
ABSTRACT
Therapeutic monoclonal antibodies against the PD-L1/PD-1 (programmed death ligand-1/programmed cell death protein-1) axis have achieved great successes in cancer treatments, but the development of small-molecule immunomodulators of the pathway has lagged far behind. We established a cellular coculture assay with two stable transfectant cell lines, a PD-L1-expressing tumor cell line PC9/PD-L1 and a PD-1-expressing T cell line Jurkat/PD-1. Western blotting analyses were used to monitor the PD-L1/PD-1 interaction and signaling. We analyzed PD-L1 glycosylation by lectin binding assay and glycosidase digestion, and examined subcellular localization of PD-L1 by immunocytochemical staining. Luciferase assay and real-time PCR were used to evaluate T cell activation in the coculture experiments. We found that coculturing of the PC9/PD-L1 cells with the Jurkat/PD-1 cells induced a lysosomal degradation of PD-1. A small-molecule PD-L1 inhibitor BMS1166 developed by Bristol-Myers Squibb inhibited the coculture-induced PD-1 degradation through a unique mechanism. BMS1166 specifically affected PD-L1 glycosylation and prevented transporting of the under-glycosylated form of PD-L1 from endoplasmic reticulum (ER) to Golgi, leading to accumulation of PD-L1 in ER. In doing so, BMS1166 blocked PD-L1/PD-1 signaling. Coculturing PD-L1-expressing cells with PD-1-expressing cells induced degradation of PD-1, which could be used as a readout to identify inhibitors of PD-L1/PD-1 signaling. The small-molecule PD-L1 inhibitor BMS1166 abolished the glycosylation and maturation of PD-L1 by blocking its exporting from ER to Golgi. Our study discovered a new strategy to identify inhibitors of the PD-L1/PD-1 signaling pathway and to develop new drugs for the treatment of cancer.
KEYWORDS:
Introduction
“Immune escape” has been recognized as one of the emerging hallmarks of cancer in recent years and defines the actions of cancer cells to avoid attack and elimination by the immune system.Citation1 Immune checkpoint molecules, serving as regulators of immune response to maintain immune tolerance to self-antigens, are often hijacked by cancer cells to achieve immune escape.Citation2,Citation3 Programmed cell death protein 1 (PD-1, also known as CD279) is one of the most widely studied inhibitory checkpoint molecules expressed on the surface of mature T lymphocytes to counteract with the T-cell receptor (TCR) signaling via ligation of its ligand, programmed death-ligand 1 (PD-L1, also known as CD274 or B7-H1), leading to T cell exhaustion, characterized by loss of differentiation, proliferation, cytokine production, and cytolytic activity.Citation4 PD-L1 has been found to be widely overexpressed in various types of cancer cells, including melanoma, lymphoma, glioblastoma, breast cancer, lung cancer, bladder cancer, kidney cancer, and ovarian cancer, and is believed to play key roles for cancer cells to escape from immune surveillance.Citation5 Blockade of the PD-L1/PD-1 axis can reactivate the anergy T cells and therefore has been considered as a major strategy to overcome the immune resistance of cancer cells.
So far, there have been six monoclonal antibodies targeting PD-L1/PD-1 axis approved by the US Food and Drug Administration (FDA) and have achieved great success in treating multiple types of cancers, especially the advanced and refractory ones.Citation6,Citation7 However, there are also some disadvantages associated with the antibody agents, such as immunogenicity, instability, high cost, limited modes of administration, and low penetration into tissues, urging the need for developing small molecular inhibitors of the PD-L1/PD-1 signaling pathways.Citation8,Citation9 Three main hot spots on the interface of PD-L1/PD-1 were identified as promising drug targets and a large number of small molecules with different structures have been synthesized to disrupt the PD-L1/PD-1 interactions.Citation5,Citation10–15 One of them, CA-170, developed by Curis/Aurigene in 2015, has entered Phase I/II clinical trials for the treatment of advanced solid tumors and lymphomas with promising results.Citation16,Citation17 Recent studies suggest that CA-170 may act through an unknown mechanism to regulate the PD-L1/PD-1 signaling instead of directly interfering with the PD-L1/PD-1 interaction.Citation17–19
Bristol-Myers Squibb has developed a series of small molecules to interfere with PD-L1/PD-1 interaction based on an in-vitro homogenous time-resolved fluorescence (HTRF) binding assay.Citation20 Compound no.1166 (hereafter BMS1166) was one of the most effective compounds with an IC50 of 1.4 nM in the HTRF assay.Citation20 Structural studies on derivatives of BMS1166 revealed that this class of molecules bound to the PD-1-interacting surface of PD-L1 and potently blocked PD-L1/PD-1 engagement in vitro.Citation21 However, whether this is the mechanism of BMS1166 in vivo has not been studied.
Asparagine-linked glycosylation (N-glycosylation) is a co- and post-translational modification to attach oligosaccharides to asparagine residues within the NXT (Asn-X-Ser/Thr) motifs of nascent peptides synthesized in the Endoplasmic Reticulum (ER).Citation22 The oligosaccharides on the proteins undergo further processing through the secretory pathway to become high-mannose-, hybrid-, or complex-type glycans.Citation23 N-glycosylation plays critical roles in many biochemical and biological events, such as protein folding, degradation, cellular localization, and protein-protein interactions.Citation24 It has been reported that most of the immune-related receptors and ligands, including PD-1 and PD-L1, are extensively glycosylated.Citation25 Four N-glycosylation sites (N35, N192, N200, and N219) have been identified at the extracellular domain of PD-L1 and the glycosylation has been confirmed to be necessary for its stability and its engagement with PD-1.Citation23,Citation25
In the present study, we established a new cellular assay to evaluate PD-L1/PD-1 interactions and to identify small molecules that interfere with the interactions. We observed that coculturing of a PD-L1-expressing cell line with a PD-1-expressing cell line induced a lysosomal-dependent degradation of PD-1. Using this assay, we revealed that the small-molecule PD-L1 inhibitor BMS1166 effectively inhibited the coculturing-induced PD-1 degradation by blocking PD-L1 exporting from ER and further glycosylation, leading to its failure to interact with PD-1 to activate its signaling. Our study provided a new method for identifying inhibitors of PD-L1/PD-1 interaction and discovered a new mechanism to block PD-L1/PD-1 signaling.
Results
Coculturing PD-L1-expressing cells with PD-1-expressing cells induced lysosomal degradation of PD-1
In order to develop a cellular assay to evaluate the interactions between PD-L1 and PD-1, we established two stable transfectant cell lines, PC-9/PD-L1, a human lung cancer PC-9 cell line overexpressing PD-L1, and Jurkat/PD-1, a human T lymphocyte Jurkat cell line overexpressing PD-1 (). The surface expressions of the two proteins were confirmed by immunofluorescence staining and flow cytometry analyses respectively (,c)).
Figure 1. Coculture of PD-L1-expressing cells with PD-1-expressing cells induced lysosomal degradation of PD-1. (a) Total cell lysates of PC-9, PC9/PD-L1, Jurkat or Jurkat/PD-1 cells were collected and processed to western blot analysis using antibodies as indicated. The anti-GAPDH antibody was used as a control for equal protein loading. (b) Confocal micrographs of PC-9 or PC9/PD-L1 cells probed with anti-PD-L1 (green) and DAPI (for nucleus, blue). Bar, 10 μm. (c) Flow cytometry analysis of PD-1 expression on cell surface of Jurkat or Jurkat/PD-1 cells after incubation with or without Alexa Fluor 488 anti-PD-1. NC, negative control. (d) PC9/PD-L1 and Jurkat/PD-1 cells were cocultured with each other for indicated time and then the whole cell lysates were processed for western blotting analysis using antibodies as indicated. The anti-GAPDH or anti-α-Tubulin antibody was used as a control for equal protein loading. The curves on the right indicated the quantitation of PD-1 or PD-L1 on the western blot relative to the control normalized by GAPDH or α-Tubulin. The relative protein levels were calculated by the gray scale scanning of AzureSpot software. (e) RT-qPCR analysis of PD-1 mRNA expression of Jurkat/PD-1 cells after coculturing with PC9/PD-L1 cells for 0 hr or 17 hr. ****p ≤ 0.001, Student’s t test. Error bars, mean±SD. (f) Jurkat/PD-1 and PC9/PD-L1 cells were treated with DMSO, 25 μM Chloroquine (CQ) or 50 nM bortezomib (BTZ) for 24 hr separately or in co-culture. The whole cell lysates were then collected and processed for western blotting analysis using antibodies as indicated. The anti-GAPDH antibody was used as a control for equal protein loading. (g) Jurkat/PD-1 cells were cocultured with PC-9 or PC9/PD-L1 for 0 hr or 10 hr, and then the whole cell lysates were collected and processed for western blotting analysis using antibodies as indicated. The anti-GAPDH antibody was used as a control for equal protein loading. (h) PC9/PD-L1 cells were pretreated with or without PD-L1 silencing RNA for 24 hr (PC9/PD-L1 (KD) or PC9/PD-L1), and then cocultured with Jurkat/PD-1 cells for another 0 hr or 17 hr. The whole cell lysates were processed for western blot using the antibodies as indicated. The anti-GAPDH or anti-α-Tubulin antibody was used as a control for equal protein loading. The histogram on the right quantified the level of PD-1 in each treatment which was normalized by GAPDH and quantified to the first treatment. ns, no significance, **p ≤ 0.005 and ****p ≤ 0.001, Student’s t test. Error bars, mean±SD.
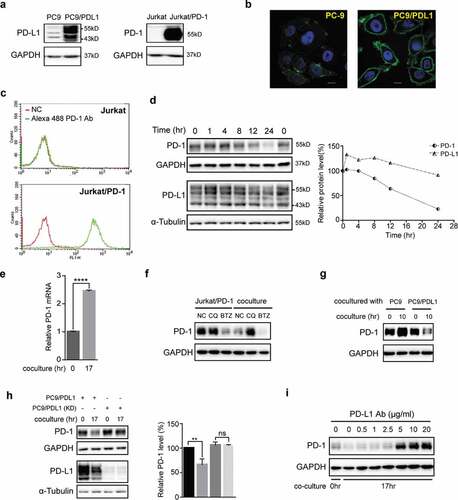
To mimic the physiological behavior of “T cells infiltrating into tumors”, the Jurkat/PD-1 cells were added to the PC9/PD-L1 cell culture after adherence to the PC9/PD-L1 cells. As shown in ), coculturing PC9/PD-L1 and Jurkat/PD-1 cells time-dependently downregulated the protein levels of PD-L1 and PD-1, especially PD-1 ()). To confirm above observations in a more physiological context, we used 12-O-Tetradecanoylphorbol-13-acetate (TPA) and ionomycin (INM) to induce the expression of the endogenous PD-1 in the normal un-transfected Jurkat cells. A similar downregulation of PD-1 was observed when we cocultured the TPA/INM-treated Jurkat cells with the PC9/PD-L1 cells (data not shown).
In order to explore the underlying mechanisms of the PD-1 reduction, we analyzed the PD-1 mRNA level in the coculture and found that the coculture did not decrease the PD-1 mRNA transcription but increased instead ()), suggesting that coculture of PC9/PD-L1 and Jurkat/PD-1 induced the degradation of PD-1 protein. Indeed, Chloroquine (CQ), a lysosome inhibitor, but not bortezomib (BTZ), a proteasome inhibitor, attenuated the coculture-induced PD-1 downregulation ()), indicating that the coculture-induced PD-1 degradation through the lysosomal pathway.
To confirm that it was PD-L1 that caused the coculture-induced PD-1 degradation, we compared the PD-1 levels in the Jurkat/PD-1 cells after coculturing with PC-9 or PC-9/PD-L1 and found that only the coculturing with PC-9/PD-L1 induced PD-1 degradation ()). Consistently, knocking-down of PD-L1 in the PC9/PD-L1 cells attenuated the coculture-induced PD-1 degradation ()). Furthermore, an anti-PD-L1 antibody (PD-L1 Ab), which blocks the PD-L1/PD-1 interaction,Citation26 was found to dose-dependently prevent the coculturing-induced PD-1 degradation ()).
Taken together, these data suggested that coculture of PC9/PD-L1 and Jurkat/PD-1 cells induced a PD-L1/PD-1 interaction-generated lysosomal degradation of PD-1, which can be used to evaluate inhibitors that target the PD-L1/PD-1 interactions.
The small-molecule PD-L1 inhibitor BMS1166 blocked PD-L1/PD-1 interaction by targeting PD-L1
Bristol-Myers Squibb has disclosed a series of small molecules that interfere with PD-L1/PD-1 interaction in the HTRF binding assay.Citation20 We tested one of their molecules, BMS1166 (Example no. 1166), in our coculture system ()). As shown in ), BMS1166 dose-dependently prevented the degradation of PD-1 after coculturing, suggesting that BMS1166 effectively inhibited the PD-L1/PD-1 interaction.
Figure 2. Small-molecule inhibitor BMS1166 blocked PD-L1/PD-1 interaction by targeting PD-L1. (a) Chemical structure of BMS1166. (b) Effects of anti-PD-L1 Ab or BMS1166 on PD-L1 and PD-1 in the coculture. PC9/PD-L1 cells were pretreated with indicated concentration of anti-PD-L1 Ab or BMS1166 for 1 hr, and then Jurkat/PD-1 cells were added in the culture for additional 0 hr or 17 hr. The whole cell lysates were collected and processed for western blotting analysis using antibodies as indicated. The anti-GAPDH antibody was used as a control for equal protein loading. The histogram below indicated the quantitation of PD-1 on the western blot relative to the control normalized by GAPDH. (c) Effects of anti-PD-L1 Ab or BMS1166 on PD-L1 and PD-1 in separately cultured cells. PC9/PD-L1 or Jurkat/PD-1 cells were treated with DMSO (NC), 10 μg/ml anti-PD-L1 Ab (Ab) or 10 μM BMS1166 for 17 hr. Then the whole cell lysates were collected and processed for western blotting analysis using antibodies as indicated. The anti-GAPDH antibody was used as a control for equal protein loading. (d) Effects of PD-L1 siRNA on PD-L1 protein. PC9/PD-L1 cells were transfected with or without PD-L1 siRNA (no. 1/2/3) for 24 hr. Total cell lysates were collected and processed for western blotting analysis using antibodies as indicated. The anti-GAPDH antibody was used as a control for equal protein loading. (e) H1975 cells were treated with indicated concentration of BMS1166 or tunicamycin (TM) for 17 hr. Total cell lysates were collected and processed for western blotting analysis using antibodies as indicated. The anti-α-Tubulin antibody was used as a control for equal protein loading. (f) H1975 cells were incubated with indicated concentration of BMS1166 or TM in the presence or absence of 100 ng/ml IFN-γ for 17 hr. Total cell lysates were collected and processed for western blotting analysis using antibodies as indicated. The anti-GAPDH antibody was used as a control for equal protein loading.
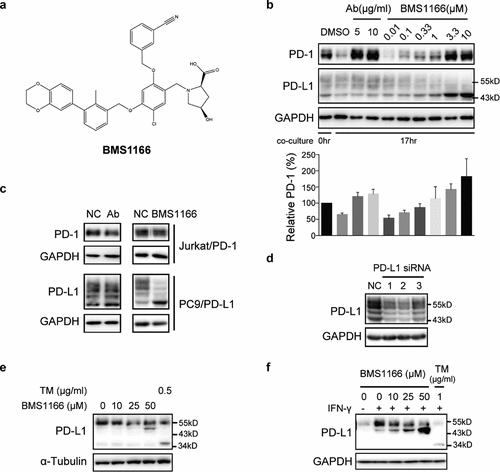
However, the effects of BMS1166 on PD-L1 differed from that of anti-PD-L1 Ab. BMS1166 reduced the higher molecular weight form but increased the lower molecular weight form of PD-L1 while the anti-PD-L1 Ab did not ()). Furthermore, neither BMS1166 nor anti-PD-L1 Ab affected the PD-1 protein in the Jurkat/PD-1 cells when the cells were cultured alone, but BMS1166 still changed the PD-L1 protein in the PC9/PD-L1 cells ()). The higher molecular weight forms of PD-L1 (~55-kDa PD-L1) were time-dependently reduced to a 43-kDa form upon BMS1166 treatment (Figure S1). Knocking down PD-L1 by three different specific siRNAs downregulated all the protein bands we detected (including the ~55-kDa and 43-kDa protein bands), confirming that they were all different forms of the PD-L1 proteins ()). Similarly, BMS1166 also changed the pattern of the constitutive and the IFN-γ-induced PD-L1 protein in the H1975 non-small-cell lung cancer cells (,f)). Taken together, these results suggested that BMS1166 acted in a different way from the anti-PD-L1 Ab in blocking the PD-L1/PD-1 interactions.
BMS1166 partially and specifically inhibited PD-L1 glycosylation
PD-L1 is a type I integral membrane glycoprotein which is extensively N-glycosylated at four conserved Asparagine residues.Citation23,Citation27 To investigate whether the alteration of PD-L1 pattern resulted from N-glycosylation inhibition by BMS1166, we treated the PC9/PD-L1 cells with tunicamycin (TM), a pan-N-linked glycosylation inhibitor, or the peptide-N-glycosidase (PNGase F), which removes the entire N-glycan structure from peptides.Citation28,Citation29 We found that all the PD-L1 protein bands on the Western blot, including the BMS1166-generated 43-kDa bands, were totally shifted to an approximately 34-kDa protein bands by either TM or the PNGase F treatment ()), close to its unmodified form,Citation27 suggesting that BMS1166 partially inhibited PD-L1 N-glycosylation.
Figure 3. BMS1166 partially and specifically inhibited PD-L1 glycosylation. (a) PC9/PD-L1 cells were treated with DMSO, 10 μM BMS1166 or 1 μg/ml tunicamycin (TM) for 17 hr, and the whole cell lysates were collected and incubated with PNGase for 1 hr at 37°C, and then processed to western blot analysis using antibodies as indicated. The anti-GAPDH antibody was used as a control for equal protein loading. (b) PC9/PD-L1 or Jurkat/PD-1 cells were treated with DMSO, 10 μM BMS1166 or indicated concentration of TM for 17 hr. Total cell lysates were collected and processed for western blot using antibodies as indicated. The anti-GAPDH antibody was used as a control for equal protein loading. (c) 4T1 cells were transfected with mPD-L1 plasmids for 24 hr and then incubated with DMSO (NC), 10 μM BMS1166 or 1 μg/ml TM overnight. Total cell lysates were collected and processed for western blot using antibodies as indicated. Anti-α-Tubulin antibody was used as a control for equal protein loading.
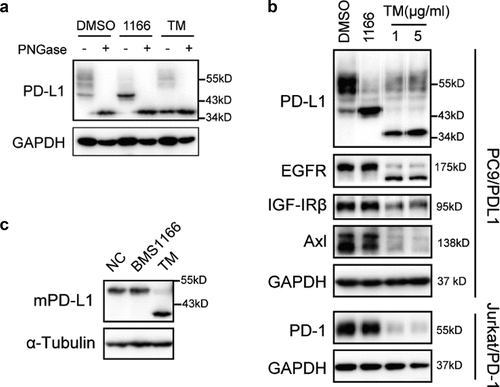
To understand the mechanisms of the partial inhibition of PD-L1 glycosylation by BMS1166, we analyzed the specificity of BMS1166 on protein glycosylation. The effect of BMS1166 was strictly human PD-L1 (hPD-L1)-specific. It did not affect other glycoproteins we tested, including the mouse PD-L1 (mPD-L1) which has 77.15% homology to the hPD-L1 (,c) and S2). Thus, BMS1166 partially but specifically inhibited the N-glycosylation of PD-L1.
BMS1166 inhibited the glycosylation of PD-L1 variants and their interactions with PD-1
N-glycosylation, except the glycosylation at amino acid N35, of PD-L1 is required for its ligand-receptor engagement.Citation25 To find out which of the glycosylation sites of PD-L1 are affected by BMS1166, we analyzed the effects of BMS1166 on the glycosylation of different PD-L1 variants, each of which had one to three of the glycosylation sites mutated,Citation23 and on their interactions with PD-1 in the coculture system ()). Mutations at the N-glycosylation sites caused varying degrees of changes in the PD-L1 glycosylation, and consequently affected their interactions with the PD-1, as indicated by the PD-1 protein levels ()). BMS1166 affected the glycosylation of all the mutated PD-L1s and almost all of their interactions with the PD-1 ()), suggesting that BMS1166 inhibited the glycosylation of all of the tested PD-L1 glycosylation sites. Among them, the interaction between the 3NQ PD-L1 mutant and PD-1 seemed to be too weak to be affected by BMS1166 (,c)).
Figure 4. BMS1166 inhibited the glycosylation of PD-L1 variants and their interactions with PD-1. (a) Schematic diagram of PD-L1 mutants used in the study. Every red “×” indicates that the asparagine was substituted to glutamine. (b) and (c) 293 cells were transfected with indicated plasmid for 24 hr. They were then cultured alone (0 hr) or cocultured with Jurkat/PD-1 cells for 17 hr (17 hr) in (b). PD-L1 WT or mutants expressing 293 cells were incubated with or without 10 μM BMS1166 for 17 hr when cocultured with Jurkat/PD-1 cells in (c). “PD-L1/TM”, 293 cells were treated with 5 μg/ml TM overnight after transfection, which in order to compare the total expression level of non-glycosylated PD-L1. Whole cell lysates were processed for western blotting analysis using antibodies as indicated. The anti-GAPDH antibody was used as a control for equal protein loading. The histogram below indicated the quantitation of PD-1 relative to the control normalized by GAPDH. *p ≤ 0.05, **p ≤ 0.005 and ***p < .001. ns, no significance. Student’s t test. Error bars, mean±SD.
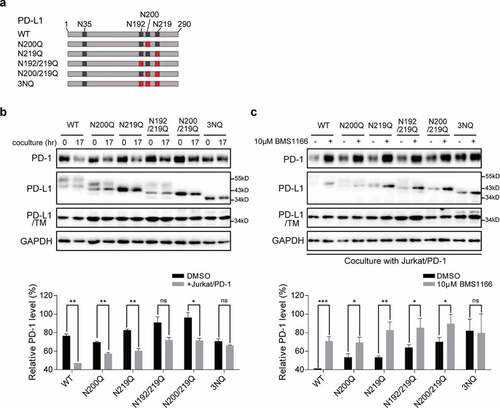
Taken together, these data suggested that PD-L1 glycosylation was important for PD-L1/PD-1 interaction and that BMS1166 specifically blocked part of the glycosylation processes of PD-L1 and inhibited its interactions with PD-1.
BMS1166 blocked PD-L1 ER-export and prevented its further glycosylation
To understand the nature of the specific and partial inhibition of PD-L1 glycosylation by BMS1166, we conducted a lectin binding assay using Concanavalin A (Con A), which specifically binds to high-mannose glycoproteins.Citation29 As shown in ), Con A preferentially bond and precipitated the BMS1166-generated 43-kDa form of PD-L1 than the higher molecular weight form or the TM-generated 34-kDa form of PD-L1, suggesting that the BMS1166-generated 43-kDa form of PD-L1 was rich in mannose. To confirm, we treated the cell lysates with Endo H, which cleaves high-mannose and some hybrid N-glycans but not the complex ones.Citation27 Upon Endo H treatment, the 43-kDa PD-L1 was converted into the 34-kDa form while the ~55-kDa forms of PD-L1 were not ()), supporting that BMS1166 blocked PD-L1 processing into complex forms.Citation27
Figure 5. BMS1166 retained PD-L1 in ER and prevented its further glycosylation without inducing ER stress. (a) PC9/PD-L1 cells were treated with DMSO, 10 μM BMS1166 or 1 μg/ml TM for 17 hr, and the whole cell lysates were collected as “input”, and part of the lysates were incubated with agarose bound with Con A overnight as “Agarose bound Con A”. All the samples were processed to western blot analysis using antibodies as indicated. (b) PC9/PD-L1 cells were treated with DMSO, 10 μM BMS1166 or 1 μg/ml TM for 17 hr, and the whole cell lysates were collected and incubated with or without Endo H enzyme for 1 hr at 37°C. All the samples were processed to western blot analysis using antibodies as indicated. The anti-GAPDH antibody was used as a control for equal protein loading. (c) and (d) PC-9 cells were transfected with PD-L1-GFP plasmids for 24 hr and then treated with DMSO or 10 μM BMS1166 for 17 hr. Fixed samples were stained with ER-tracker red (1:1000) (c) or anti-GM130 (d) and 0.1 μg/ml DAPI, and then were visualized by confocal microscopy. White arrows point to co-localization of PD-L1-GFP and ER-tracker. Bar, 10 μm. (e) PC-9 or PC9/PD-L1 cells were incubated with DMSO (NC), BMS1166 or 1 μg/ml TM for 17 hr. Whole cell lysates were processed for western blotting analysis using antibodies as indicated. Anti-β-actin antibody was used as a control for equal protein loading. (f) PC-9 or PC9/PD-L1 cells were incubated with indicated concentrations of BMS1166 for 48 hr. Cell viability was assessed by MTT assay.
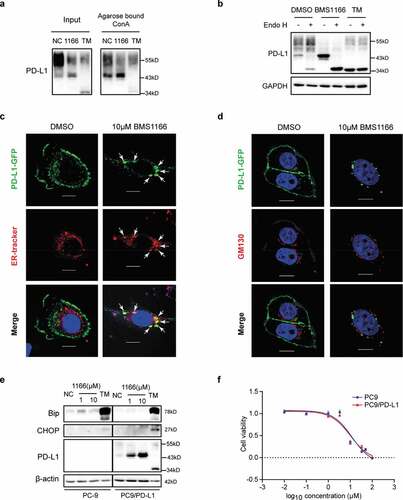
We next analyzed the effects of BMS1166 on the post-translational processing of PD-L1 using cells expressing a GFP-tagged PD-L1 (PD-L1-GFP). We found that the PD-L1-GFP was trapped in ER after BMS1166 treatment (,d)). There was no PD-L1-GFP to be found on the cell surface or in the Golgi, which was labeled by the anti-GM130 antibody,Citation30 demonstrating that BMS1166 prevented the newly-synthesized PD-L1 from transporting into Golgi to undergo further glycosylation.Citation31
As a consequence, the under-glycosylated 43-kDa form of PD-L1 in the ER underwent rapid degradation, likely through the ER-associated protein degradation (ERAD) pathway as the normal PD-L1 did (Figure S3A-C).Citation32 It had relatively short half lifetime compared to other PD-L1 forms (Figure S3A), and was ubiquitinated (Figure S3B) and degraded in proteasome, which was inhibited by bortezomib (BTZ) but not by Chloroquine (CQ) (Figure S3C). Although the binding of BMS1166 to PD-L1 trapped the protein in ER, it did not seem to induce ER stress, as indicated by the unchanged ER stress marker Bip and CHOP, and cell growth inhibition at the concentration that inhibited PD-L1 transporting (,f)), suggesting that BMS1166 might not have general cytotoxicity to cells.
Taken together, these results suggested that BMS1166 hampered the transporting of PD-L1 from ER to Golgi and its further glycosylation, leading to its degradation through proteasomes.
BMS1166 restored effector T cell function
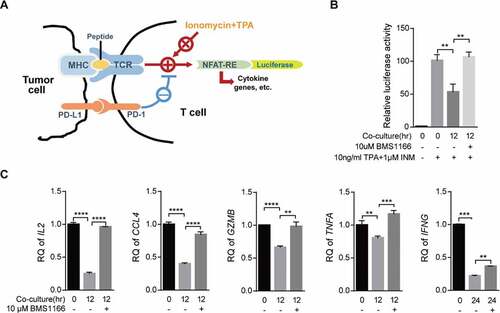
To functionally confirm that inhibition of PD-L1 exporting by BMS1166 can block the PD-L1/PD-1 signaling and activate T cells, we employed a luciferase gene reporter assay to monitor T cell activation ()). We transiently transfected the effector T cell, Jurkat/PD-1, with a luciferase reporter gene under the control of the NFAT response elements from the IL-2 gene promoter (NFAT-luc)Citation33 and stimulated the cells with a combination of INM, a Ca2+ ionophore, and TPA, an analog of diacylglycerol (DAG), which mimics the stimulation of T cells through TCR and CD28 and activates the NFAT-luc reporter gene.Citation4,Citation34 As shown in ), INM and TPA remarkably induced the luciferase expression in the Jurkat/PD-1/NFAT-luc cells. The engagement of PD-L1/PD-1 interaction by coculturing the Jurkat/PD-1/NFAT-luc cells with the PC9/PD-L1 cells suppressed the activation. However, BMS1166 completely restored the suppressed luciferase activation ()), demonstrating that BMS1166 was capable of reversing the immune suppression induced by PD-L1/PD-1 interaction, which was consistent with the previous report.Citation35 We further examined the effects of BMS1166 on the mRNA expressions of several cytokine genes (IL-2, IL2; TNF-α, TNFA; IFN-γ, INFG), chemokine genes (MIP-1β, CCL4), and cytotoxicity-related genes (Granzyme B, GZMB) downstream of TCR signaling in the INM and TPA-stimulated coculture cells and found that BMS1166 restored expressions of all of these genes suppressed by the coculture ()). Therefore, BMS1166 was able to recover T cell function by inhibiting PD-L1 processing and PD-L1/PD-1 interaction.
Figure 6. BMS1166 relieved immune suppression and activated T cell. (a) Schematic diagram of luciferase reporter gene assay. NFAT-RE, NFAT response elements. (b) and (c) PC9/PD-L1 cells were incubated with DMSO or 10 μM BMS1166 for 17 hr, and then cocultured with NFAT-luc-expressing Jurkat/PD-1 cells (b) or Jurkat/PD-1 cells (c) for 12 hr by concomitant treatment of 1 μM INM plus 10 ng/ml TPA. Cell lysates were collected to react with luciferase substrates (b) or analyzed by RT-qPCR. **, P < .01; ***, P < .001; ****, P < .0001, Student’s t test. Error bars, mean ± SD.
Discussion
We made two observations in this study. One observation was that coculturing of the PD-L1-expressing cells and the PD-1-expressing cells induced a lysosomal-dependent degradation of PD-1. This observation allowed us to use the coculture system to identify reagents that inhibited the coculture-induced PD-1 degradation and the PD-L1/PD-1 signaling. The second observation was that a small-molecule PD-L1 inhibitor BMS1166 was found to inactivate PD-L1 by blocking its exporting from ER to Golgi and inhibited PD-L1/PD-1 interaction and signaling, leading to T cell reactivation.
The PD-L1/PD-1 signaling is initiated by the interaction between PD-L1 and PD-1. The events downstream of PD-1 have been extensively studied.Citation4 However, the fate of PD-1 after PD-L1 stimulation has not been well understood. Coculture of PD-L1 expressing cells and PD-1 expressing cells have been used to evaluate therapeutic interventions targeting the PD‐1/PD‐L1 axis.Citation36 We for the first time demonstrated that PD-1 underwent internalization and a lysosomal degradation after the ligand engagement, which could be used as a novel readout to identify and evaluate inhibitors of the PD-L1/PD-1 signaling pathway. Although the function of the internalization of PD-1 is not clear at present, it is common that membrane receptors are internalized for degradation to attenuate signaling after ligand binding and signaling. It is therefore possible that the degradation of PD-1 receptor after signaling is a negative feedback of the T cell inactivation induced by the PD-L1-PD-1 interaction.
On the contrary, the ligand PD-L1 did not seem to be internalized and degraded together with PD-1 and the PD-L1 reduction was not affected by either CQ or BTZ treatment (Figure S4), suggesting that the down-regulation of PD-L1 after coculturing was different from that of PD-1 and was not a consequence of protein degradation. The function and regulation of PD-L1 need further investigation. The membrane-anchored PD-L1 is largely bound to the Jurkat/PD-1 cells after coculturing (Figure S4). It is unclear what was the function of the PD-L1 captured by the Jurkat/PD-1 cells. Some reports suggested that the acquisition of PD-L1 by T cells allowed them to induce apoptosis of adjacent T cells and therefore amplify the immunosuppressive effects.Citation37
Using the PC9/PD-L1-Jurkat/PD-1 coculture assay, we found that a small-molecule BMS1166, designed and synthesized by Bristol-Myers Squibb to disrupt the PD-L1-PD-1 interaction, blocked the coculturing-induced PD-1 degradation by an unexpected mechanism. We provide sufficient evidences to suggest that the binding of BMS1166 to PD-L1 blocked the post-translational processing of PD-L1, preventing it from directly interacting with PD-1. BMS1166 retained the newly synthesized and partially glycosylated PD-L1 in the ER and prevented its exporting from ER to Golgi and further glycosylation and maturation.
It has been reported that N-glycans play important roles in determining PD-L1 function, particularly the engagement with PD-1.Citation25,Citation38 Several small molecules, such as 2-deoxyglucose, metformin, and resveratrol, have been shown to cause abnormal PD-L1 glycosylation and induce its ER accumulation or decrease its stability.Citation39–41 Our data that the PD-L1 3NQ glycosylation mutant failed to induce PD-1 degradation in the coculture supported the essentiality of glycosylation for the function of PD-L1 ()). However, the blockade of PD-L1 ER exporting by BMS1166 was not a consequence of inhibition of PD-L1 glycosylation, because a complete loss of N-glycosylation induced by TM did not affect the ER exporting and membrane localization of PD-L1 (Figure S5). The effect of BMS1166 on PD-L1 glycosylation was the blockage of the partially glycosylated 43-kDa form of PD-L1 to be further glycosylated into the ~55-kDa form ()). The difference between the two forms was their glycan structure. The 43-kDa form was the mannose-rich type while the ~55-kDa form was the complex type (,b)), indicating that they were products of different glycosylation processes and an abnormal ER mannose trimming was possibly induced.Citation38,Citation39 Confocal microscopy analyses further confirmed that after BSM1166 treatment, PD-L1 was primarily retained in ER without entering into Golgi, where the glycans were elaborated into complex forms (,d)).Citation22 The mechanisms of how BMS1166 inhibited PD-L1 ER exporting were not clear. Skalniak et al. examined BMS1166 in co-crystallization with PD-L1 protein and found that BMS-1166 induced PD-L1 dimerization by inserting deeply into a hydrophobic cavity between two PD-L1 molecules.Citation21,Citation35 A possible PD-L1 dimer induced by BMS1166 might block its ER exporting into Golgi, therefore preventing it to be further glycosylated to become the mature ~55-kDa form. This mechanism explained the specificity of BMS1166. The specific binding and dimer formation may result in misfolding and subsequent degradation of PD-L1 (Figure S3).
The consequence of the downregulation of cell surface expression of PD-L1 in tumor cells by BMS1166 was to relieve the T cells from suppression and reactivated the effector genes downstream of PD-1Citation42 (). Among them, IL-2 is the most sensitive gene in response to PD-L1/PD-1 signaling, while IFN-γ gene is relatively insensitive.Citation43,Citation44 Our data were consistent with the previous report that IL-2 production was the most significantly recovered upon BMS1166 treatment while IFN-γ was the least affected gene ()), confirming that our coculture system reflected the physiological interactions between cancer cells and T cells.
In summary, we established a novel cellular assay for evaluating inhibitors of PD-L1/PD-1 checkpoint and demonstrated the mechanisms of a small-molecule PD-L1 inhibitor, BMS1166, in blocking the post-translational processing and maturation of PD-L1. Our study will help to identify and develop novel inhibitors of PD-L1/PD-1 signaling to combat tumor immunosuppression.
Materials and methods
Cell lines
The PC-9, PC9/PD-L1, Jurkat, Jurkat/PD-1, A375/PDL1 cells were cultured in RPMI1640 medium (Invitrogen) with 10% FBS.
The SW116-RFP cells were cultured in MEM medium (Invitrogen) with 10% FBS.
The H1975, 4T1 cells were cultured in DMEM medium (Invitrogen) with 10% FBS.
All parental cells were obtained from the American Type Culture Collection.
Reagents
The sources of chemicals, antibodies, plasmids and other reagents were as follows:
BMS1166 were provided by Prof. Dawei Ma (Shanghai Institute of Organic Chemistry, Shanghai, China). Chloroquine (# c6628, Sigma-Aldrich), bortezomib (# S1013, Selleck), anti-PD-L1 Ab for blockade (# 29E.2A3, BioLegend), PD-L1/PD-1 inhibitor 1 (#S7911, Selleck), tunicamycin (# 654380, Sigma-Aldrich), cycloheximide (Sigma-Aldrich), TPA/PMA (Sigma-Aldrich), ionomycin (Sigma-Aldrich)
Anti-GAPDH (Shanghai Kangchen), anti-PD-L1 (# PA5-20343, Life), anti-PD-1 (# PA5-20350, Life), anti-PD-L1 (# 51296, CST), anti-mouse PD-L1 Ab (# ab80276, Abcam), anti-α-Tubulin (# SC-5286, Santa Cruz Biotechnology), anti-EGFR Ab (# sc-03-G, Santa Cruz Biotechnology), anti-IGF1Rβ Ab (# 9750, CST), anti-Axl (# 4566, CST), anti-GM130 (# ab52649, abcam), anti-Bip (# 3177, CST), anti-CHOP (# 2895, CST), anti-β-actin (# P30002M, Abmart).
Plasmid: pCMV-hPD-L1, pCMV-hPD-1 and pCMV-mPD-L1-HA were purchased from Sino Biological Inc. pGL4.30-luc-NFAT (# E848A) was purchased from Promega. pCDH-PD-L1-WT, N200Q, N219Q, N192/219Q, N200/219Q or 3NQ-Flag were gifts from Prof. Mien-Chie Hung (The University of Texas, Houston, USA).
IFN-γ (Peprotech).
Coculture experiments
PC9/PD-L1 cells or other tumor cells were cultured to 90% confluence, and were then pre-treated with or without appropriate antibodies or compounds for 1 hr. Jurkat/PD-1 cells were added at the ratio of 4:1 (Jurkat/PD-1: PC9/PD-L1). For the Chloroquine and the bortezomib treatment experiments, the Jurkat/PD-1 and PC9/PD-L1 cells were cocultured at the ratio of 4:1 with DMSO, 25 μM Chloroquine, or 50 nM bortezomib for 24 hr.
Western blotting
Western blotting was performed as previously described.Citation45
Immunofluorescence analyses
Immunofluorescence analyses were performed as described previously.Citation45 For ER-tracker red staining, ER-tracker red (# C1041, Beyotime) was added in cell culture as described by the manufacturer.
RNAi and transfection
The siRNA target sequences synthesized by GenePharma were as follows:
Negative control: sense-UUC UCC GAA CGU GUC ACG UTT; antisense-ACG UGA CAC GUU CGG AGA ATT;
PD-L1 no.1 (human): sense- AG GAA GAC CUG AAG GUU CAG CAU A; antisense- AU GCU GAA CCU UCA GGU CUU CCU C ;Citation46
PD-L1 no.2 (human): sense-CCU ACU GGC AUU UGC UGA ACG CAU U; antiscense-AAU GCG UUC AGC AAA UGC CAG UAG G ;Citation46
PD-L1 no.3 (human): sense-UGA UAC ACA UUU GGA GGA GAC GUA A; antiscense-UUA CGU CUC CUC CAA AUG UGU AUC A.Citation46
SiRNAs were transfected into cells using Lipofectamine 2000 (Invitrogen), and plasmids were using Lipofectamine 3000 (Invitrogen).
Flow cytometry analysis
For surface PD-1 analysis, about 2 million cells were centrifuged and washed once with PBS, and then incubated with 10% equine serum in PBS at room temperature for 30 min. Alexa Fluor 488 anti-PD-1 (# 329935, BioLegend) was added at 1:100 dilution and incubated for another 30 min. The samples were washed twice with PBS and then analyzed by FACS Calibur (Becton Dickinson and Company).
Real-time quantitative PCR assay
Total RNA was collected for real-time quantitative PCR assay as described previously.Citation45 The sequences of primers synthesized by Life Technology are as follows:
GAPDH: Forward-ACC ACA GTC CAT GCC ATC AC; Reverse-TCC ACC ACC CTG TTG CTG;
PD-1: Forward-AAG GCG CAG ATC AAA GAG AGC C; Reverse- CAA CCA CCA GGG TTT GGA ACT G;
IL-2 (human): Forward-GTT GTT TCA GAT CCC TTT AGT TCC A; Reverse-ACA GAA CTG AAA CAT CTT CAG TGT C ;Citation47
CCL4 (human): Forward-GCT TCC TCG CAA CTT TGT GG; Reverse-GCG GAG AGG AGT CCT GAG TA ;Citation48
GZMB (human): Forward-GCA GGA AGA TCG AAA GTG CG; Reverse-TAC AGC GGG GGC TTA GTT TG ;Citation49
TNFA (human): Forward-AGC CCA TGT TGT AGC AAA CC; Reverse-GGA AGA CCC CTC CCA GAT AG ;Citation50
IFNG (human): Forward-GAA AAG CTG ACT AAT TAT TCG GTA ACT G; Reverse-GTT CAG CCA TCA CTT GGA TGA G.Citation51
Agarose bound Concanavalin A (Con A) Pulldown
Agarose bound Con A Pulldown was performed as described previously.Citation52 PC9/PD-L1 cells grown in 100 mm-dishes to 100% confluence. Agarose bound Con A (# VL-1003) was purchased from VECTOR.
Glycosylation analyses of PD-L1
Total cell lysates were collected as described in western blotting experiments.
PNGase F digestion: a mixture of 10 μl cell lysates, 10× Glyco Buffer 2, 2 μl 10% NP-40, 2 μl PNGase F (# P0704S, NEW ENGLAND BioLabs Inc.) and 4 μl H2O was incubated at 37°C for 1 hr.
Endo H digestion: the mixture with 10 μl cell lysates, 10× GlycoBuffer 3, 2 μl Endo H (# P0702S, NEW ENGLAND BioLabs Inc.) and 6 μl H2O was incubated at 37°C for 1 hr.
Statistical analyses
Data were graphically represented as mean ± SD. Statistical analyses were performed by Prism version 6.0 (GraphPad Prism Software). All experiments were replicated at least 3 times.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Ethics approval and consent to participate
Not applicable.
Authors’ contributions
Fang-fang CHEN conducted the experiments, analyzed the data and wrote the manuscript. Zheng Li and Dawei MA synthesized the compound BMS1166. Qiang YU conceived the study, analyzed the data, and wrote the manuscript.
Supplemental Material
Download ()Acknowledgments
We thank Dr. Mien-Chie Hung of The University of Texas MD Anderson Cancer Center for providing the PD-L1 plasmids.
Data availability statement
All data generated or analyzed during this study are included in this published article.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
References
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–11. doi:10.1016/j.cell.2011.02.013.
- Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–264. doi:10.1038/nrc3239.
- Wilky BA. Immune checkpoint inhibitors: the linchpins of modern immunotherapy. Immunol Rev. 2019;290(1):6–23. doi:10.1111/imr.12766.
- Boussiotis VA. Molecular and biochemical aspects of the PD-1 checkpoint pathway. N Engl J Med. 2016;375(18):1767–1778. doi:10.1056/NEJMra1514296.
- Zak KM, Kitel R, Przetocka S, Golik P, Guzik K, Musielak B, Domling A, Dubin G, Holak TA. Structure of the complex of human programmed death 1, PD-1, and its ligand PD-L1. Structure. 2015;23(12):2341–2348. doi:10.1016/j.str.2015.09.010.
- Wang X, Huang S, Zhang Y, Zhu L, Wu X. The application and mechanism of PD pathway blockade for cancer therapy. Postgrad Med J. 2018;94(1107):53–60. doi:10.1136/postgradmedj-2017-135187.
- Akinleye A, Rasool Z. Immune checkpoint inhibitors of PD-L1 as cancer therapeutics. J Hematol Oncol. 2019;12(1):92. doi:10.1186/s13045-019-0779-5.
- Zhan MM, Hu XQ, Liu XX, Ruan BF, Xu J, Liao C. From monoclonal antibodies to small molecules: the development of inhibitors targeting the PD-1/PD-L1 pathway. Drug Discov Today. 2016;21(6):1027–1036. doi:10.1016/j.drudis.2016.04.011.
- Yang J, Hu L. Immunomodulators targeting the PD-1/PD-L1 protein-protein interaction: from antibodies to small molecules. Med Res Rev. 2019;39:265–301.
- Wu L, Xiao K, Yao. W. Heterocyclic compounds as immunomodulators. In. Edited by Incyte Corporation W, DE (US). US; 2020.
- Aktoudianakis E, Cho A, Graupe M, Lad LT, Tello PAM, Medley JW, Metobo SE, Naduthambi D, Phillips BW, Simonovich. SP, et al. PD-1/PD-L1 inhibitors. In. Edited by Gilead Sciences I, Foster City, CA (US). US; 2020.
- Sasikumar PGN, Ramachandra M, Naremaddepalli SSS. Cyclic substituted-1,3,4-oxadiazole and thiadiazole compounds as immunomodulators. In: Edited by Aurigene Discovery Technologies Limited BI. Bangalore (IN): US: Aurigene Discovery Technologies Limited; 2018.
- Minghua W. Symmetric or semi-symmetric compounds useful as immunomodulators. In. Edited by Arising International LU; 2019.
- Qi C, Konkol LC, Wu L, Lajkiewicz N, He C, Xiao K, Zhu W, Li Z, Mei S, Yao W. Bicyclic heteroaromatic compounds as immunomodulators. In. Edited by Incyte Corporation W, DE (US); 2018.
- Wu L, Yu Z, Zhang F, Yao W. Pyridine derivatives as immunomodulators. In. Edited by Incyte Corporation W, DE (US); 2018.
- Li K, Tian H. Development of small-molecule immune checkpoint inhibitors of PD-1/PD-L1 as a new therapeutic strategy for tumour immunotherapy. J Drug Target. 2018;1–13.
- Musielak B, Kocik J, Skalniak L, Magiera-Mularz K, Sala D, Czub M, Stec M, Siedlar M, Holak TA, Plewka J. CA-170 - a potent small-molecule PD-L1 inhibitor or not? Molecules. 2019;24(15):2804. doi:10.3390/molecules24152804.
- Ganesan A, Ahmed M, Okoye I, Arutyunova E, Babu D, Turnbull WL, Kundu JK, Shields J, Agopsowicz KC, Xu L, et al. Comprehensive in vitro characterization of PD-L1 small molecule inhibitors. Sci Rep. 2019;9(1):12392. doi:10.1038/s41598-019-48826-6.
- Blevins DJ, Hanley R, Bolduc T, Powell DA, Gignac M, Walker K, Carr MD, Hof F, JE W. In vitro assessment of putative PD-1/PD-L1 Inhibitors: suggestions of an alternative mode of action. ACS Med Chem Lett. 2019;10(8):1187–1192. doi:10.1021/acsmedchemlett.9b00221.
- S Dm CL, Martin Scotte W, Xiaofan Z, Piyasena H, Connolly Timothy P, Ningning X, Kap-sun Y, Juliang Z, Langley DR, Tenney DJ, et al. Compounds useful as immunomodulators. In. Edited by Squibb B-M. US; 2015.
- Zak KM, Grudnik P, Guzik K, Zieba BJ, Musielak B, Dömling A, Dubin G, Holak TA. Structural basis for small molecule targeting of the programmed death ligand 1 (PD-L1). Oncotarget. 2016;7(21):30323–30335. doi:10.18632/oncotarget.8730.
- Schwarz F, Aebi M. Mechanisms and principles of N-linked protein glycosylation. Curr Opin Struct Biol. 2011;21(5):576–582. doi:10.1016/j.sbi.2011.08.005.
- Li C-W, Lim S-O, Xia W, Lee -H-H, Chan L-C, Kuo C-W, Khoo K-H, Chang -S-S, Cha J-H, Kim. T, et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat Commun. 2016;7:12632. doi:10.1038/ncomms12632.
- Lopez-Sambrooks C, Shrimal S, Khodier C, Flaherty DP, Rinis N, Charest JC, Gao N, Zhao P, Wells L, Lewis TA, et al. Oligosaccharyltransferase inhibition induces senescence in RTK-driven tumor cells. Nat Chem Biol. 2016;12(12):1023–1030. doi:10.1038/nchembio.2194.
- Li CW, Lim SO, Chung EM, Kim YS, Park AH, Yao J, Cha JH, Xia W, Chan LC, Kim T, et al. Eradication of triple-negative breast cancer cells by targeting glycosylated PD-L1. Cancer Cell. 2018;33(2):187–201e110. doi:10.1016/j.ccell.2018.01.009.
- Kataoka K, Shiraishi Y, Takeda Y, Sakata S, Matsumoto M, Nagano S, Maeda T, Nagata Y, Kitanaka A, Mizuno S, et al. Aberrant PD-L1 expression through 3ʹ-UTR disruption in multiple cancers. Nature. 2016;534(7607):402–406.
- Maher CM, Thomas JD, Haas DA, Longen CG, Oyer HM, Tong JY, Kim FJ. Small-molecule Sigma1 modulator induces autophagic degradation of PD-L1. Mole Cancer Res. 2018;16(2):243–255. doi:10.1158/1541-7786.MCR-17-0166.
- Contessa JN, Bhojani MS, Freeze HH, Rehemtulla A, Lawrence TS. Inhibition of N-linked glycosylation disrupts receptor tyrosine kinase signaling in tumor cells. Cancer Res. 2008;68(10):3803–3809. doi:10.1158/0008-5472.CAN-07-6389.
- Roth Z, Yehezkel G, Khalaila I. Identification and quantification of protein glycosylation. Int J Carbohydr Chem. 2012;2012:1–10. doi:10.1155/2012/640923.
- Nakamura N. Characterization of a cis-Golgi matrix protein, GM130. J Cell Biol. 1995;131(6):1715–1726. doi:10.1083/jcb.131.6.1715.
- Chung CY, Majewska NI, Wang Q, Paul JT, Betenbaugh MJ. SnapShot: N-glycosylation processing pathways across kingdoms. Cell. 2017;171(1):258–258e251. doi:10.1016/j.cell.2017.09.014.
- Benyair R, Ogen-Shtern N, Lederkremer GZ. Glycan regulation of ER-associated degradation through compartmentalization. Semin Cell Dev Biol. 2015;41:99–109. doi:10.1016/j.semcdb.2014.11.006.
- Wang L, Yu C, Yang Y, Gao K, Wang J. Development of a robust reporter gene assay to measure the bioactivity of anti-PD-1/anti-PD-L1 therapeutic antibodies. J Pharm Biomed Anal. 2017;145:447–453. doi:10.1016/j.jpba.2017.05.011.
- Macia F, Ga-Cz F, Im S-H, Horton HF, Byrne MC, Rao A. Transcriptional mechanisms underlying lymphocyte tolerance. Cell. 2002;109:719–731. doi:10.1016/S0092-8674(02)00767-5.
- KMZ LS, Guzik K, Magiera K, Musielak B, Pachota M, Szelazek B, Kocik J, Grudnik P, Tomala M, Krzanik S, et al. Small-molecule inhibitors of PD-1/PD-L1 immune checkpoint alleviate the PD-L1-induced exhaustion of T-cells. Oncotarget. 2017;8(42):72167–72181. doi:10.18632/oncotarget.20050.
- Tocheva AS, Lerrer S, Mor A. In vitro assays to study PD-1 biology in human T cells. Curr Protoc Immunol. 2020;130:e103.
- Gary R, Voelkl S, Palmisano R, Ullrich E, Bosch JJ, Mackensen A. Antigen-specific transfer of functional programmed death ligand 1 from human APCs onto CD8+ T cells via trogocytosis. J Immunol. 2012;188(2):744–752. doi:10.4049/jimmunol.1101412.
- Li CW, Lim SO, Xia W, Lee HH, Chan LC, Kuo CW, Khoo KH, Chang SS, Cha JH, Kim T, et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat Commun. 2016;7(12632).
- Cha JH, Yang WH, Xia W, Wei Y, Chan LC, Lim SO, Li CW, Kim T, Chang SS, Lee HH, et al. Metformin promotes antitumor immunity via endoplasmic-reticulum-associated degradation of PD-L1. Mol Cell. 2018;71(4):606–620e607. doi:10.1016/j.molcel.2018.07.030.
- Sara Verdura EC, Cortada E, Brunet J, Lopez-Bonet E, Martin-Castillo B, Bosch-Barrera J, Encinar JA, Menendez JA. Resveratrol targets PD-L1 glycosylation and dimerization to enhance antitumor T-cell immunity. Aging. 2020;12(1):8–34. doi:10.18632/aging.102646.
- Kim B, Sun R, Oh W, Kim AMJ, Schwarz JR, Lim SO. Saccharide analog, 2-deoxy-d-glucose enhances 4-1BB-mediated antitumor immunity via PD-L1 deglycosylation. Mol Carcinog. 2020;59(7):691–700. doi:10.1002/mc.23170.
- Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. 2015;15(8):486–499. doi:10.1038/nri3862.
- Fang Wei SZ, Zhengyu M, Kong H, Medvec A, Rafi Ahmed GJ, Freeman MK, Riley JL. Strength of PD-1 signaling differentially affects T-cell effector functions. PNAS. 2013;110(27):E2480–E2489. doi:10.1073/pnas.1305394110.
- Wherry EJ. T cell exhaustion. Nat Immunol. 2011;12(6):492–499. doi:10.1038/ni.2035.
- Chen F, Song Q, Yu Q. Axl inhibitor R428 induces apoptosis of cancer cells by blocking lysosomal acidification and recycling independent of Axl inhibition. Am J Cancer Res. 2018;8:1466–1482.
- Hobo W, Maas F, Adisty N, de Witte T, Schaap N, van der Voort R, Dolstra H. siRNA silencing of PD-L1 and PD-L2 on dendritic cells augments expansion and function of minor histocompatibility antigen-specific CD8+ T cells. Blood. 2010;116(22):4501–4511. doi:10.1182/blood-2010-04-278739.
- Rahman M, Nara H, Onoda T, Araki A, Li J, Hoshino T, Asao H. Cloning and characterization of an isoform of interleukin-21. FEBS Lett. 2007;581(21):4001–4009. doi:10.1016/j.febslet.2007.07.034.
- Marischen L, Englert A, Schmitt AL, Einsele H, Loeffler J. Human NK cells adapt their immune response towards increasing multiplicities of infection of Aspergillus fumigatus. BMC Immunol. 2018;19(1):39. doi:10.1186/s12865-018-0276-6.
- Liu JY, Li F, Wang LP, Chen XF, Wang D, Cao L, Ping Y, Zhao S, Li B, Thorne SH, et al. CTL- vs Treg lymphocyte-attracting chemokines, CCL4 and CCL20, are strong reciprocal predictive markers for survival of patients with oesophageal squamous cell carcinoma. Br J Cancer. 2015;113(5):747–755. doi:10.1038/bjc.2015.290.
- Feldstein AE, Werneburg NW, Canbay A, Guicciardi ME, Bronk SF, Rydzewski R, Burgart LJ, Gores GJ. Free fatty acids promote hepatic lipotoxicity by stimulating TNF-alpha expression via a lysosomal pathway. Hepatology. 2004;40(1):185–194. doi:10.1002/hep.20283.
- Bedi A, Chang X, Noonan K, Pham V, Bedi R, Fertig EJ, Considine M, Califano JA, Borrello I, Chung CH, et al. Inhibition of TGF-beta enhances the in vivo antitumor efficacy of EGF receptor-targeted therapy. Mol Cancer Ther. 2012;11(11):2429–2439. doi:10.1158/1535-7163.MCT-12-0101-T.
- Guo H, Kuang S, Song QL, Liu M, Sun XX, Yu Q. Cucurbitacin I inhibits STAT3, but enhances STAT1 signaling in human cancer cells in vitro through disrupting actin filaments. Acta Pharmacol Sin. 2018;39(3):425–437. doi:10.1038/aps.2017.99.