
ABSTRACT
Antibodies targeting the co-inhibitory receptor programmed cell death 1 (PDCD1, best known as PD-1) or its main ligand CD274 (best known as PD-L1) have shown some activity in patients with metastatic triple-negative breast cancer (TNBC), especially in a recent Phase III clinical trial combining PD-L1 blockade with taxane-based chemotherapy. Despite these encouraging findings, however, most patients with TNBC fail to derive significant benefits from PD-L1 blockade, calling for the identification of novel therapeutic approaches. Here, we used the 4T1 murine mammary cancer model of metastatic and immune-resistant TNBC to test whether focal radiation therapy (RT), a powerful inducer of immunogenic cell death, in combination with various immunotherapeutic strategies can overcome resistance to immune checkpoint blockade. Our results suggest that focal RT enhances the therapeutic effects of PD-1 blockade against primary 4T1 tumors and their metastases. Similarly, the efficacy of an antibody specific for V-set immunoregulatory receptor (VSIR, another co-inhibitory receptor best known as VISTA) was enhanced by focal RT. Administration of cyclophosphamide plus RT and dual PD-1/VISTA blockade had superior therapeutic effects, which were associated with activation of tumor-infiltrating CD8+ T cells and depletion of intratumoral granulocytic myeloid-derived suppressor cells (MDSCs). Overall, these results demonstrate that RT can sensitize immunorefractory tumors to VISTA or PD-1 blockade, that this effect is enhanced by the addition of cyclophosphamide and suggest that a multipronged immunotherapeutic approach may also be required to increase the incidence of durable responses in patients with TNBC.
Introduction
Successful tumor rejection requires the induction of robust anticancer T-cell responses.Citation1 Therapeutic targeting of the immunosuppressive pathways regulated by cytotoxic T lymphocyte-associated protein 4 (CTLA4) and programmed cell death 1 (PDCD1, best known as PD-1) has been successfully implemented in the clinical management of several malignancies. However, primary and acquired resistance to immune checkpoint inhibitors (ICIs) remain an obstacle in the majority of patients.Citation2
In breast cancer, early studies testing monoclonal antibody directed against PD-1 or its main ligand CD274 (best known as PD-L1) have shown variable but generally modest activity, which was relatively more pronounced in patients with triple-negative breast cancer (TNBC).Citation3 Recent results from the Phase III IMpassion130 clinical trial demonstrate that the addition of the PD-L1-targeting ICI atezolizumab to nab-paclitaxel increases progression-free survival (PFS) of metastatic TNBC patients.Citation4 Interim analysis also showed improved overall survival (OS) (25 mo vs. 15.5 mo) among women with PD-L1+ tumors (369/902, i.e., 41%). These results raise the question as to whether other cytotoxic inducers of immunogenic cell death (ICD)Citation5–7 could enhance the proportion of patients with breast cancer that respond to anti-PD-1/PD-L1 therapies. Both radiation therapy (RT) and cyclophosphamide have multiple immunomodulatory effects,Citation8,Citation9 encompassing the ability to induce ICDCitation6,Citation10,Citation11 and boost responses to ICIs.Citation12–15 In support of this notion, various clinical studies have shown a positive interaction between RT and antibodies targeting PD-1, PD-L1, or CTLA4 in patients with lung cancer.Citation16–18 However, multiple co-inhibitory receptors other than PD-1 or CTLA4 have been described, potentially explaining why most patients fail to respond to the combination of RT and ICIs.Citation19 While such receptors may offer alternative pathways of immunoevasion to developing tumors, they may also constitute potential targets for therapeutic intervention.Citation20,Citation21 In line with this possibility, multiple studies have demonstrated the advantage of simultaneously targeting distinct immunological checkpoints in preclinical tumor models.Citation22–24
V-set immunoregulatory receptor (VSIR, best known as VISTA) is a co-inhibitory receptor that shares structural resemblance with other members of the Ig domain-containing B7 family.Citation25 VISTA is constitutively expressed in the hematopoietic compartment, with the highest expression levels found on myeloid cells. Specifically, VISTA suppresses cytokine production by antigen-presenting cells and hence their ability to drive proliferative T cell responses.Citation26 VISTA is also expressed on (and inhibits the activity of) CD4+ T cells,Citation27 where expression overlaps with that of PD-1 and other co-inhibitory receptors.Citation26 Studies from knock-out mice indicate that VISTA and PD-1 have distinct and non-overlapping roles in the regulation of T-cell activation, which can be therapeutically targeted to achieve a synergistic anti-tumor activity.Citation28
Here, we used mouse 4T1 mammary cancer cells as a model of rapidly metastatic and poorly immunogenic TNBCCitation29 to test the hypothesis that a multipronged therapeutic strategy including ICD inducers like RT and cyclophosphamide as well as ICIs is required for the activation of robust antitumor immune responses, capable to limit metastatic dissemination and increase survival. VISTA emerged as a promising candidate, largely in line with the notion that immunosuppressive myeloid cells have been shown to constitute a large fraction of the immunological infiltrate of human breast cancer,Citation30 they are induced by focal radiotherapy and they express high levels of VISTA.
Our data suggest that optimal therapeutic responses of immunotherapy against TNBC require a multipronged approach that leverages the direct immunostimulation of focal radiotherapy while limiting lymphoid (anti-PD-1, anti-VISTA) and myeloid (cyclophosphamide) immunosuppression. These results provide the rationale for testing VISTA blockade as a component of a multipronged immunotherapeutic approach for tumors that are insensitive to radiation and PD-1 blockade.
Materials and methods
Cells and reagents
Mouse 4T1 mammary cells were grown in DMEM supplemented with 2 mol/L L-glutamine, 100 U/mL penicillin, 100 μg/mL streptomycin, 25 µmol/L 2-mercaptoethanol and 10% fetal bovine serum (Invitrogen). Cells were authenticated by morphology, growth, and pattern of metastasis in vivo and routinely screened for Mycoplasma spp. contamination with the LookOut® Mycoplasma PCR Detection Kit (Sigma-Aldrich). The InVivoMAB mouse anti-PD-1 antibody (Clone RMP1-14) was purchased from BioXCell. The anti-VISTA antibody (Clone 13F3) was generously provided by Janssen Pharmaceuticals.
Animal experiments
Six to eight-week-old wild-type female BALB/c mice were obtained from Taconic. All in vivo experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of Weill Cornell Medicine. Mice were subcutaneously (s.c.) inoculated with 0.5 × 105 4T1 cells and randomly assigned to treatment groups thirteen days later, when tumors typically achieved an average diameter of 5 mm. Focal RT was given with the Small Animal Radiation Research Platform (SARRP, from Xstrahl Ltd) in two doses of 12 Gy each on days 13 and 14 post-tumor implantation. To this aim, mice were anesthetized with isoflurane and animals assigned to radiation were placed on a dedicated tray and positioned so that only the tumor was targeted by the radiation beam by means of a 10 × 10 mm collimator. Tumors were measured every 2–3 days until euthanasia (at experimental endpoints, when tumor exceeded 5% of body weight, or if mice showed signs of pain or distress). Perpendicular tumor diameters were obtained using a Vernier caliper and total tumor volume calculated following the common ellipsoid approachCitation12,Citation31,Citation32 as longer diameter x shorter diameterCitation2 x π/6. Cyclophosphamide (100 mg/kg body weight) was given i.p. on day 9 post tumor implantation. Systemic (i.p.) checkpoint blockade using monoclonal antibodies targeting PD-1 (Clone RMP1-14, 200 μg/mouse) and/or VISTA (Clone 13F3, 10 mg/kg) was initiated the day after the last RT dose. In experiments evaluating the efficacy of treatment on metastatic dissemination, mice were euthanized on day 32 and excised lungs were fixed in 4% paraformaldehyde. Gross lung metastases were enumerated using a dissecting microscope by at least 3 observers, which were blinded to the treatment received by each specimen.
Flow cytometry
4T1 tumors were excised and digested with the Mouse Tumor Dissociation kit (Miltenyi Biotec) as per manufacturer’s instructions, and ran on a Miltenyi gentleMACS Octo Dissociator with Heaters using pre-set program (37C_m_TDK2). The resulting cell suspensions were filtered using a 40 µm cell strainer and subjected to RBC lysis. Samples were counted and stained with the Zombie Aqua Fixable Viability Dye (BioLegend) to distinguish live cells. All samples were then incubated with purified anti-mouse CD16/32 (Fc block) prior to staining. The following anti-mouse antibodies, all purchased from BioLegend, were used for immunostaining in the indicated dilutions: CD69 APC (Clone H1.2 F3) 1:100, CD4 PE/Cy5 (Clone GK1.5) 1:100, FOXP3 Alexa Fluor 488 (Clone 150D) 1:50, CD25 APC (Clone PC61) 1:100, CD45 APC-Cy7 Clone 30-F11 (1:500), CD3 BV421 Clone 145–2 C11(1:100), CD8 FITC Clone 53–6.7 (1:100), CD11b PerCP-Cy5.5 Clone M1/70 (1:200), Ly6G Alexa Fluor 488 (Clone 1A8) 1:100, Ly6C Brilliant Violet 421 (Clone HK1.4) 1:100, CD4 PerCP-Cy5.5 Clone GK1.5 (1:100). Flow data were acquired using a MACSQuant Analyzer 10 and analyzed using FlowJo version 10.1 (Tree Star).
Ex vivo IFNγ production
0.5 x 106 cells from tumor-draining lymph nodes were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum, 2 mmol/L L-glutamine, 100 U/mL penicillin, 100 µg/mL streptomycin, 25 µmol/L 2-mercaptoethanol in a 48-well plate. Feeder cells were obtained from naïve BALB/c mice using 3 × 106 irradiated (12 Gy) splenocytes pre-loaded with the tumor-associated immunodominant antigen AH-1-A5 (SPSYAYHQF) or the irrelevant peptide pMCMV (YPHFMPTNL), both from Genscript, at a final concentration of 1 µg/mL. Supernatants were collected after 48 hours and secreted IFNγ was measured using the Mouse IFN-gamma Quantikine ELISA Kit (R&D Systems).
Statistical analysis
Statistical analyses were done using GraphPad Prism v. 8. To determine significant differences in tumor volumes among treatment groups, two-way ANOVA with repeated measures and Tukey correction for multiple comparisons was utilized. For in vitro experiments, ordinary one-way ANOVA with Holm-Sidak’s posttest correction for samples with single pooled variance was employed to identify significant changes. Kruskal–Wallis test with Dunn’s correction for multiple comparisons was used to detect significant differences in lung metastases among treatment groups. The Kaplan–Meier method was used to estimate median OS and the log cumulative hazard transformation was used to derive 95% confidence limits for median OS in each arm. Differences in OS curves were compared using log-rank (Mantel-Cox) test with correction for multiple pairwise comparisons. All reported p values are two-sided and statistical significance is defined as p< .05.
TCGA analysis
Patients with TNBC (n = 116) were identified in The Cancer Genome Atlas (TCGA) public database (https://cancergenome.nih.gov/). Differentially expressed genes (DEGs) between the VSIRhigh and VSIRlow groups were determined using the LIMMA-R package.Citation33 Hierarchical clustering analysis was conducted using the ComplexHeatmap package, based on the Pearson distance and complete clustering method.Citation34 The MCP-counter R package and “metagene” markers were used to estimate the relative abundance of tissue-infiltrating immune cell populations.Citation35,Citation36 Functional and enrichment analysis of DEGs was performed using the ClusterProfiler.Citation37 Survival analysis was performed using the Survival and Survminer R packages, based on log-rank tests. The prognostic value of continuous variables was assessed using median cutoffs. Correlation was analyzed by the Spearman’s correlation approach and visualized by using the corrplot package in R. GSEA analyses were performed using the fgsea package in R and loading gene set analysis was conducted using MSigDB gene sets H, from msigdbr R package.Citation38 R version 3.6.0 was used for all in silico studies.
Results
Focal RT elicits local and systemic anticancer effects in the context of multiple immune checkpoint blockade
The mouse mammary carcinoma 4T1 model is a well-characterized model of cold, highly metastatic, and immunotherapy-resistant mammary tumor, mimicking the behavior of aggressive TNBC in humans.Citation29,Citation39–41 Treatment of 4T1 tumors established in syngeneic BALB/c mice with ICIs targeting CTLA4 and/or PD-1 is ineffective.Citation29 We have previously shown that RT directed to primary 4T1 tumors enables responsiveness to CTLA4- or PD-1-targeting ICI by inducing T cells that are able to reject the irradiated tumor and reduces metastatic dissemination to the lungs.Citation12,Citation42 Although mice treated with RT plus ICIs experience increased OS as compared to mice receiving ICIs alone, they ultimately succumb to disease progression, suggesting the presence of additional barriers limiting tumor rejection. One such barrier may be represented by MDSCs, which are abundant in the 4T1 microenvironment,Citation29,Citation43 and are known to mediate robust immunosuppressive effects both mice and humansCitation44 prompting interest in developing therapeutic strategies to target them.Citation45
Since myeloid cells have been shown to express high levels of the VISTA,Citation25 we asked whether targeting VISTA could improve responses to RT and PD-1 blockade in the 4T1 model. As monotherapy, neither VISTA nor PD-1 blockade limited the progression of 4T1 tumors established in BALB/c mice (Suppl. Fig. 1A,B). Conversely, both the VISTA-targeting and the PD-1-targeting ICI significantly improved the local control of 4T1 tumors receiving 2 focal RT doses of 12 Gy each, on two consecutive days ()) and reduced the number of lung metastases (Suppl. Fig. 1 C). Local tumor control rates achieved with the RT plus VISTA blockade were comparable to those observed with RT plus PD-1 blockade. However, dual VISTA/PD-1 blockade failed to further improve local or systemic tumor control rates achieved with RT plus PD-1 or VISTA blockade () and Suppl. Fig. 1 C). These results lend further support to the notion that RT can be used to sensitize immunoresistant tumors to ICIs but do not suggest a benefit for dual VISTA and PD-1 blockade.
Figure 1. Anti-tumor effect of dual PD1/VISTA blockade in 4T1 tumor-bearing mice requires radiotherapy and pre-treatment with cyclophosphamide
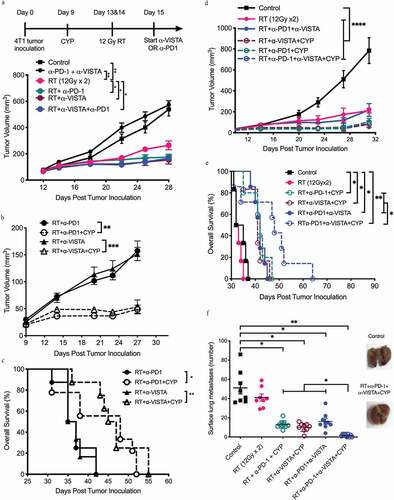
Next, we asked whether the responses obtained with RT plus VISTA blockade could be further improved by cyclophosphamide, a chemotherapeutic agent with broad immunomodulatory propertiesCitation6 that has been successfully exploited in preclinical studies as a therapeutic partner for vaccine-based and other immunotherapeutic approaches.Citation46–48 We thus tested the effect of a single low dose of cyclophosphamide (100 mg/kg) given a few days prior to RT, based on a treatment schedule that was previously shown to induce durable anti-tumor immunity along with a temporary decrease in regulatory T (TREG) cells in 4T1-bearing mice treated with RT and a Toll-like receptor 7 (TLR7) agonist.Citation47 In our model, treatment with cyclophosphamide alone neither delayed tumor growth or OS, nor did it improve therapeutic responses to RT (Suppl. Fig. 1D,E). However, when combined with RT plus PD-1 (p= .0014) or VISTA (p= .0003) blockade cyclophosphamide significantly improved tumor control and OS. ()).
Next, we tested a multipronged immunotherapeutic strategy involving cyclophosphamide, RT as well as PD-1- and VISTA-targeting ICIs. The effect of cyclophosphamide pre-administration on tumor control in mice treated with radiation plus dual PD-1/VISTA blockade was comparable to that achieved by radiation plus either checkpoint blockade (mean tumor volume at day 31: 103.53 ± 12.79 mm3 in cyclophosphamide plus RT plus PD-1 blockade, 82.1 ± 12.6 mm3 in cyclophosphamide plus RT plus VISTA blockade vs. 81.3 ± 17.7 mm3 in cyclophosphamide plus RT plus dual PD-1/VISTA blockade, p= .9550) ()). However, mice treated with cyclophosphamide plus RT and dual PD-1/VISTA blockade experienced a significantly longer median OS as compared to all other mice (median survival: 48 days for cyclophosphamide plus RT plus VISTA and PD-1 blockade vs. 42 days for cyclophosphamide plus RT plus PD-1 blockade, p= .048; and 41 days for cyclophosphamide plus RT plus VISTA blockade, p= .0495.) ()). Importantly, cyclophosphamide was required for survival extension, as median OS in mice treated with RT plus dual PD-1/VISTA blockade was significantly shorter (42 days, p= .0351).
As the survival of 4T1 tumor-bearing mice is mainly dictated by metastatic spread to the lungs,Citation39 we set to evaluate metastatic lung burden prior to overt symptoms of respiratory distress. Mice treated with cyclophosphamide plus RT and dual PD-1/VISTA blockade had significantly fewer lung metastases as compared to all other groups, with one-third of these animals free of metastases at 32 days after tumor inoculation (mean number of metastases: 13.1 ± 1.2 for cyclophosphamide plus RT plus PD-1 blockade, 10.11 ± 1.4 for cyclophosphamide plus RT plus VISTA blockade vs. 1.7 ± 0.47 for cyclophosphamide plus RT and dual PD-1/VISTA blockade, p= .0002) ()).
We further tested whether the timing of cyclophosphamide administration could affect its beneficial effects on systemic tumor control. Machiels and coworkers had previously demonstrated that optimal antitumor immune responses were achieved when cyclophosphamide was given a few days before a GM-CSF-secreting whole-cell vaccine.Citation48 On the other hand, improved tumor responses to RT have been demonstrated when cyclophosphamide was given concurrently to irradiation.Citation49 Thus, we compared the administration of cyclophosphamide 4 days before RT (day 9) vs. concurrent with the first RT dose (day 13) (Suppl. Fig. 2A). No difference in efficacy (neither on tumor growth nor on metastatic dissemination) could be observed when cyclophosphamide was delivered according to different schedules in the context of RT plus dual PD-1/VISTA blockade (Suppl. Fig. 2B,C). Overall, these findings support an essential role for low-dose cyclophosphamide to maximize the ability of RT plus dual PD-1/VISTA to control the progression and metastatic dissemination of 4T1 tumors.
Cyclophosphamide in combination with RT and dual PD-1/VISTA blockade enables the priming of tumor-specific CD8+ T cells coupled with MDSC depletion
To understand the mechanisms underlying the improved control of lung metastases in 4T1 tumor-bearing mice treated with cyclophosphamide plus RT and dual PD-1/VISTA blockade, we analyzed the tumor immune infiltrate at day 18, 3 days after administration of the first ICI dose ()). The flow cytometry-assisted analysis of immune cells isolated from 4T1 tumors demonstrated that RT was required, but not sufficient, to drive robust tumor infiltration by CD8+ T cells. Indeed, a significant increase in intratumoral CD8+ T cells was observed in animals treated with RT plus PD-1 blockade (p= .022) or RT plus VISTA blockade (p= .024), but not RT alone. The combination of cyclophosphamide plus RT and dual PD-1/VISTA blockade induced the largest augmentation in tumor-infiltrating CD8+ T cells, which expressed increased levels of the activation marker CD69 ()). The fraction of tumor-infiltrating CD8+ T cells expressing high levels of PD-1, which is a marker terminally activated/exhausted T cells,Citation50 was reduced in mice treated with cyclophosphamide plus RT and single or dual VISTA/PD-1 blockade, as well as mice treated with RT plus dual VISTA/PD-1 blockade in the absence of cyclophosphamide ()). We next investigated tumor-specific CD8+ T cell responses in tumor-draining lymph nodes. Notably, interferon gamma (IFNG, best known as IFN-γ) secretion by tumor-infiltrating CD8+ T cells exposed to the CD8 epitope AH-1-A5, which is derived from the envelope of an endogenous retrovirus expressed by 4T1 cells,Citation51,Citation52 was markedly increased only in mice treated with cyclophosphamide plus RT and dual PD-1/VISTA blockade (p< .005) ()). Thus, in the 4T1 model of TNBC, only a multipronged immunotherapeutic strategy comprising cyclophosphamide, RT and two ICIs elicits abundant tumor infiltration by activated CD8+ T cells plus robust priming of tumor-specific immunity.
Figure 2. Changes in tumor infiltration by CD8+ T cells induced following the different treatments. 4T1 tumor-bearing mice were treated as described in ). On day 18 post 4T1 cell injection, tumors were excised and digested and single-cell suspensions analyzed by flow cytometry. (a) A multi-step gating strategy was employed to identify CD8+ TILs in dissociated tumors. Positive populations were identified based on negative staining on fluorescence-minus-one (FMO) controls. (b) Percentage of CD8+ T cell infiltration. (c) CD69 expression in CD8+ T cells and (d) percentage of CD8+ TILs expressing PD-1. (b-d, *p< .05, **p< .005, ***p< .0005, ****p< .0001, one-way ANOVA). (e) Effect of treatment on tumor-specific IFN-γ response in tumor-draining lymph nodes. Dissociated cell suspensions were incubated with peptides (tumor-specific epitope AH-1-A5) or irrelevant peptide pMCMV and IFN-γ production measured 48 hours later by ELISA. Each symbol represents the response of an individual mouse to tumor epitope AH-1-A5 after subtraction of the background response to pMCMV (****p< .0005, one-way ANOVA). (f) Percentage of CD4 + T cell infiltration. (g) Proportion of CD4+CD25+FOXP3+ regulatory T (TREG) cells in the CD4 compartment. (h) Expression of CD25 among effector CD4+ T cells. (*p< .05, one-way ANOVA)
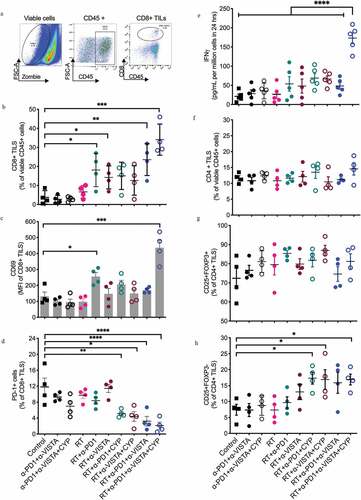
Analysis of the CD4 compartment revealed no significant changes in total CD4+ T cells in any of the treatment groups ()). Similarly, the proportion of TREG cells, which constituted ~70% of all CD4+ T-cells in untreated 4T1 tumors, was not significantly altered by treatment ()). However, activated CD4+ effectors, identified by interleukin 2 receptor subunit alpha (IL2RA, best known as CD25) expression and forkhead box P3 (FOXP3) lack of expression, were increased in the tumors of mice treated with cyclophosphamide plus RT and PD-1, VISTA or dual PD-1/VISTA blockade ()). Cyclophosphamide has previously been shown to temporarily decrease TREG cells.Citation47,Citation48 As we failed to observe such a decrease in intratumoral TREG cells in mice received cyclophosphamide for 9 days (data not shown), we asked whether TREG cells could have been depleted earlier, shortly after cyclophosphamide administration. To address this question, TREG cells were analyzed in the spleen and tumor of mice treated with cyclophosphamide and/or RT at different time points: (1) 3 days after cyclophosphamide administration (day 12), (2) at completion of RT (day 15), and (3) at day 20 (Supp. Fig 2D). This analysis revealed a mild but significant decrease in TREG cells in both the spleen and tumor of mice treated with RT plus cyclophosphamide at day 15, but TREG cells quickly rebounded to baseline levels by day 20 (Suppl. Fig. 2E,F).
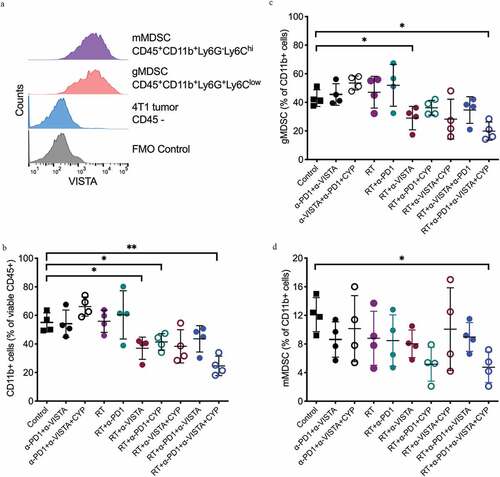
To gain more insights into the mechanisms underlying the development of antitumor immunity in mice treated with cyclophosphamide plus RT and dual PD-1/VISTA blockade, we next analyzed MDSCs. Differential expression of lymphocyte antigen 6 complex, locus G (Ly6G) and lymphocyte antigen 6 complex, locus C (Ly6C) on intratumoral CD11b+ cells defines the two major MDSC subsets: monocytic MDSCs (mMDSCs, Ly6G−Ly6Chi) and granulocytic MDSCs (gMDSCs, Ly6G+Ly6Clow).Citation53,Citation54 Both MDSC subsets infiltrating 4T1 tumors expressed comparable levels of VISTA ()). In untreated mice, CD11b+ myeloid cells comprised approximately 55% of all tumor-infiltrating CD45+ cells ()), ~40% of which were granulocytic MDSCs ()). In the absence of RT and regardless of cyclophosphamide treatment, dual PD-1/VISTA blockade did not alter the abundance of tumor-infiltrating CD11b+ cells. Similarly, RT employed as a standalone treatment did not significantly impact tumor infiltration by CD11b+ myeloid cells ()). Conversely, RT combined with VISTA (but not PD-1) blockade led to a significant decrease in tumor-infiltrating CD11b+ cells ()), particularly in the granulocytic MDSC compartment ()). Addition of cyclophosphamide and a PD-1-targeting ICI to RT plus VISTA blockade did not further decrease the proportion of tumor-infiltrating gMDSCs. Finally, also in the absence of VISTA blockade, the combination of cyclophosphamide with RT and PD-1 blockade significantly reduced CD11b+ myeloid cells as compared to control conditions ()). Of note, RT was critical to achieve gMDSC depletion even in the context of cyclophosphamide plus dual PD-1/VISTA blockade (mean %: 53.55 ± 2.44 for cyclophosphamide plus dual PD-1/VISTA blockade vs. 19.9 ± 3.18 for cyclophosphamide plus RT and dual PD-1/VISTA blockade, p= .0001), suggesting that RT induces key changes in the tumor microenvironment that are required for VISTA blockade to deplete gMDSCs.
Figure 3. Changes in tumor infiltration by MDSC induced following the different treatments. 4T1 tumor-bearing mice were treated as described in Figure 1A. On day 18 post 4T1 cell inoculation, tumors were excised and digested and single-cell suspensions analyzed by flow cytometry. Samples were gated on viable CD45+ cells. (a) Representative histograms show surface expression of VISTA in monocytic and granulocytic MDSCs. Gates were drawn based on negative staining of fluorescence-minus-one (FMO) control. (b) Percentage of CD11b+ cells in 4T1 tumors of mice in each treatment group. (c) Percentage of granulocytic MDSCs among CD11b+ cells. (d) Percentage of monocytic MDSCs among CD11b+ cells. (*p< .05, **p< .005, one-way ANOVA)
Impact of VISTA on the immune infiltrate of breast cancer patients
To investigate the translational value of our findings, we took advantage of The Cancer Genome Atlas (TCGA) public patient dataset, which contains annotated bulk transcriptomic data for 116 patients with immunohistochemistry-confirmed TNBC. First, we interrogated whether VISTA expression levels would be indicative of increased immune infiltration by T cells, based on Spearman correlation on genes the encode phenotypic markers preferentially (although not exclusively) expressed by these immune effector cells. We found that VSIR levels positively correlate with general markers of the T cell compartment (e.g., CD3E), with markers of specific T cell populations (e.g., CD4, CD8A, FOXP3), as well as with co-inhibitory T cell receptors (e.g., CTLA4, HAVCR2, LAG3, PDCD1) ()). Based on our previous observations in the ovarian setting,Citation21 we postulated that such an immunological configuration would be associated with improved OS. However, VSIR levels did not influence the OS of patients with TNBC from the TCGA, neither when patients were stratified based on median VSIR levels ()), nor when VSIR was assessed as a continuous variable (HR: 1.34; 95% CI: 0.84;2.15; p= .22).
Figure 4. VISTA expression in human TNBC correlates with immunoactivation and immunoregulatory gene signature. (a) Correlation between VSIR gene expression and expression levels of eight selected immune genes in TNBC (TCGA-BRCA) cohort. The correlation coefficient is displayed. (b) Overall survival of 116 TNBC patients from the TCG-BRCA database stratified based on median expression level of VSIR gene. (c) Relative expression levels of gene sets associated with T-cells, CD8 cells, Cytotoxic cells, B cells, natural killer (NK) cells, TH1 cells, monocytes, myeloid dendritic cells, neutrophils, regulatory T (TREGS) cells, myeloid-derived suppressor cells (MDSC) and macrophages between VSIR LOW and VSIR HIGH patients from TNBC (TCGA-BRCA) cohort. Box plots: lower quartile, median, upper quartile; whiskers, minimum, maximum. (d) Unsupervised hierarchical clustering of differentially expressed genes that were significantly changed (adjusted p-value <0.05) in VSIR HIGH versus VSIR LOW patients in TNBC (TCGA-BRCA) cohort. (e) GSEA Bar plot for enriched GSEA HALLMARK categories. Only categories with adjusted p < .05 were considered significant. Bar plot depicting the normalized enrichment scores of the most positively (green) and negatively (red) enriched categories in VSIR HIGH patients. (f) Correlation between VSIR gene expression and expression levels of immunoregulatory genes for TNBC cohort in the TCGA-BRCA dataset. The correlation coefficient is displayed. (g) Relative expression levels of immunoregulatory genes between VSIR LOW and VSIR HIGH group of patients from TNBC (TCGA-BRCA) cohort. Box plots: lower quartile, median, upper quartile; whiskers, minimum, maximum
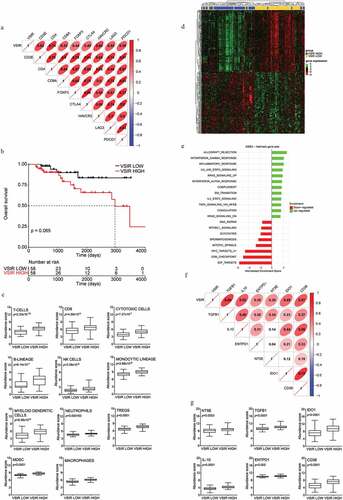
We thus hypothesized that other immunological features of the tumor microenvironment of patients with TNBC from the TCGA could be relevant. We, therefore, tested the relative abundance of multiple immune cell subsets in patients with higher-than-median (VSIRhigh) versus lower-than-median (VSIRlow) VSIR levels by harnessing the MCPcounter R package, which is based on gene signatures that identify specific immune cell populations.Citation35 As compared to their VSIRlow counterparts, VSIRhigh tumors were enriched not only in lymphoid cells encompassing T cells, CD8+ T cells, cytotoxic lymphocytes, TREG cells, B cells, and NK cells (largely replicating the results of our Spearman correlation analysis), but also in cells from the monocytic lineage, myeloid dendritic cells, MDSCs, macrophages, and neutrophils ()). Consistent with these findings, the unsupervised hierarchical clustering of patients with TNBC from the TCGA based on the 400 most differentially expressed genes between VSIRhigh and VSIRlow tumors, identified two major patient clusters that were almost precisely determined by VISTA status ()), and were largely defined by signatures of immunological competence (VSIRhigh vs. VSIRlow) ()). Thus, VSIRhigh TNBCs stand out as tumors with a complex lymphoid and myeloid infiltrate.
We next tested whether immunosuppressive features of the myeloid immune infiltrate would correlate with VISTA levels in this patient subset. We found that VSIR levels correlate (to variable degrees) with the abundance of TGFB1 and IL10 (coding for two cytokines with robust immunosuppressive activity), ENTPD1 and 4NTE (which code for two ectonucleotidases involved in the generation of the immunoregulatory metabolite adenosine),Citation55,Citation56 IDO1 (encoding an intracellular enzyme involved in the degradation of tryptophan, which is required for optimal T cell activity, and the synthesis of kynurenine, which is immunosuppressive)Citation57,Citation58 as well as CD38 (which codes for another extracellular enzyme with immunoregulatory activity)Citation59 ()). In line with this notion, VSIRhigh TNBCs significantly differed from their VSIRlow counterparts in the relative abundance of each of these transcripts taken individually and in association, conveying a global signature of myeloid immunosuppression ()).
With the caution imposed by the transcriptomic analysis of a single patient cohort, the immunological signature we documented in VSIRhigh TNBCs lend further support to our preclinical findings indicating that optimal therapeutic response to radiation therapy plus VISTA inhibitors may require not only immune checkpoint blockers to offset immunosuppression in the lymphoid compartment, but also strategies to target myeloid immunosuppression (cyclophosphamide).
Discussion
The success of ICIs that target CTLA4 and PD-1 for the management of an ever-growing list of malignancies underlies the key relevance of immunosuppressive pathways that prevent T cells from effectively recognizing and killing their neoplastic counterparts.Citation60 However, while durable responses to ICI-based immunotherapy have been documented in a fraction of patients with solid tumors, most patients fail to respond to ICI when employed as single agents. Efforts to increase response rate by simultaneously blocking the two co-inhibitory receptors CTLA4 and PD-1 have been successful in patients with melanoma and lung cancer, but at the expense of increased toxicity.Citation61–63 In addition, DNA-damaging agents with immunostimulatory effects, such as focal RT, have been shown to synergize with ICIs in some patient populations.Citation17,Citation18,Citation64 These findings demonstrate that combining multiple immunotherapies with non-overlapping mechanisms of action may constitute a valuable strategy to increase response rate to ICI-based immunotherapy.
PD-1 and VISTA regulate immune responses via non-overlapping pathways, and the concurrent targeting of PD-1 and VISTA has been shown to improve the control of mouse CT26 colorectal carcinomas as compared to either agent employed as monotherapy.Citation28 However, we found that 4T1 tumors are refractory to VISTA blockade alone as well as to dual PD-1/VISTA blockade (Supp Fig. 1). Prior work by Le Mercier and colleagues has demonstrated that antibody-mediated VISTA blockade limits the growth of various mouse tumors, at least in part by depleting MDSCs.Citation65 In our study, we used the same antibody clone used by Le Mercier and collaborators,Citation65 pointing to the highly immunosuppressive microenvironment established by growing 4T1 tumors as to the reason for limited monotherapeutic activity. Consistent with this notion, VISTA blockade was able to deplete MDSCs in the microenvironment of 4T1 tumors only when given with RT ().
Moreover, VISTA administration significantly improved the control of irradiated 4T1 tumors and metastatic dissemination, an effect that was comparable to the PD-1 blockade. Dual PD-1/VISTA blockade failed to further improve tumor control in this setting (). However, when low-dose cyclophosphamide was administered before RT, we observed a significant improvement in tumor control and OS in mice treated with RT plus PD-1 or VISTA blockade, and that combination of all four therapies (cyclophosphamide, RT, PD-1 blockade, VISTA blockade) further extended OS resulting in almost complete control of lung metastases, independently of the time of administration of cyclophosphamide ( and Suppl. Fig. 2).
In the absence of immunotherapy, cyclophosphamide did not increase RT-mediated tumor control (Suppl. Fig. 1), suggesting a role for the immunomodulatory effects of cyclophosphamide in the improved tumor responses enabled by ICIs. Such effects have generally been linked to the depletion of intratumoral TREG cells, which in rodents are more sensitive to cyclophosphamide than conventional T cells.Citation66 There was a small and temporary reduction in TREG cells in the spleen and tumor of 4T1 tumor-bearing mice treated with RT and cyclophosphamide, but TREG cells represented the majority of the CD4 compartment in tumors exposed to various combinations of RT and ICIs regardless of cyclophosphamide ( and Suppl. Fig. 2). Thus, it is unlikely that the ability of cyclophosphamide to dramatically enhance the priming of tumor-specific CD8+ T cells in mice treated with RT and dual PD-1/VISTA blockade () originates from TREG cell depletion. Cyclophosphamide has also been shown to promote the activation of cytotoxic CD8+ T cells and TH1/TH17 polarization in CD4+ T cells,Citation8 at least in part linked to the ability of cyclophosphamide to reshape the intestinal microbiota.Citation67 Thus, it is conceivable that the improved anti-tumor T cell responses observed in mice treated with cyclophosphamide plus RT and dual PD-1/VISTA blockade may reflect at least some degree of systemic immunomodulation by cyclophosphamideCitation68 coupled to (1) MDSC depletion by cyclophosphamide and (2) de-repression of the effector phase of the immune response in the tumor microenvironment by VISTA and PD-1 blockade.
With the caveats associated with a retrospective transcriptomic study based on a relatively small patient cohort, our preclinical findings are supported by the fact that the microenvironment of patients with TNBC from the TCGA database containing high VSIR levels is enriched in gene signatures pointing to a robust myeloid immunosuppression (). Moreover, CD68+ macrophages have recently been identified as an important reservoir of VISTA-expressing cells in prostate and pancreatic tumors,Citation69,Citation70 suggesting a key role for VISTA in the myeloid tumor microenvironment. Of note, RT can drive robust tumor infiltration by myeloid cells, as shown by a study in non-metastatic prostate cancer patients,Citation71 and multiple pre-clinical work suggesting that RT generates a broad and complex effect on recruitment, removal, reorganization, repolarization and/or representation of tumor-infiltrating myeloid cells.Citation72,Citation73 In prostate tumors, treatment with fractionated low-dose RT led to elevated levels of macrophage colony-stimulating factor 1 (CSF1), a key cytokine driving the systemic accumulation of MDSCs.Citation74 In this context, RT-induced DNA damage was shown to mediate the nuclear translocation of ABL proto-oncogene 1, non-receptor tyrosine kinase (ABL1), and consequent Csf1 transactivation. On the other hand, indolent type I interferon secretion by RT has been implicated in MDSC recruitment via C-C motif chemokine receptor 2 (CCR2),Citation75 which not only supports TREG infiltration upon RT but also has been proposed to represent a biomarker for cyclophosphamide sensitivity.Citation76,Citation77 Most importantly, selective targeting of these axes, either by small molecule inhibitors of the CSF1 receptor or CCR2 antagonists, has defined a new therapeutic partnership to increase patient response to RT. Our study suggests that VISTA blockade stands out as an additional pathway through which the detrimental effects of myeloid (and potentially TREG) cell accumulation driven by RT can be overcome.
In conclusion, our data suggest that the immunological rejection of tumors that are resistant to ICIs may require treatments that act at multiple levels, encompassing not only the robust activation of ICD (and hence an increased availability of tumor-associated antigens and danger signals), as effectively elicited by RT, but also the neutralization of immunosuppressive circuitries involving lymphoid and myeloid compartments, as mediated by multiple ICIs and cyclophosphamide, respectively. Moreover, these findings suggest that tumor types with prominent MDSC-dependent immunosuppression may benefit from combinatorial therapies that also target this compartment. Additional studies are required to translate these observations into clinical trials.
Disclosure of Potential Conflicts of Interest
M.H. and J.F. are full-time employees of Sotio. L.G. declares research funding from Lytix, and Phosplatin, and speaker and/or advisory honoraria from Boehringer Ingelheim, Astra Zeneca, OmniSEQ, The Longevity Labs, Inzen, the Luke Heller TECPR2 Foundation. S.D. has received prior honorarium for consulting from AstraZeneca, AbbVie, Lytix Biopharma, EMD Serono, Eisai Inc., Cytune Pharma, Regeneron, and research grants from Nanobiotix, and Lytix Biopharma. S.C.F. has received prior honorarium for consulting/speaker from AstraZeneca, Merck, Regeneron, Bayer, Serono, and research funding from Varian, Merck, Bristol Meyer Squibb. All other authors have no conflicts of interests to disclose. As per standard operations at Oncoimmunology, LG has been excluded from all steps of editorial evaluation of the present article.
Authors contributions
Concept and design: K.A.P., S.D., S.C.F. Data acquisition: K.A.P., C.D., J.F., M.H. Data analysis and interpretation: K.A.P., M.H., J.F, L.G., S.D. Writing and/or review of manuscript: K.A.P., L.G., S.D., S.C.F. Support and infrastructure: J.F., S.C.F. Approval of the manuscript: All authors.
Supplemental Material
Download ()Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
References
- Galluzzi L, Chan TA, Kroemer G, Wolchok JD, López-Soto A. The hallmarks of successful anticancer immunotherapy. Sci Transl Med. 2018;10:1–14.
- Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. 2017;168:707–10.
- Emens LA. Breast cancer immunotherapy: facts and hopes. Clin Cancer Res. 2018;24:511–520.
- Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H, Diéras V, Hegg R, Im SA, Shaw Wright G, et al. Atezolizumab and nab-paclitaxel in advanced triple-negative breast cancer. N Engl J Med. 2018;379:2108–2121.
- Ma Y, Kepp O, Ghiringhelli F, Apetoh L, Aymeric L, Locher C, Tesniere A, Martins I, Ly A, Haynes NM, et al. Chemotherapy and radiotherapy: cryptic anticancer vaccines. Semin Immunol. 2010;22:113–124.
- Galluzzi L, Vitale I, Warren S, Adjemian S, Agostinis P, Martinez AB, Chan TA, Coukos G, Demaria S, Deutsch E, et al. Consensus guidelines for the definition, detection and interpretation of immunogenic cell death. J Immunother Cancer. 2020;8:e000337.
- Rodriguez-Ruiz ME, Vitale I, Harrington KJ, Melero I, Galluzzi L. Immunological impact of cell death signaling driven by radiation on the tumor microenvironment. Nat Immunol. 2020;21:120–134.
- Sistigu A, Viaud S, Chaput N, Bracci L, Proietti E, Zitvogel L. Immunomodulatory effects of cyclophosphamide and implementations for vaccine design. Semin Immunopathol. 2011;33:369–383.
- Demaria S, Golden EB, Formenti SC. Role of local radiation therapy in cancer immunotherapy. JAMA Oncol. 2015;1:1325–1332.
- Golden EB, Frances D, Pellicciotta I, Demaria S, Helen Barcellos-Hoff M, Formenti SC. Radiation fosters dose-dependent and chemotherapy-induced immunogenic cell death. Oncoimmunology. 2014;3:e28518.
- Rodriguez-Ruiz ME, Buque A, Hensler M, Chen J, Bloy N, Petroni G, Sato A, Yamazaki T, Fucikova J, Galluzzi L. Apoptotic caspases inhibit abscopal responses to radiation and identify a new prognostic biomarker for breast cancer patients. Oncoimmunology. 2019;8:e1655964.
- Demaria S, Kawashima N, Yang AM, Devitt ML, Babb JS, Allison JP, Formenti SC. Immune-mediated inhibition of metastases after treatment with local radiation and CTLA-4 blockade in a mouse model of breast cancer. Clin Cancer Res. 2005;11:728–734.
- Deng L, Liang H, Burnette B, Beckett M, Darga T, Weichselbaum RR, Fu YX. Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice. J Clin Invest. 2014;124:687–695.
- Dovedi SJ, Adlard AL, Lipowska-Bhalla G, McKenna C, Jones S, Cheadle EJ, Stratford IJ, Poon E, Morrow M, Stewart R, et al. Acquired resistance to fractionated radiotherapy can be overcome by concurrent PD-L1 blockade. Cancer Res. 2014;74:5458–5468.
- Pilones KA, Vanpouille-Box C, Demaria S. Combination of radiotherapy and immune checkpoint inhibitors. Semin Radiat Oncol. 2015;25:28–33.
- Shaverdian N, Lisberg AE, Bornazyan K, Veruttipong D, Goldman JW, Formenti SC, Garon EB, Lee P. Previous radiotherapy and the clinical activity and toxicity of pembrolizumab in the treatment of non-small-cell lung cancer: a secondary analysis of the KEYNOTE-001 phase 1 trial. Lancet Oncol. 2017;18:895–903.
- Antonia SJ, Villegas A, Daniel D, Vicente D, Murakami S, Hui R, Yokoi T, Chiappori A, Lee KH, de Wit M, et al. Durvalumab after chemoradiotherapy in stage III non-small-cell lung cancer. N Engl J Med. 2017;377:1919–1929.
- Formenti SC, Rudqvist NP, Golden E, Cooper B, Wennerberg E, Lhuillier C, Vanpouille-Box C, Friedman K, Ferrari de Andrade L, Wucherpfennig KW, et al. Radiotherapy induces responses of lung cancer to CTLA-4 blockade. Nat Med. 2018;24:1845–1851.
- Sharma P, Allison JP. Dissecting the mechanisms of immune checkpoint therapy. Nat Rev Immunol. 2020;20:75–76.
- Zarour HM. Reversing T-cell Dysfunction and Exhaustion in Cancer. Clin Cancer Res. 2016;22:1856–1864.
- Fucikova J, Rakova J, Hensler M, Kasikova L, Belicova L, Hladikova K, Truxova I, Skapa P, Laco J, Pecen L, et al. TIM-3 dictates functional orientation of the immune infiltrate in ovarian cancer. Clin Cancer Res. 2019;25:4820–4831.
- Vanpouille-Box C, Formenti SC. Dual transforming growth factor-β and programmed death-1 blockade: a strategy for immune-excluded tumors? Trends Immunol. 2018;39:435–437.
- Tauriello DVF, Palomo-Ponce S, Stork D, Berenguer-Llergo A, Badia-Ramentol J, Iglesias M, Sevillano M, Ibiza S, Cañellas A, Hernando-Momblona X, et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature. 2018;554:538–543.
- Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, Kadel EE III, Koeppen H, Astarita JL, Cubas R, et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. 2018;554:544–548.
- Nowak EC, Lines JL, Varn FS, Deng J, Sarde A, Mabaera R, Kuta A, Le Mercier I, Cheng C, Noelle RJ. Immunoregulatory functions of VISTA. Immunol Rev. 2017;276:66–79.
- Wang L, Rubinstein R, Lines JL, Wasiuk A, Ahonen C, Guo Y, Lu LF, Gondek D, Wang Y, Fava RA, et al. VISTA, a novel mouse Ig superfamily ligand that negatively regulates T cell responses. J Exp Med. 2011;208:577–592.
- Flies DB, Han X, Higuchi T, Zheng L, Sun J, Ye JJ, Chen L. Coinhibitory receptor PD-1H preferentially suppresses CD4⁺ T cell-mediated immunity. J Clin Invest. 2014;124:1966–1975.
- Liu J, Yuan Y, Chen W, Putra J, Suriawinata AA, Schenk AD, Miller HE, Guleria I, Barth RJ, Huang YH, et al. Immune-checkpoint proteins VISTA and PD-1 nonredundantly regulate murine T-cell responses. Proc Natl Acad Sci U S A. 2015;112:6682–6687.
- Mosely SI, Prime JE, Sainson RC, Koopmann JO, Wang DY, Greenawalt DM, Ahdesmaki MJ, Leyland R, Mullins S, Pacelli L, et al. Rational selection of syngeneic preclinical tumor models for immunotherapeutic drug discovery. Cancer Immunol Res. 2017;5:29–41.
- Markowitz J, Wesolowski R, Papenfuss T, Brooks TR, Carson WE 3rd. Myeloid-derived suppressor cells in breast cancer. Breast Cancer Res Treat. 2013;140:13–21.
- Pilones KA, Charpentier M, Garcia-Martinez E, Daviaud C, Kraynak J, Aryankalayil J, Formenti SC, Demaria S. Radiotherapy cooperates with IL15 to induce antitumor immune responses. Cancer Immunol Res. 2020;8:1054–1063.
- Tomayko MM, Reynolds CP. Determination of subcutaneous tumor size in athymic (nude) mice. Cancer Chemother Pharmacol. 1989;24:148–154.
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47.
- Gu Z, Eils R, Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 2016;32:2847–2849.
- Becht E, Giraldo NA, Lacroix L, Buttard B, Elarouci N, Petitprez F, Selves J, Laurent-Puig P, Sautès-Fridman C, Fridman WH, et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 2016;17:218.
- Angelova M, Charoentong P, Hackl H, Fischer ML, Snajder R, Krogsdam AM, Waldner MJ, Bindea G, Mlecnik B, Galon J, et al. Characterization of the immunophenotypes and antigenomes of colorectal cancers reveals distinct tumor escape mechanisms and novel targets for immunotherapy. Genome Biol. 2015;16:64.
- Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–287.
- Korotkevich G, Sukhov V, Sergushichev A. Fast gene set enrichment analysis. bioRxiv. 2019. 060012.
- Pulaski BA, Ostrand-Rosenberg S. Mouse 4T1 breast tumor model. Curr Protoc Immunol. Chapter 20: Unit20.2. 2001.
- Aslakson CJ, Miller FR. Selective events in the metastatic process defined by analysis of the sequential dissemination of subpopulations of a mouse mammary tumor. Cancer Res. 1992;52:1399–1405.
- Savas P, Salgado R, Denkert C, Sotiriou C, Darcy PK, Smyth MJ, Loi S. Clinical relevance of host immunity in breast cancer: from TILs to the clinic. Nat Rev Clin Oncol. 2016;13:228–241.
- Vanpouille-Box C, Diamond JM, Pilones KA, Zavadil J, Babb JS, Formenti SC, Barcellos-Hoff MH, Demaria S. TGFβ is a master regulator of radiation therapy-induced antitumor immunity. Cancer Res. 2015;75:2232–2242.
- Sinha P, Clements VK, Bunt SK, Albelda SM, Ostrand-Rosenberg S. Cross-talk between myeloid-derived suppressor cells and macrophages subverts tumor immunity toward a type 2 response. J Immunol. 2007;179:977–983.
- Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–174.
- Liu Y, Wei G, Cheng WA, Dong Z, Sun H, Lee VY, Cha SC, Smith DL, Kwak LW, Qin H. Targeting myeloid-derived suppressor cells for cancer immunotherapy. Cancer Immunol Immunother. 2018;67:1181–1195.
- Taieb J, Chaput N, Schartz N, Roux S, Novault S, Ménard C, Ghiringhelli F, Terme M, Carpentier AF, Darrasse-Jèze G, et al. Chemoimmunotherapy of tumors: cyclophosphamide synergizes with exosome based vaccines. J Immunol. 2006;176:2722–2729.
- Dewan MZ, Vanpouille-Box C, Kawashima N, DiNapoli S, Babb JS, Formenti SC, Adams S, Demaria S. Synergy of topical toll-like receptor 7 agonist with radiation and low-dose cyclophosphamide in a mouse model of cutaneous breast cancer. Clin Cancer Res. 2012;18:6668–6678.
- Machiels JP, Reilly RT, Emens LA, Ercolini AM, Lei RY, Weintraub D, Okoye FI, Jaffee EM. Cyclophosphamide, doxorubicin, and paclitaxel enhance the antitumor immune response of granulocyte/macrophage-colony stimulating factor-secreting whole-cell vaccines in HER-2/neu tolerized mice. Cancer Res. 2001;61:3689–3697.
- Zachariae C, Overgaard J. Interactions of radiation, cyclophosphamide and nimorazole in a C3H mammary carcinoma in vivo. Int J Radiat Oncol Biol Phys. 1986;12:1445–1448.
- Thommen DS, Schumacher TN. T cell dysfunction in cancer. Cancer Cell. 2018;33:547–562.
- Scrimieri F, Askew D, Corn DJ, Eid S, Bobanga ID, Bjelac JA, Tsao ML, Allen F, Othman YS, Wang SC, et al. Murine leukemia virus envelope gp70 is a shared biomarker for the high-sensitivity quantification of murine tumor burden. Oncoimmunology. 2013;2:e26889.
- Pilones KA, Aryankalayil J, Babb JS, Demaria S. Invariant natural killer T cells regulate anti-tumor immunity by controlling the population of dendritic cells in tumor and draining lymph nodes. J Immunother Cancer. 2014;2:37.
- Gabrilovich DI. Myeloid-derived suppressor cells. Cancer Immunol Res. 2017;5:3–8.
- Youn JI, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol. 2008;181:5791–5802.
- Allard B, Allard D, Buisseret L, Stagg J. The adenosine pathway in immuno-oncology. Nat Rev Clin Oncol. 2020;17:611–629.
- Wennerberg E, Spada S, Rudqvist NP, Lhuillier C, Gruber S, Chen Q, Zhang F, Zhou XK, Gross SS, Formenti SC, et al. CD73 blockade promotes dendritic cell infiltration of irradiated tumors and tumor rejection. Cancer Immunol Res. 2020;8:465–478.
- Martinek J, Wu TC, Cadena D, Banchereau J, Palucka K. Interplay between dendritic cells and cancer cells. Int Rev Cell Mol Biol. 2019;348:179–215.
- Lee YS, Radford KJ. The role of dendritic cells in cancer. Int Rev Cell Mol Biol. 2019;348:123–178.
- Kennedy BE, Sadek M, Elnenaei MO, Reiman A, Gujar SA. Targeting NAD(+) synthesis to potentiate CD38-based immunotherapy of multiple myeloma. Trends Cancer. 2020;6:9–12.
- Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science. 2018;359:1350–1355.
- Boutros C, Tarhini A, Routier E, Lambotte O, Ladurie FL, Carbonnel F, Izzeddine H, Marabelle A, Champiat S, Berdelou A, et al. Safety profiles of anti-CTLA-4 and anti-PD-1 antibodies alone and in combination. Nat Rev Clin Oncol. 2016;13:473–486.
- Hellmann MD, Ciuleanu TE, Pluzanski A, Lee JS, Otterson GA, Audigier-Valette C, Minenza E, Linardou H, Burgers S, Salman P, et al. Nivolumab plus ipilimumab in lung cancer with a high tumor mutational burden. N Engl J Med. 2018;378:2093–2104.
- Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, Segal NH, Ariyan CE, Gordon RA, Reed K, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369:122–133.
- Gray JE, Villegas A, Daniel D, Vicente D, Murakami S, Hui R, Kurata T, Chiappori A, Lee KH, Cho BC, et al. Three-year overall survival with durvalumab after chemoradiotherapy in stage III NSCLC-update from PACIFIC. J Thorac Oncol. 2020;15:288–293.
- Le Mercier I, Chen W, Lines JL, Day M, Li J, Sergent P, Noelle RJ, Wang L. VISTA regulates the development of protective antitumor immunity. Cancer Res. 2014;74:1933–1944.
- Ghiringhelli F, Larmonier N, Schmitt E, Parcellier A, Cathelin D, Garrido C, Chauffert B, Solary E, Bonnotte B, Martin F. CD4+CD25+ regulatory T cells suppress tumor immunity but are sensitive to cyclophosphamide which allows immunotherapy of established tumors to be curative. Eur J Immunol. 2004;34:336–344.
- Viaud S, Saccheri F, Mignot G, Yamazaki T, Daillère R, Hannani D, Enot DP, Pfirschke C, Engblom C, Pittet MJ, et al. The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science. 2013;342:971–976.
- Vanpouille-Box C, Alard A, Aryankalayil MJ, Sarfraz Y, Diamond JM, Schneider RJ, Inghirami G, Coleman CN, Formenti SC, Demaria S. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat Commun. 2017;8:15618.
- Gao J, Ward JF, Pettaway CA, Shi LZ, Subudhi SK, Vence LM, Zhao H, Chen J, Chen H, Efstathiou E, et al. VISTA is an inhibitory immune checkpoint that is increased after ipilimumab therapy in patients with prostate cancer. Nat Med. 2017;23:551–555.
- Blando J, Sharma A, Higa MG, Zhao H, Vence L, Yadav SS, Kim J, Sepulveda AM, Sharp M, Maitra A, et al. Comparison of immune infiltrates in melanoma and pancreatic cancer highlights VISTA as a potential target in pancreatic cancer. Proc Natl Acad Sci U S A. 2019;116:1692–1697.
- Nickols NG, Ganapathy E, Nguyen C, Kane N, Lin L, Diaz-Perez S, Nazarian R, Mathis C, Felix C, Basehart V, et al. The intraprostatic immune environment after stereotactic body radiotherapy is dominated by myeloid cells. Prostate Cancer Prostatic Dis. 2020.
- Vatner RE, Formenti SC. Myeloid-derived cells in tumors: effects of radiation. Semin Radiat Oncol. 2015;25:18–27.
- Ahn GO, Tseng D, Liao CH, Dorie MJ, Czechowicz A, Brown JM. Inhibition of Mac-1 (CD11b/CD18) enhances tumor response to radiation by reducing myeloid cell recruitment. Proc Natl Acad Sci U S A. 2010;107:8363–8368.
- Xu J, Escamilla J, Mok S, David J, Priceman S, West B, Bollag G, McBride W, Wu L. CSF1R signaling blockade stanches tumor-infiltrating myeloid cells and improves the efficacy of radiotherapy in prostate cancer. Cancer Res. 2013;73:2782–2794.
- Liang H, Deng L, Hou Y, Meng X, Huang X, Rao E, Zheng W, Mauceri H, Mack M, Xu M, et al. Host STING-dependent MDSC mobilization drives extrinsic radiation resistance. Nat Commun. 2017;8:1736.
- Loyher PL, Rochefort J, Baudesson de Chanville C, Hamon P, Lescaille G, Bertolus C, Guillot-Delost M, Krummel MF, Lemoine FM, Combadiere C, et al. CCR2 influences T regulatory cell migration to tumors and serves as a biomarker of cyclophosphamide sensitivity. Cancer Res. 2016;76:6483–6494.
- Mondini M, Loyher PL, Hamon P, Gerbe de Thore M, Laviron M, Berthelot K, Clemenson C, BL S, Combadiere C, Deutsch E, et al. CCR2-dependent recruitment of tregs and monocytes following radiotherapy is associated with tnfalpha-mediated resistance. Cancer Immunol Res. 2019;7:376–387.