citation13–Citation16
The lack of targeted therapies for triple-negative breast cancers (TNBC) have prompted the use of immunotherapy for this subtype, especially for patients who do not respond to first-line chemotherapy. Compared to other breast cancer subtypes, TNBC has a higher degree of tumor-infiltrating lymphocytes (TILs) suggesting a higher degree of immunogenicity.Citation17,Citation18 However, there are lower levels of TILs at metastatic sites compared to the primary breast tumorCitation19 and therefore metastases may be less responsive to immunotherapy.
We previously found that breast tumors grown in the lungs of mice were less responsive to αPD-1/αCTLA4 and an agonist antibody treatment trimAb (αDR5, αCD40, and α4-1BB) compared to tumors grown elsewhere.Citation20 Comparisons between 67NR lung and mammary fat pad (MFP) tumors revealed that these tumors had distinct TMEs and differences in the immune cells required for tumor control. The current study aimed to investigate potential cross-talk between tumors in different locations when present simultaneously to more accurately represent metastatic disease. We investigated 67NR primary MFP tumors and metastatic lung tumors and assessed their influence on each other when present simultaneously in the context of immunotherapy.
Methods
Cell lines and mice
BALB/c mouse breast carcinoma cell line 67NR was provided by Professor Robin Anderson (Olivia Newton John Cancer Center, Victoria, Australia). Tumor cells were cultured at 37°C, 10% CO2 in DMEM media (Gibco, Massachusetts, USA) supplemented with 10% heat-inactivated FCS, 2 mM glutamine, 1 mM sodium pyruvate, 0.1 mM nonessential amino acids, 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 100 U ml−1 penicillin and 100 μg ml−1 streptomycin. All lines tested negative for mycoplasma (Peter MacCallum Cancer Center Genotyping Core Facility).
BALB/c mice were purchased from the Walter and Eliza Hall Institute for Medical Research (Victoria, Australia). Mice were used between 6 and 20 weeks of age and were housed in the Peter MacCallum Cancer Center animal facility under PC2 pathogen-free conditions. All animal work was approved by the Peter MacCallum Cancer Center Animal Experimentation Ethics Committee under the ethics numbers E498 and E582.
Mouse models
For the mammary gland tumor model, cells were injected into the inguinal (fourth) mammary gland with 4 × 105 67NR cells in a cell suspension volume of 20 μl with a Hamilton syringe and 27 gauge needle and insulin syringe. Mice were anaesthetized using controlled isoflurane supplemented with oxygen using an anesthetic machine through a nose cone, and injections were performed under anesthesia. This tumor model was referred to as MFP. Tumor growth was monitored at least twice weekly using digital callipers and end point was when tumors reached a size of 150 mm2.
To produce lung metastases, 4 × 105 67NR tumor cells were injected intravenously (IV) into the tail vein in restrained mice using a cell suspension volume of 200 μl with an insulin syringe and 26 gauge needle. Health of the mice was monitored daily by both researchers and animal technicians and end point was when mice appeared sick, ruffled, and had heavy breathing due to lung tumor burden. Upon end point, mice were euthanized and autopsied to confirm tumor burden and photographed.
For dual tumor models, mice were first injected with tumor cells IV directly followed by injection in the MFP following the protocols outlined above.
Therapeutic and depletion antibodies for in vivo experiments
All therapeutic anti-mouse antibodies were injected intra-peritoneally in 200 μl PBS. Antibody against asialo-GM1 (rabbit polyclonal) was purchased from Wako Pure Chemical Industries, Ltd., Japan. Antibodies against 4–1BB (3H3), CD40 (FGK4.5), DR5 (MD5.1) CTLA4 (9H10), PD-1 (RPM1-14), CD8 (YTS 169.4) or isotype control antibody (2A3) were purchased from BioXcell, New Hampshire, USA. Each dose of trimAb consisted of a mixture of 50 μg αDR5, 25 μg αCD40 and 25 μg α4-1BB. One dose of αPD-1/αCTLA4 consisted 200 μg αPD-1 and 150 μg αCTLA4. Mice were dosed every 3–4 days with a total of 4 doses for trimAb and 2 doses for αPD-1/αCTLA4. All depletion antibodies against CD8 (200 μg per dose) or asialo-GM1 (10 μl per dose as per manufacturer recommendations) were injected at day −1 and 0 (tumor cell injection date) with two repeated doses spaced three to 4 days apart before the experiment end date at 7 days post-tumor injection.
Lung metastases counts
To visualize and count lung metastases, mice were culled and lungs and tracheas were exposed. Using a 10 mL syringe and 23 gauge needle, india ink (15% diluted in dH2O) was carefully injected through the trachea into the lungs until the whole lungs expanded and filled with india ink. Lungs were then removed, washed in PBS 3–5 times before incubating in Fekete’s solution (4.5% glacial acetic acid, 9% formalin, 64% ethanol in dH2O) for a minimum of 12 hours. After incubation in Fekete’s solution, lungs were washed in PBS 3 to 5 times and photographed. Metastases in each lobe were counted under a dissection microscope with the numbers per lobe added together for the total number of metastases per lung. At least one replicate experiment per data set was blinded to eliminate researcher bias.
Nanostring assay and analysis
To isolate RNA, tumors were carefully microdissected to avoid surrounding healthy tissue and snap frozen in liquid nitrogen. Samples were disrupted and homogenized in 600 μl RTL buffer (Qiagen, Netherlands) per 30 mg of tumor tissue with two 2 mm sterile metal beads using the Fast Prep-24 Sample Preparation system (M.P. Biomedicals, California, USA). RNA was extracted using RNeasy mini kit (Qiagen, Netherlands) as per manufacturers’ instruction. A nanostring assay was performed using the nCounter Mouse Pan Cancer Immune profiling 770-plex panel. Briefly, 100 ng of RNA was hybridized and run on each panel using the nCounter FLEX Analysis System as per the manufacturer’s instructions by the Molecular Genomics Facility at the Peter MacCallum Cancer Center.
For analysis, downstream normalization and gene quantification were performed using nSolver Analysis Software v4.0. Raw log2 normalized data was further analyzed using for data visualization and principle component analysis using Perseus computational platform (version 1.5.6) from the Max Planck Institute of Biochemistry.Citation21
Flow cytometry
Tumor tissues were harvested, stained, and analyzed as previously described.Citation20 The following antibodies were used: CD3 BV605 (Biolegend, 17A2), CD4 BUV805 (BD, GK1.5), CD8 BUV737 (BD, 53–6.7), CD11b BV711 (Biolegend, M1/70), CD11c BV785 (Biolegend, N418), CD19 BV785 (BD, 6D5), CD27 APC (BD, LG.7F9), CD44 FITC (BD, 1M7), CD45.2 APC Cy7 (eBioscience, 104), CD49b FITC (BD, DX5), CD62L BV785 (Biolegend, MEL-14), CD69 PE Cy7 (Biolegend, H1.2F3), CD103 PE (Biolegend, 2E7), CD206 FITC (Biolegend, C068C2), CTLA4 PE (BD, UC10-4F10-11), FoxP3 e450 (eBioscience, FJK16S), F4/80 BV421 (BD, T45-2342), IFNγ APC (Biolegend, XMG1.2), Ly6C PE Cy7 (Biolegend, HK1.4), Ly6G BV605 (Biolegend, 1A8), MHCII APC (eBioscience, M5/144.15.2), PD-1 BUV395 (BD, J43), TNFα Pacific Blue (Biolegend, MP6-XT22) and the viability dye Fixable Yellow (Invitrogen).
Ex vivo T cell stimulation assay
Tumors were processed to single-cell suspension using the same protocol for flow cytometry experiments. One-tenth of the single-cell suspension was stimulated with 10−6 M gp70 peptide, 1 × 105 67NR cells or no stimulation control as indicated in the presence of 1:1000 dilution of GolgiPlug and 1:1500 dilution of GolgiStop (BD, New Jersey, USA) for 5 hours. The Murine Leukaemia Virus gp70 (423–431) peptide was synthesized by Mimotopes at >95% Purity (Melbourne, Australia). Before staining with CD3, CD8, IFNγ, and TNFα antibodies cells were fixed and permeabilized using the eBioscience kit (California, USA) as per manufacturer’s instructions. Samples were run on a Symphony cytometer (BD, New Jersey, USA) and analyzed using FlowJo software V10.
Statistical analysis
Statistical analyses in this study for all experiments (except NanoString analyses) were performed using GraphPad Prism (version 7). Details of replicates and exact statistical test are stated in the figure legends. Data is displayed as the mean ± standard error of the mean (SEM), unless otherwise stated.
Results
The presence of a MFP tumor improves response of lung tumors to immunotherapy
In the current study, we developed a model with responsive and resistant tumors present simultaneously to investigate if there was cross-talk between tumors that could impact on the overall response to immunotherapy. We utilized the BALB/c syngeneic 67NR breast cancer cell line, which is a derivative of the 4T1 TNBC line that does not spontaneously metastasize.Citation22 This allows for precise control of tumor location, thereby eliminating metastases to non-target locations. We previously demonstrated disparate TMEs and immunotherapy responses in 67NR MFP and lung tumors.Citation20 Therefore, use of this model was ideal to investigate whether cross-talk between primary and metastatic tumors can influence immunotherapy responses.
BALB/c mice were injected with 4 × 105 67NR cells in the MFP in one site (MFP), in contralateral MFPs (MFP + MFP), the MFP, and intravenously (IV) to produce lung metastases (MFP + Lung) and IV alone (Lung) (). For dual tumor models, 4 × 105 67NR cells were injected in each site. Mice with dual MFP tumors were used as a control for the increased tumor burden in mice with concomitant MFP and lung tumors. Approximately 10 days after tumor injection (when MFP tumors were 30–50 mm2), mice were treated with αPD-1/αCTLA4 or trimAb and response to therapy was monitored by MFP tumor growth and development of respiratory stress for lung tumors.
We observed similar patterns of MFP tumor growth regardless of the presence of another tumor and this was observed in both treated and non-treated mice (,c). Interestingly, assessment of the survival of mice bearing lung tumors revealed that mice with concomitant MFP tumors treated with αPD-1/αCTLA4 or trimAb had enhanced survival compared to treated mice with lung tumors alone (,e). In fact, the survival of treated MFP + Lung mice was comparable to treated MFP + MFP mice and suggests that the presence of a responding MFP tumor enhances immunotherapy responses of the non-responding metastatic lung tumors.
Figure 1. Presence of MFP tumors reduces lung tumor burden and enhances immunotherapy responses of lung tumor-bearing mice. (a) BALB/c mice were injected with 4 × 105 67NR tumor cells per injection in one MFP, contralateral MFPs (MFP + MFP), MFP and IV (MFP + Lung) and IV only (Lung). When MFP tumors were 30–50 mm2, mice were treated with 2 doses of 200 μg αPD-1 and 150 μg αCTLA4, 4 doses of 50 μg αDR5, 25 μg αCD40 and 25 μg α41BB (trimAb) therapy or 200 μg 2A3 rat IgG2 isotype control antibody. (b,c) Growth of MFP tumors in MFP tumor-bearing mice as described in A treated with either αPD-1/αCTLA4 (b) or trimAb (c). Data represented as mean ± SEM, representative of 2–3 independent experiments of n = 5–8 mice per group. Tukey’s multiple comparison test. (d,e) Survival of mice as described in (a) treated with either αPD-1/αCTLA4 (d) or trimAb (e). After 100 days mice were culled and tumor eradication was confirmed via autopsy. Data representative of 2 independent experiments, n = 5–8 mice per group. Mantel-Cox test. (f,g) Lungs of lung tumor-bearing mice were harvested and metastases enumerated during treatment (f) at 7 days post-treatment or before treatment (g) 10 days post tumor injection. (f) Representative experiment (n = 3 mice per group) (g) Pooled data from 3 independent experiments (n = 4–11 mice per group, per experiment). Data points represent lungs from individual mice. Unpaired t-test. Data (excluding (d) and (e)) represents mean ± SEM. ns P ≥ 0.05; *P < .05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001.
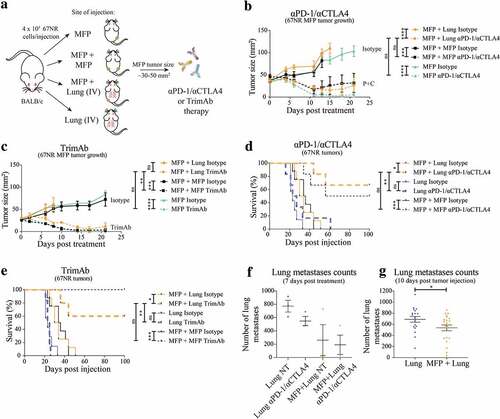
Consistent with the overall survival of these mice, we found that at 7 days post-treatment there was a trend toward decreased lung metastases in MFP + Lung mice compared to Lung mice (, Supplementary Figure 1a). Intriguingly, this decrease applied to non-treated MFP + Lung mice and there was only a small decrease in the number of lung metastases upon treatment in this group. To investigate this further, we enumerated lung metastases in mice at 10 days post-tumor injection when treatment with immunotherapies had not yet commenced. In these experiments, we again observed a decrease in metastases in mice with concomitant MFP tumors (, Supplementary Figure 1B). During the process of MFP tumor injection, mice are anaesthetized using isoflurane, and the differential use of anesthetic may have impacted on lung metastases upon IV injection of tumor cells.Citation23 However, we found that the administration of isoflurane to mice directly after IV injection with tumor cells did not result in decreased lung metastases numbers compared to IV injected mice with no isoflurane administration (Supplementary Figure 2). Thus, our study demonstrated that the presence of a MFP tumor decreased lung tumor burden compared to mice without MFP tumors, even before the onset of immunotherapy. The increase in survival observed in these mice may reflect this difference in tumor burden, and the existing difference may be further enhanced by administration of αPD-1/αCTLA4 or trimAb therapies.
T cell infiltration is enhanced in the lungs of MFP tumor-bearing mice
Previously we found that resistant 67NR lung tumors had decreased infiltration and activation of T cells, decreased activation of NK cells, and increased infiltration of MDSCs reflective of an immunosuppressive TME.Citation20 Given our previous observation and the impact of the TME on immunotherapy responses, we next investigated the TMEs of MFP and lung tumors when present alone or when a tumor in the opposite site was present simultaneously. We performed a Nanostring assay with the nCounter Mouse Pan Cancer Immune profiling 770-plex panel on MFP or lung tumors 14 days after tumor injection. MFP and lung 67NR tumors had distinct immune gene expression profiles as shown by PCA (), consistent with transcriptomic analysis in our previous study.Citation20 We did not observe any significant global changes in transcription of immune-related genes when comparing lung tumors in mice with or without concomitant MFP tumors. Similarly, there were limited differentially expressed genes in MFP tumors of mice with or without simultaneous lung tumor growth.
Figure 2. T cell phenotype within the lung TME is altered by the presence of a MFP tumor. (a) MFP and/or lung 67NR tumors from BALB/c MFP + MFP, MFP + Lung or Lung mice were harvested 14 days post-injection as outlined in and depicted in the key. RNA was sequenced with the Nanostring Immune gene panel. Principal component analysis (PCA) plot reveals variation between expression profiles of samples. (b,c) MFP and/or lung 67NR tumors from BALB/c MFP + MFP, MFP + Lung or Lung mice were harvested 7 days after commencement of αPD-1/αCTLA4 and TILs analyzed by flow cytometry as shown in Supplementary Figure 3. Data points represent individual tumors, pooled from 2 independent experiments with n = 5 mice per group per experiment. Bars represent mean ± SEM. Mann-Whitney test. *P < .05; **P ≤ 0.01.
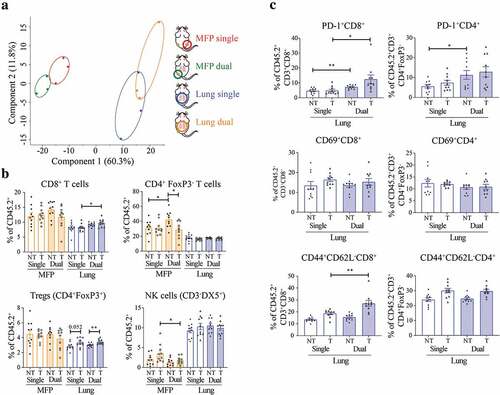
To specifically analyze changes in the immune TME, we analyzed infiltrating immune cells by flow cytometry (Supplementary Figure 3). At 10 days post-tumor injection, before therapeutic intervention, we observed minimal changes by flow cytometry of infiltrating immune cells in MFP and lung tumors present alone or simultaneously (Supplementary Figure 4). The only significant difference observed at this time point was an increase in Ly6C+ myeloid cells in the lungs of mice with simultaneous MFP tumors. Next, we performed the same flow cytometry analysis on MFP and lung tumors harvested at 7 days post-initiation with αPD-1/αCTLA4 therapy (, Supplementary Figure 5). We found a significant increase in CD8+ T cell infiltration in treated lung tumors from mice with an additional MFP tumor, consistent with the enhanced response to immunotherapy in this group (). Additionally, there was an increase in Treg infiltration in lungs of treated compared to non-treated mice, regardless of the presence of a MFP tumor. Within the T cell compartment, there was an increase in PD-1+ CD8+ and CD4+ T cells in the lungs of mice with concomitant MFP tumors (). However, there was no difference in expression of the early activation marker CD69 on T cells. There was an increase in CD44+CD62L− CD8+ T cells, which can indicate effector memory status, in the treated lung tumors of mice with co-existing MFP tumors compared to mice with lung tumors alone. The increase in PD-1+ could demonstrate activation of these cells and combined with the increase in CD44+CD62L− CD8+ T cells may indicate a more effective anti-tumor T cell response in the lungs of mice with concomitant MFP tumors. Together, immune cell profiling indicated that the presence of a responding MFP tumor enhanced the CD8+ T cell response in previously non-responding lung tumors, which is consistent with the enhanced response to immunotherapy in these mice.
In addition to the changes in T cells, we observed decreased infiltration of macrophages in treated lung tumors of mice with concomitant MFP tumors. This decrease in total macrophages was accompanied by a decrease in the percentage of CD206+ macrophages (Supplementary Figure 5), which are known to be immunosuppressive within the TME.Citation24 Similar to our analysis, 10 days post-tumor injection, there was an increase in Ly6C+ myeloid cells in the lungs of mice with MFP tumors, but this increase was abolished following αPD-1/αCTLA4 therapy. This decrease in CD206+ macrophages and Ly6C+ myeloid cells may contribute to a less immunosuppressive TME in the lungs of treated mice with simultaneous MFP tumors. Indeed, the balance of myeloid cells within breast tumors has demonstrated importance to the response to αPD-1/αCTLA4.Citation25 There were various changes observed in MFP tumors when comparing mice with or without additional lung tumors (, Supplementary Figure 5). We observed changes in frequencies of CD4+ T cells, NK cells, B cells, macrophages, Ly6G,+ and Ly6C+ myeloid cells between MFP tumors of mice in different treatment and tumor groups. As there were no significant changes to tumor growth or immunotherapy response in MFP tumors, it is currently unclear what roles these differences play in tumor progression.
Effect of MFP tumors on lung metastases is CD8+ T cell dependent
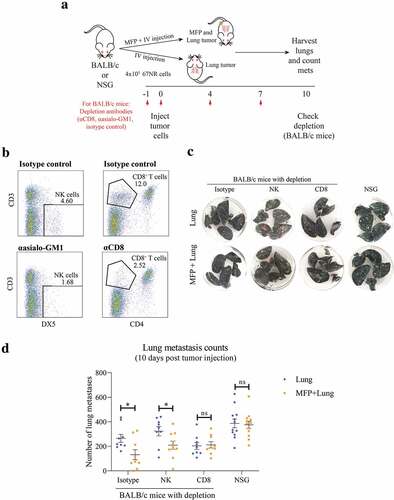
To further investigate the mechanism by which the presence of a MFP tumor impacts on lung metastases, we conducted a series of experiments using immunodeficient mouse models (). BALB/c mice depleted of CD8+ T cells or NK cells and NSG mice, which are deficient in T, B, and functional NK cells, were injected with MFP and lung tumors, or lung tumors alone (). Mice receiving depletion antibodies were successfully depleted of NK cells and CD8+ T cells (). As observed previously, we found that isotype treated mice with MFP and lung tumors had decreased lung metastases compared to mice with lung tumors alone (,d). This decrease was observed in mice depleted of NK cells but was ablated in CD8+ T cell depleted and NSG mice. Therefore, the decrease in lung metastases observed in MFP tumor-bearing mice is CD8+ T cell dependent.
Figure 3. Decreased lung tumor burden in MFP tumor-bearing mice is CD8+ T cell dependent. (a) Experimental overview. BALB/c mice were injected with depletion antibodies against CD8+ (CD8+ T cell depletion) or asialo-GM1 (NK cell depletion) before injection with 67NR tumor cells in the MFP and IV (MFP + Lung) or IV only (Lung), with repeated dosing of depletion antibodies as indicated. The same tumor injections were performed in NSG mice, without depletion antibodies. Mice were culled 10 days after tumor injection for lung metastasis enumeration and depletion check. (b) Representative plot of depletion check where spleens of BALB/c mice in (a) were harvested, after 4 doses of depletion antibodies, and stained with antibodies indicated for flow cytometry. (c,d) Representative images (c) and enumerated metastases in lungs of BALB/c and NSG mice as described in (a) Data points represent whole lungs from individual mice from 2 independent experiments with mean ± SEM (n = 4–6 mice per group, per experiment). Unpaired t-test. ns P ≥ 0.05; *P < .05.
Lung tumors of mice with concomitant MFP tumors have increased tumor-specific T cells
To further investigate CD8+ T cells within lung tumors, we stimulated cells from lungs of mice with or without concomitant MFP tumors ex vivo with 67NR tumor cells or the Murine Leukemia Virus envelope glycoprotein 70 (gp70 peptide), known to be expressed in 67NR cells and presented by MHCI.Citation26 Following stimulation in the presence of GolgiPlug and GolgiStop, we analyzed IFNγ and TNFα expression in CD8+ T cells (). The results from this assay demonstrated that there were significantly increased IFNγ+ CD8+ T cells from the lungs of mice with concomitant MFP tumors when stimulated (,c). Although not significant, CD8+ T cells from the lungs of MFP + Lung mice tended to have increased TNFα expression however this trend was also observed in the unstimulated control suggesting that it is not specific to stimulation with tumor-specific antigen (). Lastly, we observed a significant increase in the IFNγ+TNFα+ CD8+ T cells stimulated with 67NR tumor cells when these T cells were isolated from the lungs of MFP + Lung mice (). Together, these results highlight that the presence of a MFP tumor enhances the infiltration of tumor-specific CD8+ T cells to lung tumors. This result aligns with our observation that the decrease in lung metastases in MFP tumor-bearing mice was CD8+ T cell dependent and the increase in PD-1+ and CD44+CD62L− CD8+ T cells in lungs of mice with MFP tumors.
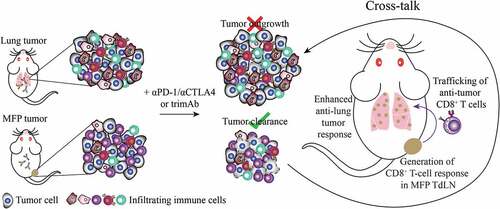
Given the enhanced survival of immunotherapy treated mice with dual MFP and lung tumors, we propose a model whereby tumor-specific CD8+ T cells are generated through the immunogenic location at the MFP, likely in the TdLN. These T cells then traffic throughout the body and infiltrate lung metastases where they mediate tumor regression in the lungs ().
Figure 4. Lungs of mice with concomitant MFP and lung tumors have increased frequencies of tumor reactive CD8+ T cells. (a) Experimental overview. BALB/c mice were injected with 4 × 105 67NR cells in the MFP and IV (MFP + Lung) or IV only (Lung). Lungs were harvested 10 days after tumor injection, processed into single-cell suspension and stimulated in the presence of GolgiPlug and GolgiStop with 10−6 M gp70 peptide, 1 × 105 67NR tumor cells or unstimulated as a negative control. (b–e) Representative plots and frequencies of CD8+ T cells expressing IFNγ, TNFα or both cytokines assessed by flow cytometry from samples as described in (a). Data points represent individual mice from 2 independent experiments with mean ± SEM (n = 6 mice per group, per experiment). Unpaired t-test. *P < .05; **P ≤ 0.01.
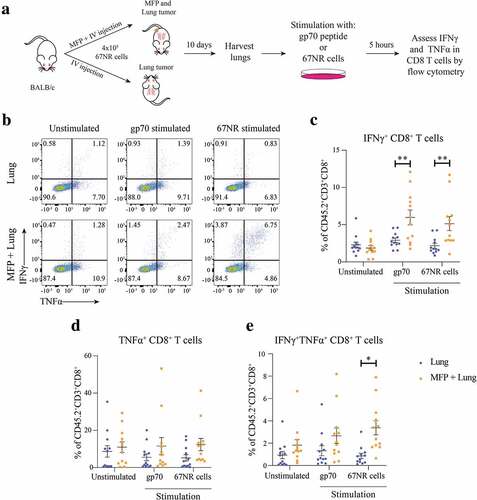
Figure 5. Model of cross-talk between MFP and lung tumors. In isolation, treatment of 67NR MFP tumors with immunotherapy generates an anti-tumor response able to completely eradicate tumors. In contrast 67NR lung tumors are unable to mount an effective anti-tumor immune response. When present simultaneously, lung tumors could be eradicated in a CD8+ T cell-specific manner. Therefore, we propose a model where a tumor-specific CD8+ T cell response is generated in the MFP TdLN and these T cells enter into circulation where they can be recruited to the lung TME, resulting in enhanced anti-lung tumor responses.
Discussion
Immunotherapy has become a major treatment modality for cancer, especially for advanced metastatic patients. Tumors in various anatomical sites have been shown to have distinct TMEs and responses to therapy.Citation13–Citation15 In metastatic disease, these tumors are linked through the blood and lymphatic circulatory networks.Citation5 Despite evidence in specific contexts, such as abscopal responses and the pre-metastatic niche, there is limited research into how distal tumors in various anatomical sites influence each other.Citation2,Citation3,Citation5 Here, we utilized murine models of cancer to investigate the consequences to immunotherapy responses in mice bearing tumors with discrepant TMEs and immunotherapeutic outcomes.
Our experiments revealed that mice with both MFP and lung tumors had enhanced survival compared with mice that had only lung tumors when treated with αPD-1/αCTLA4 or trimAb. Mice with MFP and lung tumors growing simultaneously had less lung metastases numbers compared to mice with lung tumors only. Furthermore, the survival of mice with MFP and lung tumors treated with αPD-1/αCTLA4 or trimAb was similar to mice with dual MFP tumors, which led to complete tumor eradication in a proportion of mice. This result is similar to a study comparing the differential metastatic potential between EMT6 and 4T1 breast cancer lines which revealed that, in the less metastatic EMT6 line, IV injection of tumor cells resulted in lung metastases only without the presence of an EMT6 tumor in the MFP.Citation27 Likewise, a B16 melanoma model demonstrated that SC melanoma was required for the response of intracranial tumors to αPD-1/αCTLA4.Citation12 Together, these studies reveal a role for cross-talk between tumors influencing tumor growth and therapy responses in mice.
We observed an increase in CD8+ T cell total frequency, PD-1+ positivity, and CD44+CD62L−CD8+ T cell frequency in lung tumors treated with αPD-1/αCTLA4 in mice with concomitant MFP tumors compared to mice with only lung tumors. Consistent with this increase in activated, effector memory-like CD8+ T cells, we found that depletion of CD8+ T cells abolished the reduction in lung metastases in mice with concomitant MFP tumors. Although NK cells have been implicated in metastasis formation,Citation28 we found that they had no significant impact on the influence from MFP tumors in our model. After further investigation into lung infiltrating CD8+ T cells, we revealed that mice with concomitant MFP tumors had more tumor-antigen specific CD8+ T cells compared to mice without MFP tumors. Encouragingly, results from the study of 4T1 and EMT6 metastasis are consistent with our result that depletion of CD8+ T cells, but not NK cells, abolished the anti-metastatic effect in mice with resected EMT6 tumors.Citation27
Although our study provided some insight into a form of immunological cross-talk between tumors, the exact mechanisms and molecules involved should be investigated in future study. Previous studies have implicated cytokines such as IL-6 and G-CSFCitation27 and adhesion molecules on tumor vasculatureCitation12 in impacting the CD8+ T cell response to metastases in the presence of a primary tumor. An unexplored mechanism could involve type I IFN signaling, which is known to be important in anti-tumor immunity, therapy responses, and implicated in metastasis.Citation29–Citation31 Recent evidence suggests a role for IFNAR1 in cytotoxic T cell anti-tumor responsesCitation32,Citation33 and a role for type I IFNs in immunogenic cell death,Citation34,Citation35 which can be induced by cytotoxic lymphocytes.Citation36 The 67NR tumor line expresses high levels of the interferon regulatory factor, IRF7, in cultureCitation30 and this may be maintained in the MFP tumor setting, enhancing local and systemic anti-tumor immune responses. Indeed, a study of dual tumor models found that intra-tumoral injection of STING agonists could induce abscopal responses in distant tumors in an IFNAR dependent mechanism.Citation37
Our results additionally reflect the findings from studies comparing neoadjuvant and adjuvant immunotherapies in breast cancer models. Studies of the 4T1 model have demonstrated that immunotherapy administration before surgical resection of a primary breast tumor leads to decreased lung metastases and enhanced survival of mice compared to mice treated with immunotherapy in the adjuvant setting.Citation38,Citation39 As in our model, the presence of the primary tumor was required to generate a sufficient anti-tumor T cell response. Perhaps in some settings, it is appropriate to delay the resection of certain tumors that have the potential to promote immune responses in order to enhance systemic anti-tumor immunity. Indeed, clinical trials comparing the efficacy neoadjuvant and adjuvant immunotherapy have demonstrated success in the neoadjuvant setting.Citation40–Citation43
Together, our data suggests that a tumor-specific CD8+ T cell response is generated preferentially in the presence of a MFP tumor. These tumor reactive T cells are then able to enter into circulation and traffic to the lungs where they can participate in tumor destruction (). In the setting where only lung tumors were present, the immunosuppressive TME is inhibitory for mounting a sufficient anti-tumor T cell response. In order to fully understand metastatic disease, tumor models that examine various tumor locations are necessary, especially for common locations of metastasis. Our results have potential implications for therapeutic decision-making in the clinic, where it may be appropriate to delay removing immunogenic tumors that enhance systemic immunity to improve overall response.
Declaration of interest
The authors report no conflict(s) of interest.
Supplemental Material
Download ()Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
References
- Weigelt B, Peterse JL, Van ‘T Veer LJ. Breast cancer metastasis: markers and models. Nat Rev Cancer. 2005;5(8):591–10. doi:10.1038/nrc1670.
- Paget S. The distribution of secondary growths in cancer of the breast. Cancer Metastasis Rev. 1989;8:98–101.
- Demaria S, Ng B, Devitt ML, Babb JS, Kawashima N, Liebes L, Formenti SC. Ionizing radiation inhibition of distant untreated tumors (abscopal effect) is immune mediated. Int J Radiat Oncol Biol Phys. 2004;58(3):862–870. doi:10.1016/j.ijrobp.2003.09.012.
- Spitzer MH, Carmi Y, Reticker-Flynn NE, Kwek SS, Madhireddy D, Martins MM, Engleman EG, Prestwood TR, Chabon J, Bendall SC. Systemic immunity is required for effective cancer immunotherapy. Cell. 2017;168:487–502 e415. doi:10.1016/j.cell.2016.12.022.
- Oliver AJ, Darcy PK, Trapani JA, Kershaw MH, Slaney CY. Cross-talk between tumors at anatomically distinct sites. The FEBS Journal. 2020. doi:10.1111/febs.15316.
- Bronte V, Chappell DB, Apolloni E, Cabrelle A, Wang M, Hwu P, Restifo NP. Unopposed production of granulocyte-macrophage colony-stimulating factor by tumors inhibits CD8+ T cell responses by dysregulating antigen-presenting cell maturation. J Immunol. 1999;162:5728–5737.
- Veglia F, Perego M, Gabrilovich D. Myeloid-derived suppressor cells coming of age. Nat Immunol. 2018;19:108–119. doi:10.1038/s41590-017-0022-x.
- Tung KH, Ernstoff MS, Allen C, Shu S. A review of exosomes and their role in the tumor microenvironment and host-tumor “Macroenvironment”. J Immunol Sci. 2019;3:4–8. doi:10.29245/2578-3009/2019/1.1165.
- Zuckerman NS, Yu H, Simons DL, Bhattacharya N, Carcamo-Cavazos V, Yan N, Dirbas FM, Johnson DL, Schwartz EJ, Lee PP. Altered local and systemic immune profiles underlie lymph node metastasis in breast cancer patients. Int J Cancer. 2013;132(11):2537–2547. doi:10.1002/ijc.27933.
- Walker R, Poleszczuk J, Pilon-Thomas S, Kim S, Anderson A, Czerniecki BJ, … Enderling H. Immune interconnectivity of anatomically distant tumors as a potential mediator of systemic responses to local therapy. Sci Rep. 2018;8:9474. doi:10.1038/s41598-018-27718-1.
- Devaud C, John LB, Westwood JA, Yong CS, Beavis PA, Schwendener RA, … Kershaw MH. Cross-talk between tumors can affect responses to therapy. Oncoimmunology. 2015;4:e975572. doi:10.4161/2162402X.2014.975572.
- Taggart D, Andreou T, Scott KJ, Williams J, Rippaus N, Brownlie RJ, … Lorger M. Anti-PD-1/anti-CTLA-4 efficacy in melanoma brain metastases depends on extracranial disease and augmentation of CD8(+) T cell trafficking. Proc Natl Acad Sci U S A. 2018;115:E1540–E1549. doi:10.1073/pnas.1714089115.
- Oliver AJ, Lau PKH, Unsworth AS, Loi S, Darcy PK, Kershaw MH, Slaney CY. Tissue-dependent tumor microenvironments and their impact on immunotherapy responses. Front Immunol. 2018;9:70. doi:10.3389/fimmu.2018.00070.
- Horton BL, Fessenden TB, Spranger S. Tissue site and the cancer immunity cycle. Trends Cancer. 2019;5:593–603. doi:10.1016/j.trecan.2019.07.006.
- Pao W, Ooi CH, Birzele F, Ruefli-Brasse A, Cannarile MA, Reis B, Reed JC, Schubert DA, Hatje K, Pelletier N. Tissue-specific immunoregulation: a call for better understanding of the “immunostat” in the context of cancer. Cancer Discov. 2018;8:395–402. doi:10.1158/2159-8290.CD-17-1320.
- Oliver AJ, Darcy PK, Kershaw MH, Slaney CY. Tissue-specific tumour microenvironments are an emerging determinant of immunotherapy responses. Journal of Thoracic Disease. 2020. doi:10.21037/jtd.2020.03.64.
- Savas P, Salgado R, Denkert C, Sotiriou C, Darcy PK, Smyth MJ, Loi S. Clinical relevance of host immunity in breast cancer: from TILs to the clinic. Nat Rev Clin Oncol. 2016;13(4):228–241. doi:10.1038/nrclinonc.2015.215.
- Miller LD, Chou JA, Black MA, Print C, Chifman J, Alistar A, Marincola FM, Zhou X, Bedognetti D, Hendrickx W. Immunogenic subtypes of breast cancer delineated by gene classifiers of immune responsiveness. Cancer Immunol Res. 2016;4:600–610. doi:10.1158/2326-6066.CIR-15-0149.
- Savas P, Virassamy B, Ye C, Salim A, Mintoff CP, Caramia F, Loi S, Byrne DJ, Teo ZL, Dushyanthen S. Single-cell profiling of breast cancer T cells reveals a tissue-resident memory subset associated with improved prognosis. Nat Med. 2018;24:986–993. doi:10.1038/s41591-018-0078-7.
- Oliver AJ, Davey AS, Keam SP, Mardiana S, Chan JD, von Scheidt B, … Slaney CY. Tissue-specific tumor microenvironments influence responses to immunotherapies. Clin Transl Immunology. 2019;8:e1094. doi:10.1002/cti2.1094.
- Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, … Cox J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods. 2016;13:731–740. doi:10.1038/nmeth.3901.
- Eckhardt BL, Parker BS, van Laar RK, Restall CM, Natoli AL, Tavaria MD, Anderson RL, Sloan EK, Moseley JM, Anderson RL. Genomic analysis of a spontaneous model of breast cancer metastasis to bone reveals a role for the extracellular matrix. Mol Cancer Res. 2005;3:1–13.
- Li R, Huang Y, Lin J. Distinct effects of general anesthetics on lung metastasis mediated by IL-6/JAK/STAT3 pathway in mouse models. Nat Commun. 2020;11(1):642. doi:10.1038/s41467-019-14065-6.
- Devaud C, Westwood JA, John LB, Flynn JK, Paquet-Fifield S, Duong CP, Kershaw MH, Pegram HJ, Stacker SA, Achen MG. Tissues in different anatomical sites can sculpt and vary the tumor microenvironment to affect responses to therapy. Mol Ther. 2014;22:18–27. doi:10.1038/mt.2013.219.
- Kim IS, Gao Y, Welte T, Wang H, Liu J, Janghorban M, Zhang XH, Niu Y, Goldstein A, Zhao N. Immuno-subtyping of breast cancer reveals distinct myeloid cell profiles and immunotherapy resistance mechanisms. Nat Cell Biol. 2019;21:1113–1126. doi:10.1038/s41556-019-0373-7.
- Scrimieri F, Askew D, Corn DJ, Eid S, Bobanga ID, Bjelac JA, Huang AY, Allen F, Othman YS, Wang SCG. Murine leukemia virus envelope gp70 is a shared biomarker for the high-sensitivity quantification of murine tumor burden. Oncoimmunology. 2013;2:e26889. doi:10.4161/onci.26889.
- Ouzounova M, Lee E, Piranlioglu R, El Andaloussi A, Kolhe R, Demirci MF, Korkaya H, Asm I, Chadli A, Hassan KA. Monocytic and granulocytic myeloid derived suppressor cells differentially regulate spatiotemporal tumour plasticity during metastatic cascade. Nat Commun. 2017;8:14979. doi:10.1038/ncomms14979.
- Gorelik E, Wiltrout RH, Okumura K, Habu S, Herberman RB. Role of NK cells in the control of metastatic spread and growth of tumor cells in mice. Int J Cancer. 1982;30(1):107–112. doi:10.1002/ijc.2910300118.
- Guerin MV, Regnier F, Feuillet V, Vimeux L, Weiss JM, Bismuth G, Bercovici N, Guilbert T, Thoreau M, Finisguerra V. TGFbeta blocks IFNalpha/beta release and tumor rejection in spontaneous mammary tumors. Nat Commun. 2019;10:4131. doi:10.1038/s41467-019-11998-w.
- Brockwell NK, Rautela J, Owen KL, Gearing LJ, Deb S, Harvey K, Parker BS, Zanker D, Chan C-L, Cumming HE. Tumor inherent interferon regulators as biomarkers of long-term chemotherapeutic response in TNBC. NPJ Precis Oncol. 2019;3:21. doi:10.1038/s41698-019-0093-2.
- Zitvogel L, Galluzzi L, Kepp O, Smyth MJ, Kroemer G. Type I interferons in anticancer immunity. Nat Rev Immunol. 2015;15(7):405–414. doi:10.1038/nri3845.
- Lu C, Klement JD, Ibrahim ML, Xiao W, Redd PS, Nayak-Kapoor A, … Liu K. Type I interferon suppresses tumor growth through activating the STAT3-granzyme B pathway in tumor-infiltrating cytotoxic T lymphocytes. J Immunother Cancer. 2019;7:157. doi:10.1186/s40425-019-0635-8.
- Katlinski KV, Gui J, Katlinskaya YV, Ortiz A, Chakraborty R, Bhattacharya S, Fuchs SY, Beiting DP, Girondo MA, Peck AR. Inactivation of interferon receptor promotes the establishment of immune privileged tumor microenvironment. Cancer Cell. 2017;31:194–207. doi:10.1016/j.ccell.2017.01.004.
- Galluzzi L, Kroemer G. Calreticulin and type I interferon: an unsuspected connection. Oncoimmunology. 2017;6(3):e1288334. doi:10.1080/2162402X.2017.1288334.
- Vacchelli E, Sistigu A, Yamazaki T, Vitale I, Zitvogel L, Kroemer G. Autocrine signaling of type 1 interferons in successful anticancer chemotherapy. Oncoimmunology. 2015;4:e988042. doi:10.4161/2162402X.2014.988042.
- Galluzzi L, Petroni G, Kroemer G. Immunogenicity of cell death driven by immune effectors. J Immunother Cancer. 2020;8(1):e000802. doi:10.1136/jitc-2020-000802.
- Sivick KE, Desbien AL, Glickman LH, Reiner GL, Corrales L, Surh NH, McWhirter SM, Vu UT, Francica BJ, Banda T. Magnitude of therapeutic STING activation determines CD8(+) T cell-mediated anti-tumor immunity. Cell Rep. 2018;25:3074–3085 e3075. doi:10.1016/j.celrep.2018.11.047.
- Liu J, Blake SJ, Yong MC, Harjunpaa H, Ngiow SF, Takeda K, Teng MW, O’Donnell JS, Allen S, Smyth MJ. Improved efficacy of neoadjuvant compared to adjuvant immunotherapy to eradicate metastatic disease. Cancer Discov. 2016;6:1382–1399. doi:10.1158/2159-8290.CD-16-0577.
- Brockwell NK, Owen KL, Zanker D, Spurling A, Rautela J, Duivenvoorden HM, Parker BS, Caramia F, Loi S, Darcy PK. Neoadjuvant interferons: critical for effective PD-1-based immunotherapy in TNBC. Cancer Immunol Res. 2017;5:871–884. doi:10.1158/2326-6066.CIR-17-0150.
- Amaria RN, Reddy SM, Tawbi HA, Davies MA, Ross MI, Glitza IC, Wargo JA, Lewis C, Hwu W-J, Hanna E. Neoadjuvant immune checkpoint blockade in high-risk resectable melanoma. Nat Med. 2018;24:1649–1654. doi:10.1038/s41591-018-0197-1.
- Cloughesy TF, Mochizuki AY, Orpilla JR, Hugo W, Lee AH, Davidson TB, Prins RM, Ellingson BM, Rytlewski JA, Sanders CM. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat Med. 2019;25:477–486. doi:10.1038/s41591-018-0337-7.
- Pelster MS, Amaria RN. Neoadjuvant immunotherapy for locally advanced melanoma. Curr Treat Options Oncol. 2020;21(2):10. doi:10.1007/s11864-020-0700-z.
- Forde PM, Chaft JE, Smith KN, Anagnostou V, Cottrell TR, Hellmann MD, Pardoll DM, Yang SC, Jones DR, Broderick S. Neoadjuvant PD-1 blockade in resectable lung cancer. N Engl J Med. 2018;378:1976–1986. doi:10.1056/NEJMoa1716078.