
ABSTRACT
Prostaglandin E2 (PGE2), an arachidonic acid pathway metabolite produced by cyclooxygenase (COX)-1/2, has been shown to impair anti-tumor immunity through engagement with one or more E-type prostanoid receptors (EP1-4). Specific targeting of EP receptors, as opposed to COX-1/2 inhibition, has been proposed to achieve preferential antagonism of PGE2–mediated immune suppression. Here we describe the anti-tumor activity of MF-766, a potent and highly selective small-molecule inhibitor of the EP4 receptor. EP4 inhibition by MF-766 synergistically improved the efficacy of anti-programmed cell death protein 1 (PD-1) therapy in CT26 and EMT6 syngeneic tumor mouse models. Multiparameter flow cytometry analysis revealed that treatment with MF-766 promoted the infiltration of CD8+ T cells, natural killer (NK) cells and conventional dendritic cells (cDCs), induced M1-like macrophage reprogramming, and reduced granulocytic myeloid-derived suppressor cells (MDSC) in the tumor microenvironment (TME). In vitro experiments demonstrated that MF-766 restored PGE2-mediated inhibition of lipopolysaccharide (LPS)-induced tumor necrosis factor (TNF)-α production in THP-1 cells and human blood, and PGE2-mediated inhibition of interleukin (IL)-2-induced interferon (IFN)-γ production in human NK cells. MF-766 reversed the inhibition of IFN-γ in CD8+ T-cells by PGE2 and impaired suppression of CD8+ T-cells induced by myeloid-derived suppressor cells (MDSC)/PGE2. In translational studies using primary human tumors, MF-766 enhanced anti-CD3-stimulated IFN-γ, IL-2, and TNF-α production in primary histoculture and synergized with pembrolizumab in a PGE2 high TME. Our studies demonstrate that the combination of EP4 blockade with anti-PD-1 therapy enhances antitumor activity by differentially modulating myeloid cell, NK cell, cDC and T-cell infiltration profiles.
Introduction
Cancer immunotherapy, which uses the body’s own immune system to recognize and attack tumors, is revolutionizing the standard of cancer care.Citation1 Immunotherapeutic strategies, including enhancement of co-stimulators, blockade of inhibitory immune checkpoints, cancer vaccines, oncolytic virotherapy, and adoptive cell therapies (ACT), have been shown to be effective anti-cancer therapeutic modalities in clinical trials.Citation2,Citation3 The approval of anti-cytotoxic T-lymphocyte–associated protein 4 (CTLA-4) antibody and anti-programmed cell death protein 1/programmed cell death ligand 1 (PD-1/PD-L1) antibodies by the US Food and Drug Administration (FDA) in the past few years has enabled significant breakthroughs in the field.Citation3,Citation4 However, immune checkpoint inhibitors (ICIs) only benefit a small subset of patients and the majority of patients do not achieve durable responses. High tumor heterogeneity and immunosuppressive mechanisms within the tumor microenvironment (TME) remain critical barriers to effective anti-cancer therapies.Citation5–7 Therefore, it is important to develop novel drugs and combination strategies to reprogram the TME and overcome primary and acquired resistance to immunotherapy.
One of the major hallmarks of immunotherapy resistance is the accumulation of metabolic immunosuppressive molecules, such as prostaglandin E2 (PGE2) in the TME.Citation8 PGE2 is a bioactive lipid metabolite derived from arachidonic acid by the actions of several key limiting enzymes, including cyclooxygenases (COXs; constitutively active COX-1 and inducible COX-2) and PGE synthases.Citation9 PGE2 has been shown to play a key role in inflammation and tumorigenesis.Citation9–11 Overexpression of COX-2 and increased PGE2 production have been reported in human colorectal cancer (CRC),Citation12 pancreatic cancer,Citation13 non-small–cell lung cancer (NSCLC),Citation14,Citation15 breast,Citation16 prostate,Citation17 liver,Citation18 bladder,Citation19 stomach, and esophageal cancers.Citation20 Notably, PGE2 has demonstrated pro-tumorigenic activity by promoting tumor cell proliferation, migration, invasiveness, and tumor-associated angiogenesis,Citation8,Citation10 increasing immune-suppressor function of myeloid-derived suppressor cells (MDSC) and tumor-associated macrophages (TAMs),Citation21 and decreasing natural killer (NK) cellCitation22 and T-cell effector function.Citation23 PGE2 affects target cells through its binding to a family of G protein-coupled receptors: E-type prostanoid receptors 1–4 (EP1-4).Citation24 EP2 and EP4, which increase intracellular cAMP and activate the protein kinase A pathway upon binding to PGE2, have been shown to exhibit a strong association with cancer development.Citation25,Citation26 Disruption of COX-2/PGE2 signaling using non-steroidal anti-inflammatory drugs (NSAIDs) and specific COX-2 inhibitors has been tested in the clinic since the early 2000s.Citation27,Citation28 Inhibition of COX-2/PGE2 was shown to decrease the incidence of colorectal adenoma,Citation29 improve chemotherapy resistance in bladder cancer, metastatic breast cancer, NSCLC, and cervical cancer, and enhance sensitization of other anti-tumor drugs in renal cell carcinoma (RCC) and melanoma.Citation30–32 Additionally, the combination of COX-2 inhibitors and anti-PD-1 monoclonal antibodies has been shown to improve anti-tumor activity in preclinical models of cancer.Citation33,Citation34 However, dose-limiting toxicities and adverse effects, such as gastric ulcers and myocardial infarction have been observed.Citation35–37 Specific targeting of PGE2 signaling via antagonism of its receptors may therefore provide a potentially safer approach to relieve the immunosuppressive effects of PGE2 in the TME.
EP4 is mainly expressed on tumor cells, myeloid cells, and lymphocytes, and has been reported to impair innate and adaptive anti-tumor responses.Citation38 Preclinical studies in breast and bladder models demonstrate that antagonism of EP4 could limit tumor growth and metastasis.Citation38–40 In vitro studies suggest that EP4 is a common mechanism to negatively regulate interferon (IFN)-γ production and cytolytic effector (CTL) functions in both T cellsCitation39 and NK cells.Citation22 Moreover, myeloid cells such as MDSCs and TAMs within the TME promote tumor development, exhibit immunosuppressive activity, and can decrease the efficacy of ICIs.Citation40 Blockade of EP4 by E-7046 impairs tumor-promoting MDSC differentiation, M2 macrophage polarization, and T-regulatory-cell–derived immunosuppression in both preclinical and phase I clinical studies.Citation21,Citation41 Therefore, EP4 represents an attractive immunotherapy target for functionally reprogramming suppressive components in the TME.
In this study, we evaluated the immunomodulatory effects of MF-766, a potent and selective EP4 small molecule inhibitor,Citation42 both in vitro and in vivo. MF-766 reverted PGE2-suppression of type 1 cytokine production (such as IFN-γ and tumor necrosis factor [TNF]-α) in purified lymphocytes and myeloid cells. In syngeneic tumor mouse models, we observed potent anti-tumor activity of MF-766 and anti-PD-1 in combination, which was characterized by increased infiltration of CD8+ T-cell and NK cells, and decreased infiltration of MDSCs in the TME, respectively. Moreover, in purified CD8+ T cells and primary human tumor histocultures, MF-766 synergized with pembrolizumab to increase type 1 cytokines in a PGE2 high TME, suggesting its potential use in combination with anti-PD-1 in the clinic.
Materials and methods
Reagents and antibodies
EP4 antagonist MF-766 was generated by Merck & Co., Inc (Kenilworth NJ, USA).Citation42 For in vitro studies, EP4 antagonist was dissolved in dimethyl sulfoxide (DMSO, Sigma-Aldrich, St. Louis, MO) to form a 10 mM stock solution and further diluted for specific experiments. MF-766 was formulated with 10% Tween 80 for in vivo experiments. Pembrolizumab, a humanized monoclonal immunoglobulin (Ig)G4 antibody against PD-1 and IgG4 isotype, was generated by Merck & Co., Inc., Kenilworth NJ, USA. mDX400, a murine version of a rat anti-mouse PD-1 antibody with a mutated D265A mouse IgG1 Fc and the isotype control antibody mouse anti-hexon IgG1 27F11 were generated by Merck & Co. Inc., Kenilworth NJ, USA. Lipopolysaccharide (LPS) (Sigma-Aldrich, St. Louis, MO) and PGE2 (Cayman Chemical, Ann Arbor, MI) were prepared and used according to the manufacturer’s instructions. Anti-human CD3 (Clone OKT3), anti-human CD28 (Clone CD28.2), and eBioscienceTMFoxp3/Transcription Factor Staining Buffer Set were from Thermo Fisher Scientific (Waltham, MA). Human granulocyte macrophage-colony stimulating factor (GM-CSF) and human interleukin (IL)-4 were from R&D Systems (Minneapolis, MN, USA). Mouse GM-CSF, mouse IL-4, and human IL-2 were from PeproTech (Rocky Hill, NJ). Cell staining buffer was from Biolegend (San Diego, CA). Brilliant stain buffer and fixable viability dyes (FVS510 or FVS780) were from BD Biosciences (San Diego, CA). All flow cytometry antibodies were purchased from Biolegend or BD Biosciences (San Diego, CA).
Cell line and primary cells
THP-1 (Monocyte), CT26 (BALB/c mouse colon adenocarcinoma), EMT6 (BALB/c mouse mammary carcinoma), and 4T1 (BALB/c mouse mammary carcinoma) cell lines were obtained from American Type Culture Collection (ATCC, Manassas, VA). All cell lines were verified as being free of microbial contamination using an IMPACT I PCR test and genetically authenticated via CellCheck (IDEXX Laboratories) and maintained in standard cell culture media according to ATCC. Cells growing in an exponential growth phase with greater than 95% viability were harvested and counted for tumor inoculation.
Cryopreserved human PBMCs were either directly purchased from HemaCare (CAT# PB009C-1, Northridge, CA) or were prepared by density centrifugation with Ficoll from Leukopaks (HemaCare, CAT# PB001LCLP, Northridge, CA). All the human studies were approved by the Western Institutional Review Board (WIRB)–compliant IRB at Merck & Co., Inc., Kenilworth, NJ, USA.
Mice
Female mice 6 to 8 weeks of age were purchased from The Jackson Laboratory (C57BL/6 J strain) (Bar Harbor, ME) or Taconic (BALB/cAnN) (New York NY, USA). All animal procedures/protocols were approved by the Institutional Animal Care and Use Committee of Merck & Co., Inc. (Kenilworth, NJ, USA) in accordance with the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) guidelines.
In vivo experiments
Mice were injected subcutaneously with CT26 (0.5 x 106), EMT6 (0.5 x 106), or 4T1 (0.3 x 106) cells in the lower right flank. MF-766 and anti-PD-1 treatment started when the average tumor size reached 100 mm3 (75–125 mm3). Any tumor growing intradermally (ID) or intramuscularly (IM) was not enrolled. MF-766 30 mg/kg or its vehicle control was administered orally daily for 21 days (QDx21). mDX400 or its isotype control was administered intraperitoneally at 10 mg/kg, every 4 days for 4 doses (Q4Dx4). Tumor volume was calculated using the formula 0.5 × length × width2, where length was the longer dimension. The relative tumor volume (RTV) was calculated using the following formula: RTV = (tumor volume on measured day)/(tumor volume on day 0). The tumor growth inhibition ratio (TGI, %) was calculated using the following formula: TGI (%) = [1 − (RTV of the treated group)/(RTV of the control group)] × 100 (%). Tumor volumes were monitored for 60 days post dose initiation. The rechallenge study in the CT26 model used the complete response (CR) mice from the previous study, and the same number/age Balb/c naïve mice were inoculated with CT26 tumor cells (0.5 x 106) in 0.1 mL serum-free Dulbecco’s modified eagle medium (DMEM) for tumor development. Tumor measurement was conducted until day 22 (day 111 for CR mice from their first CT26 inoculation). Tail blood or facial vein blood was collected from mice in satellite groups for pharmacokinetic (PK) analysis as needed. PK parameter measurement of MF-766 was described in a previous publication.Citation42–44
Ex vivo tissue analysis
For analysis of immune cell population infiltration, fresh mouse tumors were first weighed and then enzymatically digested using a tumor dissociation kit (Miltenyi Biotec, Bergisch Gladbach, Germany) by gentleMACS. The mixture was then filtered through 70-μm cell strainers to generate single-cell suspensions. The single-cell suspension from tumors was then washed and counted before use.
Flow cytometry
Two to 3 million cells were incubated with fixable viability dyes (FVS510 or FVS780), followed by blocking with FcγIII/RII-blocking antibody (BD Biosciences, San Diego, CA) for 10 minutes on ice. Cells then were stained with fluorochrome-conjugated surface antibodies, which were prepared in cell staining buffer and brilliant stain buffer (1:1) for 30 minutes on ice. The surface antibody cocktail included CD45 (Clone 30-F11), CD11b (Clone M1/70), CD8 (Clone 53–6.7), Ly6G (Clone 1A8), F4/80 (Clone BM8), Ly6C (Clone AL-21), I-A/I-E (Clone M5/114.15.2), CD11c (Clone N418), CD3e (Clone 145–2 C11), CD4 (Clone GK1.5), CD49b (Clone DX5), CD335 (Clone 29A14), CD25 (Clone PC61), PD-1 (Clone RMP1-30), and PD-L1 (Clone MIH5). For detection of intracellular expression of FOXP3, cells were permeabilized and fixed with eBioscience™ Foxp3/Transcription Factor Staining Buffer Set (Thermo Fisher Scientific, Waltham, MA) according to the manufacturer’s instructions and then incubated with FOXP3 antibody (Clone MF-14) for 30 minutes. Stained samples were acquired on a BD Fortessa cytometer with DIVA software (BD Biosciences, San Diego, CA), and the data were analyzed using FCS Express (De Novo Software, Pasadena, CA). In flow cytometry studies, gates were drawn on CD45+ leukocytes by doublet exclusion (forward scatter height versus forward scatter area; side scatter height versus side scatter area) and dead cell exclusion using fixable viability dyes (FVS510 or FVS780). Gates were then drawn on CD3+ T cell population, followed by CD4+ and CD8+ populations. CD3−CD335+CD49b+ gated on CD45+ population defined NK cells. Further myeloid populations including gMDSC/neutrophils (CD45+CD11b+Ly6G+), macrophage (CD45+CD11b+MHCII+ F4/80+ Ly6C−) and DC (CD45+CD11c+MHCII+ F4/80− Ly6C−) were drawn. Fluorescence minus one, unstained, and isotype controls were included for assessment of surface and intracellular proteins. The percentage of each cell subset within viable CD45+ cells was calculated.
PGE2 measurement using high-performance liquid chromatography in tumors
Mouse tumors were flash-frozen and homogenized in three volumes per weight of artificial plasma (4 g bovine albumin in 100 mL PBS) for analysis. Known concentrations of PGE2 were spiked to artificial plasma to prepare calibration standards and QCs. A 200 µL aliquot of acetonitrile containing 1.5 ng/mL of d4-PGE2 as internal standard (IS) was added to each 50 μL sample (calibration standard, QCs, tumor lysate) to precipitate the proteins. Samples were mixed by vortex for homogeneity, and then subjected to centrifugation at 3000 rpm (1460 g) for 5 minutes. The supernatant (200 µL) was transferred into clean 96-well deep-well plates and injected into the LC-MS/MS system. Detection was performed using a triple quadrupole tandem mass spectrometer (API5000 Applied Biosystems) equipped with an electrospray interface (ESI), operating in negative ionization mode. Separation was performed on a Waters Acquity HSS T3 (2.1 mm x 50 mm, 1.8 μm) column at room temperature with an injection volume of 5 μL. The mobile phase consisting of solvent A (0.1% formic acid in water) and solvent B (0.1% formic acid in acetonitrile) was delivered at a flow rate of 750 μL/min. Chromatographic data were collected and integrated by MultiQuant 3.0.1 data analysis program. Peak area ratios of the analyte to IS were utilized for the construction of the calibration curve. A weighting of 1/x2 (least-squares linear regression analysis, where x is the concentration of a given standard) was used for curve fit. Concentrations in unknown samples were calculated from the best-fit equation (y = mx + b), where y is the peak area ratio. The regression equation for the calibration curve was also used to back-calculate the measured concentration at each quality control level, and the results were compared with the theoretical concentration to obtain the accuracy expressed as a percentage of the theoretical value. Accuracy was defined as the degree of deviation of the determined value from the nominal value: [(measured value − nominal value)/nominal value] × 100.
Gene expression analysis by real-time quantitative PCR
Total RNA was isolated from cells using the MagMAX mirVana total RNA isolation kit or Arcturus PicoPure RNA Isolation Kit, according to the manufacturer’s instructions (Thermo Fisher Scientific, Waltham, MA). DNase-treated total RNA was reverse transcribed using QuantiTect Reverse Transcription (Qiagen, Germantown, MD) according to the manufacturer’s protocol. A 20X primer mix was purchased from Thermo Fisher Scientific (Waltham, MA). Gene-specific pre-amplification was performed on 10 ng cDNA according to the manufacturer’s instructions (Fluidigm Biomark, Foster City, CA). Real-time quantitative PCR was then performed on the Fluidigm Biomark using 20X Taqman primer assays (Thermo Fisher Scientific, Waltham, MA) with Taqman Fast Universal PCR Master Mix with no AmpErase UNG. Samples and primers were run on either a 48.48 Dynamic Array or 96.96 Dynamic Array per the manufacturer’s instructions (Fluidigm, Foster City, CA). Ubiquitin concentrations were measured in a separate reaction and used to normalize the data by the ΔCt method. (Using the mean cycle threshold value for ubiquitin and the gene of interest for each sample, the equation 1.8 ^ (Ct ubiquitin minus Ct gene of interest) x 104 was used to obtain the normalized values.) The average fold change (FC) of treated vs untreated samples was calculated, and nominal t test analysis was performed to determine P values. For the evaluation of the reference signatures in the human dataset, the results shown are in whole or part based on data generated by the TCGA Research Network: https://www.cancer.gov/tcga. The heatmap compares pairwise Spearman correlation coefficients evaluated in solid cancer tumor samples after adjusting for cancer type. Gene expression in individual samples was based on log10 (0.01+ FPKM) for protein-coding genes after global normalization by within-sample 75th percentile.
Mouse tumors were excised and snap-frozen in liquid nitrogen and stored at −80 C until RNA isolation. For tumor tissue RNA isolation, organs were homogenized into RNA STAT-60 (Tel-Test Inc., Friendswood, TX) using a polytron homogenizer, then total RNA extracted according to the manufacturer’s instructions. After isopropanol precipitation, total RNA was re-extracted with phenol:chloroform:isoamyl alcohol (25:24:1) (Sigma-Aldrich, St. Louis, MO) using phase-lock light tubes (Thermo Fisher Scientific, Waltham, MA). RNA sequencing was performed using the Truseq stranded total RNA RiboZero library preparation kit (Catalog #: RS-122-2201) according to the manufacturer’s instructions (Illumina, San Diego, CA). The resulting cDNA libraries were sequenced on an Illumina (HiSeqTM 4000) using a 50-base paired-end run. Cleaned reads were aligned to the Mouse.B38 genome reference using the Omicsoft Aligner (Qiagen, Germantown, MD) with a maximum of two allowed mismatches. Gene level raw counts, and FPKM, were determined by the OSA algorithm as implemented in Omicsoft Array Studio (version 10.0.1.118) and using Ensembl.R93 gene models. Approximately 90% of reads across all samples mapped to the reference genome (corresponding to between 40 and 160 million reads).
Human tumor histoculture
Fresh human tumor samples were procured from various commercial sources within 24 hours of surgery. Because all tissue samples used in this study were existing, de-identified samples, the research was exempt from seeking Internal Review Board approval under §46.101, b. After trimming off visible fat tissue, the tumor tissues were cut into thin (~400 µm) slices and cultured at the air–liquid interface at 37°C in a 5% CO2 incubator in DMEM-supplemented 10% fetal bovine serum, 100 µg/mL penicillin, and 100 µg/mL streptomycin. Alternatively, single-cell suspensions of tumor samples were prepared by digesting finely minced tumor pieces at 37°C in DMEM with 100 mg/mL type 1 collagenase and 10,000 U/mL DNase I, followed by filtration through a 70-µm strainer, and washed in DMEM complete medium. A total 0.1 × 106 cells/well were used for dissociated tumor culture. All the human sample protocols comply with WIRB, Western Institutional Review Board at Merck & Co., Inc. (Kenilworth, NJ, USA).
Natural killer cell activation
Human NK cells purchased from HemaCare were pretreated either with vehicle control (DMSO) or MF-766 for 1 h and then stimulated with 50 ng/mL IL-2 with and without PGE2 for an additional 18 hours at 37°C, 5% CO2. Cell culture conditioned media were then collected for cytokine measurements using Meso Scale Discovery kits according to the manufacturer’s protocol.
In vitro activity of MF-766 in suppressive myeloid cell and T-cell co-culture assay
CD14+ monocytes were purified from frozen human peripheral blood mononuclear cells (PBMCs) isolated from healthy volunteer donors using anti-CD14 microbeads according to the manufacturer’s instructions (Stemcell Technologies, Vancouver, BC, Canada). The purified monocytes were cultured in complete cell culture medium (RPMI-1640 with 10% heat-inactivated fetal bovine serum, 2 mM L-glutamine, 50 units/mL each of penicillin/streptomycin) in the presence of human GM-CSF (20 ng/mL), human IL-4 (500 U/mL), PGE2 (10 nM), and MF-766 (10, 1, 0.1 and 0.01 μM) in 6-well low binding plates at 2 × 106 cells/well for 7 days at 37°C. The differentiated suppressive myeloid cells as described above were then co-cultured with purified autologous CD8+ T cells (1 × 105 cells/well, Stemcell Technologies, Vancouver, BC, Canada) at a 1:4 ratio in 96-well U-bottom plates in the presence of MF-766 (10, 1, 0.1, and 0.01 µM) or vehicle control. T-cell proliferation was induced with anti-CD3/CD28 beads (Thermo Fisher Scientific, Waltham, MA) and 100 U/mL human IL-2 and incubated at 37°C for 3 days. Meso Scale Discovery kits were used to determine IFN-γ concentrations in culture supernatants.
In vitro activity of MF-766 and pembrolizumab combination in human CD8+ T cells
Autologous CD8+ T cells were purified from frozen human PBMCs isolated from healthy volunteer donors using a CD8+ T-cell separation kit according to the manufacturer’s instructions (Stemcell Technologies, Vancouver, BC, Canada). The purified CD8+ T cells (1 × 105 cells/well) were cultured in complete cell culture medium in 96-well plates, in the presence of titrated concentrations of MF-766 starting at 3 μm, 3-fold dilutions ± PGE2 (10 nM). Anti-PD-1 antibody pembrolizumab or human IgG4 isotype control was added at a single concentration of 20 ug/mL. T-cell activation was induced with anti-CD3/CD28 beads (Thermo Fisher Scientific, Waltham, MA) and 100 U/mL IL-2 and incubated at 37°C for 24 hours. Meso Scale Discovery kits were used to measure IFN-concentrations in culture supernatant according to the manufacturer’s protocol.
THP-1 and ex vivo whole blood assays
THP-1 cells were obtained from ATCC (Manassas, VA). Fresh healthy whole blood samples were collected in sodium heparin BD vacutainers (Becton, Dickinson and Company, Franklin Lakes, NJ) on assay day while CRC patient blood (HemaCare, Northridge CA, USA) was collected 1 day before assay and shipped to the laboratory overnight. The fresh blood was obtained from normal healthy donors who provided their written informed consent and were enrolled in the WIRB-compliant Blood Donor Program at Merck & Co., Inc, Boston, MA, USA. THP-1 cells or the blood samples were pre-treated with MF-766 for 1 hour before being stimulated with the final concentration of 1 μg/mL LPS in the presence or absence of PGE2 for 18 to 20 hours. After incubation, samples were centrifuged, and the supernatant was collected for TNF-α determination using Meso Scale Discovery kits.
Dendritic cell activation
Bone marrow (BM) cells were flushed from the femurs of C57/BL6 mice and differentiated in
complete cell culture medium (RPMI) with recombinant 20 ng/mL mouse GM-CSF and 20 ng/mL mouse IL-4 for 6 days to generate immature dendritic cells (imDCs). Human dendritic cells (DCs) obtained from HemaCare and mouse imDCs generated in-house were pretreated with MF-766 for 1 hour and then stimulated with 1 μg/mL LPS in the presence or absence of PGE2. After 18 to 24 hours of incubation, media were collected for cytokine measurement using Meso Scale Discovery kits.
Cytokine analysis using Meso Scale Discovery kits
Secreted cytokines from T lymphocytes, NK cells, THP-1 cells, whole blood, and DCs in the supernatant were determined by a V-plex pro-inflammatory panel 1 mouse or human kit from Meso Scale Discovery (Gaithersburg, MD, USA). The mouse kit measures IFN-γ, IL-1β, IL-2, IL-4, IL-5, IL-6, IL-10, IL-12p70, KC/GRO, and TNF-α, while the human kit measures IFN-γ, IL-1β, IL-2, IL-4, IL-6, IL-8, IL-10, IL-12p70, IL-13, and TNF-α. Meso Scale Discovery plates were analyzed on the MS2400 imager (Meso Scale Discovery, Gaithersburg, MD, USA). All assays and analyses were performed according to the manufacturer’s instructions.
Statistical analysis: Data were plotted as mean±SD or mean±SEM. Paired two-tailed Student’s t-test or one-way ANOVA with Dunnett test (compared to vehicle control or MF-766 untreated group) was applied to assess the statistical significance of differences between multiple treatment groups. Data analysis was performed using GraphPad Prism (GraphPad Software, La Jolla, CA). Differences were considered at P values <.05, *P < .05, **P < .01, ***P < .001, and ****P < .0001.
Results
EP4 gene expression is prevalent in many types of human tumors
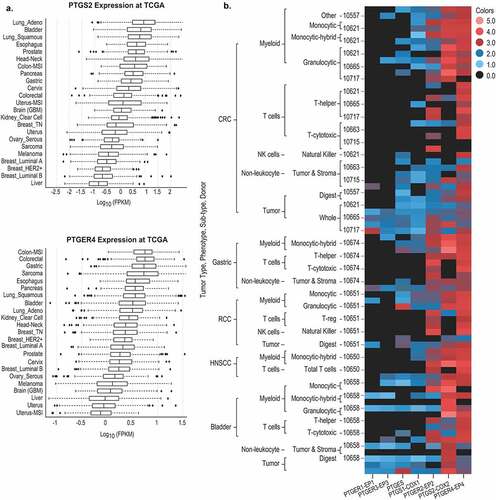
Previous studies have demonstrated that COX-2/PGE2 signaling contributes to tumor invasiveness in a variety of human cancers.Citation8 In multiple tumors, PTGER4 appears to be one of the most highly correlated COX-2/PGE2 pathway genes associated with resistance to anti-PD-1,Citation45–47 suggesting that this isoform may be particularly important in driving immunomodulatory COX-2/PGE2 signaling in the TME. To evaluate whether this finding is more broadly relevant across human cancers, PTGS2 and PTGER4 mRNA expression was assessed using RNA sequencing data from The Cancer Genome Atlas (TCGA). Both PTGS2 and PTGER4 were expressed in the majority of human cancer types, with higher expression in colorectal, esophageal, pancreatic, bladder, and lung tumors compared to other tumor types such as sarcoma and melanoma (). Tumor cell subset analysis revealed that COX-2 expression correlated with granulocytic myeloid-derived suppressor cell (gMDSCs) and mast cell infiltration in most tumors (Fig S1A, B). Examination of major immune cell populations, including NK cells, neutrophils, macrophages, monocytes, stromal cells, CD8+ T cells, and tumor cells revealed that EP4 was broadly expressed in multiple cell types across different tumors (Fig S1A, B). These results suggest that EP4 antagonism may exert pleiotropic effects within the TME with a broader mechanism of action (MOA) than previously appreciated. Since EP4 is expressed on immune and tumor cells (Fig S1A, B), high concentrations of PGE2 resulting from COX-1/2 or mouse PGE synthase-1 overexpression may inhibit the infiltration and activation of lymphocytes within the TME. To study expression patterns of COX-1/2, microsomal PGE synthase, and EP1-4, we conducted analysis in various immune cell populations sorted from healthy PBMC, CRC, RCC, gastric, head and neck squamous cell carcinoma (HNSCC), and bladder cancer. The comparative analysis demonstrated that EP4 is widely expressed in immune and tumor cells, whereas expression of COX-2 and EP2 is variable across tumor types (). This expression pattern was maintained in PBMC samples from healthy human volunteers although neutrophils and B cells exhibited lower expression levels of EP2 (Fig S1C). Collectively, these observations suggest that EP4 represents a therapeutically relevant oncology target within the PGE2/EP4 signaling pathway.
Figure 1. Gene expression analysis of EP4 and COX-2 in tumor types
Previous studies have demonstrated that EP4 antagonists such as MF-498,Citation43 MF-310,Citation44 and MF-766Citation42 can attenuate joint inflammation in experimental rodent models of arthritis. MF-766 represents a second-generation EP4 antagonist and is characterized by high potency and selectivity for the treatment of inflammatory pain.Citation42 The molecular structure of MF-766 is depicted in . The binding affinity, selectivity, potency in whole-cell assays, in-vitro profile, and PK properties of MF-766 have been previously described.Citation42 Although MF-766 demonstrated comparable maximum efficacy to celecoxib and NSAIDs in a rat AIA model of chronic inflammation, it was greater than 200-fold more potent when compared to other COX-2 inhibitors, including celecoxib and MF-tricyclic.Citation42,Citation48 We evaluated the PK properties of MF-766 in mice (). As shown, MF-766 exhibited good elimination kinetics (clearance after 24 hours), efficient and high oral bioavailability (Cmax at 15.03 ± 4.37 mM for 30 mpk at 0.5 hours; Cmax 24.05 ± 6.01 mM for 100 mpk at 0.5 hours) and high systemic exposure (AUC at 57.55 ± 10.75 mM*h for 30 mpk and AUC at 255.10 ± 97.3 mM*h for 100 mpk). These desirable PK properties further supported the evaluation of MF-766 in experimental mouse models of cancer.
Figure 2.. MF-766 restores PGE2-inhibited type 1 cytokine IFN-γ production in lymphocytes
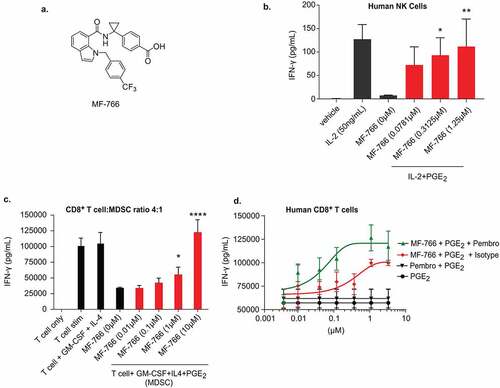
Table 1. Pharmacokinetic parameters of MF-766 in mice
MF-766 reverses PGE2-suppressed type 1 cytokine IFN-γ production in natural killer cells
NK cells play a requisite role in cancer elimination by means of mediating cytotoxicity against cancer cells, and secreting type 1 cytokines, such as IFN-γ.Citation22,Citation49 It is now appreciated that NK cell function is compromised by PGE2-mediated signaling, and that antagonism of EP4 inhibits tumor metastasis by enhancing NK cell anti-tumor activity. PGE2 has been shown to exert its inhibitory effects on NK cells mainly through EP4 dependent signals.Citation22,Citation50 Therefore, we examined the effects of MF-766 on the capacity of NK cells to secrete the canonical Th1 cytokine IFN-γ. Human NK cells were pretreated with MF-766 for 1 hour, and then stimulated with 50 ng/mL of IL-2 in the presence or absence of PGE2 to induce cytokine production overnight. Secretion of IFN-γ was suppressed by PGE2 and the inhibition by PGE2 was reversed by pretreatment with MF-766 in a dose-dependent manner (). Importantly, NK cell viability was not affected by any of the conditions in (Fig S2A).
MF-766 reduces the suppressive capacity of PGE2-induced myeloid-derived suppressor cell function
The EP4 receptor is expressed on multiple cell types, including suppressive myeloid cells (SMCs), and signaling through PGE2-EP4 directs MDSC and TAM differentiation, which can subsequently impair T-cell function.Citation21 To assess the capacity of MF-766 to reverse inhibitory effects of tumor-promoting myeloid cells in the TME, MDSCs cells were generated via an in vitro approach. Human monocytes were cultured in the presence of GM-CSF, IL-4, and PGE2, according to a previous report.Citation21 The suppressive effects of MDSCs were assessed by their capacity to inhibit IFN-production from anti-CD3/CD28 antibody-coated bead-stimulated autologous CD8+ T cells cultured at a 1:4 T cell to MDSC ratio. PGE2 increased the inhibitory activity of GM-CSF/IL-4–induced DCs/MDSCs and suppressed autologous cytokine IFN-releases in response to T-cell receptor (TCR) stimulation. This inhibitory effect was reversed by MF-766 in a dose-dependent manner at a 1:4 ratio (MDSC: T cell) as compared to DMSO-treated control cells ().
Combination activity of MF-766 and pembrolizumab in human CD8+ T cells
Tumor-derived PGE2 plays an important role in suppressing CTL function and survival in patients receiving cancer immunotherapy.Citation39 PGE2 has also been shown to suppress CTL survival, IFN-γ production, and cytotoxicity in vitro.Citation51 Treatment of tumor-bearing mice with COX-2 inhibitors and PD-1 monoclonal antibodies improves anti-tumor immunity.Citation34 The biological actions of PGE2/EP4 have also been shown to be involved in the support of exhausted CTL maintenance.Citation39 To evaluate whether the combination of EP4 inhibition and anti-PD-1 is additive in reversing CD8+ T-cell suppression caused by PGE2, CD8+ T cells were cultured in the presence of PGE2 and MF-766 alone or in combination with pembrolizumab. Neither PGE2 nor MF-766 addition affected CD8+ T cell viability in these studies (Fig S2B). PGE2 suppressed IFN-production in response to TCR stimulation (). In PGE2-suppressed T-cell cultures, MF-766 treatment alone reversed PGE2-mediated inhibition of IFN-γ production from CD8+ T cells, which was enhanced by the addition of pembrolizumab ().
MF-766 potentiates the Th1 program in T cells from histocultures using human primary tumors
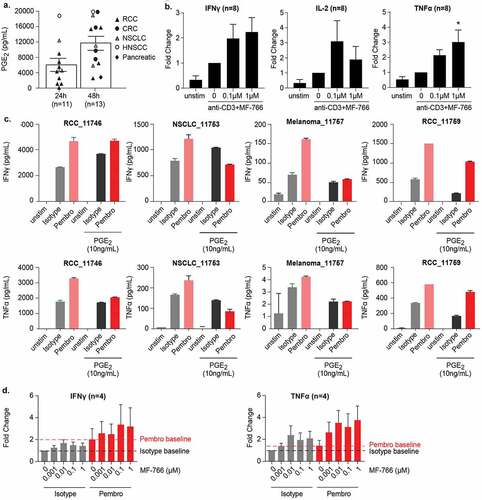
We further investigated the capacity of MF-766 to potentiate Th1 responses using surgically resected, fresh human primary tumor specimens. Examination of a large collection of primary tumors arising from the kidney, colon, lung, head and neck, and pancreas demonstrated that tumor-derived extracellular PGE2 accumulated in histoculture supernatants (). As shown in , the relative amount of PGE2 reached 6000 pg/ml after 24-hour culture, while the PGE2 concentration in 48-hour culture samples was 12000 pg/ml, respectively. Furthermore, we found that treatment of MF-766 could increase anti-CD3–stimulated IFN-γ, IL-2, and TNF-α secretion in the histoculture system (). To evaluate combinatorial effects of EP4 inhibition with pembrolizumab, anti-CD3–stimulated primary tumor digests were cultured in the presence or absence of a spike in PGE2. While pembrolizumab enhanced IFN-γ and TNF-α secretion across all donors, this enhancement was attenuated in the presence of PGE2 in 3/4 and 4/4 donors for IFN-γ and TNF-α production, respectively (). Importantly, when the tumor digest culture was treated with MF-766, the inhibitory effects of PGE2 were impaired in a dose-dependent manner, especially in pembrolizumab treated samples, although this augmentation was not statistically significant due to donor variation ().
Figure 3. MF-766 potentiates IFN-γ and TNF-α production in human primary tumors
MF-766 reverses PGE2-mediated inhibition of TNF-α production in myeloid cells
PGE2 was shown to significantly inhibit cytokine production in monocytes and human blood.Citation51,Citation52 To study the effects of MF-766 on monocytes, we first evaluated the human THP-1 monocyte cell line. While LPS stimulated TNF-α production in THP-1 cells, PGE2 significantly decreased TNF-α levels without affecting cell viability (Fig S2C, S3A), and TNF-α production was fully reversed by pretreatment with MF-766 (). Next, we pretreated healthy human whole blood with MF-766 and then stimulated TNF-α production with LPS alone or in the presence of PGE2. PGE2 dose dependently inhibited LPS-stimulated TNF-α production (Fig S3B), and MF-766 partially reversed the suppression from PGE2 (). In addition to healthy blood, we also used commercial CRC patient blood to perform the same assay, in which PGE2 dose dependently inhibited LPS-stimulated TNF-α (Fig S3C) and MF-766 partially restored PGE2-suppressed TNF-α production ().
Figure 4. MF-766 Restores PGE2-suppressed TNF-α production in myeloid cells
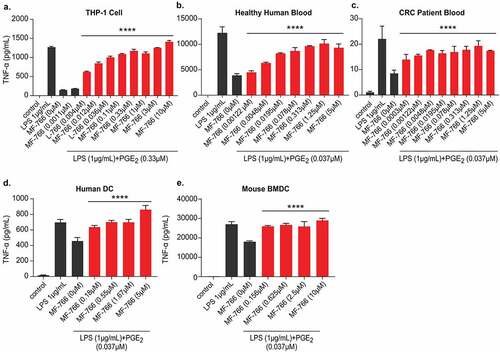
In addition to monocytes, PGE2 was also shown to inhibit TNF-α release from bone marrow-derived dendritic cells (BMDCs) stimulated with LPS.Citation53 To assess the effects of MF-766 on DCs, in-house generated BMDCs or commercially acquired human monocyte-derived DCs were examined. The results ( and ) demonstrated that PGE2 suppressed ~30% of TNF-α production without affecting cell viability (Fig S3C, S3D) when compared to LPS alone, and this suppression was fully restored by treatment with MF-766.
Anti-tumor efficacy of MF-766 in preclinical models
High concentrations of PGE2 were reported to be increased in the tumor microenvironment.Citation24,Citation45 To evaluate PGE2 concentrations in mouse tumors, we analyzed whole tumor PGE2 concentrations across 11 commonly used syngeneic tumor models. Among them, EMT6, RENCA, LL2, and CT26 exhibited high PGE2 concentrations, up to 20 nmol/g in whole tumors, whereas 4T1 had the lowest levels of PGE2 (, left panel). Consistent with these results, internal bulk RNA-sequencing data from whole tumors demonstrated a similar pattern of COX-2 and EP4 gene expression in these tumor models, whereas COX-2 and EP4 expression was undetectable in 4T1 and CT26 cell lines (, right panel). Based on tumor PGE2 concentrations () and differential response profiles to PD-1 blockade (Georgiev et al., in preparation), three mouse syngeneic tumor models (CT26, EMT6, 4T1) were selected for in vivo evaluation of MF-766. CT26 and EMT6 syngeneic tumor models are partially responsive to anti-PD-1 blockade, whereas 4T1 is intrinsically resistant (Georgiev et al., in preparation). Therefore, the anti-tumor efficacy of MF-766 was evaluated as monotherapy or in combination with the surrogate mouse anti-PD-1 antibody muDX400 in these syngeneic tumor models. As shown in , all tumors progressed on MF-766 monotherapy, with some tumors displaying slightly delayed growth kinetics as compared to vehicle-treated controls (, Fig S2A). Similarly, most CT26 tumors progressed on muDX400 monotherapy, albeit at a slower rate than isotype treated controls, with only 2 out of 12 (17%) complete responses (CRs) observed. In contrast, MF-766 + muDX400 combination therapy resulted in 7 out of 12 (58%) CRs (, Fig S4A). Additionally, tumor volume reduction was 49% and 71% (p > .05) in MF-766 and muDX400 monotherapy groups, respectively, while MF-766 + muDX400 combination treatment significantly reduced tumor volumes by 89% (p < .05) as compared to the vehicle/isotype treated group (). Finally, animals with CRs to muDX400 or MF-766 + muDX400 combination therapy were re-challenged by CT-26 tumor cells. Naïve mice developed tumors while no tumors were observed in CR mice, indicating long-term protective immunity following re-challenge (). To evaluate whether this combination effect could be extended to other syngeneic models, the same study was designed and conducted in the EMT6 () and 4T1 models (). Despite exhibiting higher expression levels of PTSG2 and PTGER4, MF-766 did not show single-agent activity in the EMT6 model (). Moreover, the muDX400 group alone and the MF-766 + muDX400 combination group had four CRs, indicating that the addition of MF-766 did not change the overall CR rate ( and Fig S4B). However, individual tumors in the combination group exhibited delayed kinetics and tumor volumes were significantly reduced by 86% in the combination group as compared to vehicle/isotype controls ( and Fig S4B). In contrast, MF-766 showed moderate single-agent efficacy in the 4T1 model with 39% TGI, whereas the MF-766 + muDX400 combination group did not exhibit enhanced anti-tumor activity as compared to vehicle/isotype controls ().
Figure 5. MF-766 improves anti-tumor activity in the setting of PD-1 blockade in multiple syngeneic models
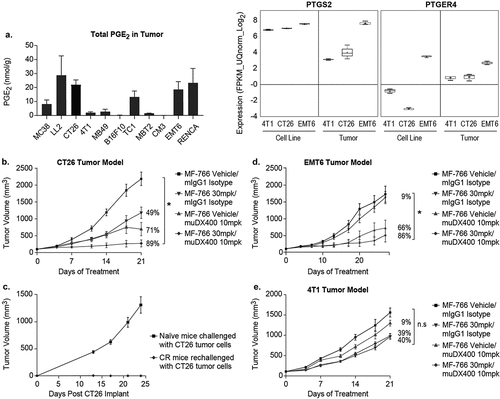
MF-766 modulates immune cell infiltration in vivo
To identify potential mechanisms driving the efficacy of MF-766 + muDX400 combination therapy, immune infiltrates D8 post-therapy in CT-26/EMT6 tumors were examined by multiparameter flow cytometry. As shown in left panel, CT26 tumor weight showed a trend toward reduction in size at day 8 in the MF-766 and MF-766+ muDX400 combination group although the difference did not reach statistical significance. Consistent with enhanced anti-tumor activity (), absolute numbers of tumor-infiltrating CD45+ leukocytes per mg tumor were significantly increased in the MF-766 and MF-766+ muDX400 combination therapy groups ( right). Among CD45+ cells, MF-766 monotherapy resulted in a significant enrichment of CD3+ T cells, both as frequency of CD45+ T cells and absolute numbers in the tumor (Fig S5A), which consisted primarily of CD8+ T cells () but not CD4+ T cells (Fig S5B). Both the percentage of NK cells in the CD45+ fraction and the absolute number of NK cells per mg tumor were significantly enriched with MF-766 monotherapy and in combination with DX400 (). In contrast, neither tumor weight (Fig S5C left) nor total tumor-infiltrating CD45+ leukocytes per mg tumor (Fig S5C right) at day 8 changed following MF-766 monotherapy or MF-766+ muDX400 combination treatment in the EMT6 model. In addition, single-agent treatment with MF-766 did not significantly increase CD8+ (Fig S5D) or NK cell infiltration into tumors (Fig S5E). Although the combination of MF-766 + muDX400 moderately increased the percentage of NK cells among the CD45+ fraction in tumors (Fig S5E left), the absolute numbers of NK cells and CD8+ T cells in tumors trended toward an increase but were not significantly different (Fig S5D-E right). These results reveal that lymphocyte composition, including NK and CD8 T cell infiltration, is differentially modulated by MF-766 therapy in CT-26 and EMT6 TMEs, respectively.
Figure 6.. MF-766 modulates immune cell infiltration in the TME
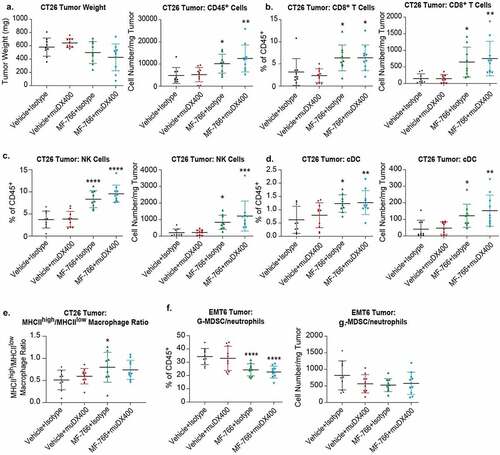
To gain additional insights into discrete changes occurring in the CT26 and EMT6 TME, we extended our analysis to examine a set of canonical immune cell populations, including immunostimulatory dendritic cells, tumor-associated macrophages (TAMs), and MDSCs. Surprisingly we observed a remarkable increase of CD8+ DCs (), which are critical for cross-presentation of tumor antigens,Citation54 after MF-766 single-agent treatment in the CT26 () but not EMT6 model (data not shown). Additionally, we found that MF-766 increased the MHCIIhigh/low macrophage ratio, suggesting it could stimulate favorable macrophage activation in CT26 () but not EMT6 tumors (data not shown). These findings are aligned with the superior anti-tumor activity of MF-766 as a monotherapy in CT-26 but not EMT6 syngeneic tumor model. Moreover, the stimulatory effects of MF-766 on macrophage activation are consistent with previous reports.Citation21,Citation41 In EMT6 tumors, however, MF-766 single-agent treatment significantly decreased the percentage of gMDSCs ( left) although the decrease in absolute numbers of gMDSCs per mg EMT6 tumor did not reach statistical significance ( right).
Discussion
Cancer is a group of diseases the development of which largely depends on the cross-talk between the tumor and its microenvironment, in which the immune system plays a key role.Citation55 Manipulation of suppressive factors in the TME to overcome primary and secondary resistance is critical for the development of effective cancer immunotherapies.Citation40,Citation46,Citation47,Citation56 Here, we demonstrate that PGE2-EP4 signaling plays a key role in negatively regulating immune cell function using various in-vitro functional assays and in-vivo tumor models. Tumor-producing COX-2/PGE2 exerts pleiotropic effects via EP4 including direct inhibition of CD8+ T cells and indirect inhibition via generation of immunosuppressive MDSCs. Both effects were reversed with MF-766 treatment in-vitro. NK cell trafficking coincides with the recruitment of classical DCs, which are thought to be recruited into the TME by an NK-dependent production of CCL5 and XCL1.Citation45,Citation54 Consistent with these studies, we demonstrate that MF-766 increased NK cell accumulation in CT26 tumors and that this coincided with increased numbers of CD8+ cross-presenting DCs and CD8+ T cells (). In vitro analysis of human NK cells demonstrated that PGE2 suppressed high-dose IL-2 induced IFN-γ production and this effect was reversed by MF-766. MF-766 also reverted PGE2-mediated inhibition of inflammatory cytokine production by monocytes. In addition to paracrine signaling, PGE2 exerts autocrine effects by upregulating the expression of CD44 and MMP-2 in human lung cancer cells in COX-2-dependent manners.Citation57
Tumor-derived PGE2 demonstrates the complex interplay between tumor and tumor-associated immune components, which can be regulated by EP4 antagonists.
Our findings are consistent with previous reports demonstrating that pharmacological blockade of EP4 by E-7046 can reverse PGE2-induced myeloid immunosuppression and M2-macrophage polarization.Citation21,Citation41 Similarly, we showed that MF-766 reverted PGE2-mediated suppression of DCs. This effect may increase the functions of antigen-presenting cells, indicating a possible role for MF-766 in promoting lymphocyte priming and is supported by our observations of massive cDC expansion following MF-766 monotherapy in the CT26 model. Moreover, MF-766 reverted PGE2-mediated suppression of IFN-γ production in NK cells and CD8+ T cells in-vitro. However, we observed variation in the effects of MF-766 on NK cell and CD8+ T-cell responses across different donors, which may be explained by the differential expression of EP2 and EP4 in human donors. Both EP2 and EP4 have been reported to contribute to PGE2-mediated suppression of IFN-γ production in T lymphocytesCitation58 and NK cells.Citation22,Citation49 We surmise that when EP2 expression is dominant in donor NK and CD8+ T cells, blocking EP4 alone is not sufficient to fully reverse the suppression induced by PGE2. Although MF-766 could fully restore PGE2-mediated inhibition of TNF-α production in the THP-1 cell line, it only exerted partial reversal in ex vivo whole blood, which may be also due to donor and cell-type variation in EP2 expression. Notably, the roles of EP2 in immunity, chronic inflammation, angiogenesis, metastasis, and cancer immunotherapy resistance additionally suggest that EP2 could be a novel therapeutic target for cancer.Citation22,Citation25,Citation58–60 Therefore, dual antagonism of EP2 and EP4 receptors may be required for optimal reversal of PGE2-mediated immune suppression in certain donors that appear reliant on both EP2 and EP4 pathways.
In addition to promoting the acquisition of an immunosuppressive phenotype in myeloid cells, EP4 signaling was shown to contribute to CTL dysfunction.Citation39 The COX-2/PGE2 axis impairs CTL effector function and promotes CTL exhaustion in human cancers by suppressing type 1 IFN-dependent immune cell activation.Citation39 Evidence shows that inhibition of this signaling pathway may enhance CTL effector function.Citation39,Citation45 In our study, MF-766 reversed PGE2-mediated suppression of NK and CD8+T cell cytokine production in vitro. Although MF-766 monotherapy treatment significantly increased infiltration of NK and CD8+ T cells in the CT26 syngeneic tumor model, single-agent treatment induced modest anti-tumor activity. In contrast, combined inhibition of PD-1 and EP4 resulted in robust anti-tumor activity characterized by a 58% CR rate in the CT26 model. This observation is in line with reports that combined blockade of PD-1 and COX-2/PGE2 pathways enhances CTL cell function in a synergistic manner.Citation39,Citation58 Taken together, our results suggest that inhibition of EP4 in combination with ICI may represent an attractive strategy for the immunotherapy of cancer, particularly in settings of ICI acquired resistance. Combination of EP4 inhibitors, including AN0025AN0025 (E-7046; NCT04432857, https://clinicaltrials.gov/ct2/show/NCT04432857?term=E-7046&draw=2&rank=3) and ONO-4578 (NCT03155061, https://clinicaltrials.gov/ct2/show/NCT03155061?term=ono-4578&draw=2&rank=1) with anti-PD-1 are already underway. However, combination strategies should not be limited to anti-PD-1 agents, especially in the context of poorly immunogenic tumors lacking T cell infiltration. This notion is supported by the lack of activity of MF-766 in combination with anti-PD-1 in the “cold” 4T1 mammary carcinoma model.Citation61 Interestingly, we observed modest single-agent activity with MF-766, which is consistent with previous reports demonstrating elicitation of partial responses in 4T1 breast and Pan02 pancreatic syngeneic tumors following treatment with the E-7046 EP4 inhibitor, as well as in a phase I clinical trial.Citation41 The importance of NSAIDs as adjuvants to current chemotherapy/radiation regimens has been reported,Citation30,Citation32,Citation62 and serves as a rationale for the evaluation of EP4 antagonism in combination with chemotherapy or other immune-based therapies. Evaluation of preoperative radiotherapy and E-7046 in rectal cancer is currently being conducted (NCT03152370, https://clinicaltrials.gov/ct2/show/NCT03152370?term=E-7046&draw=2&rank=2).
Developing response-predictive clinical biomarkers for EP4 inhibition and ICI resistance will be paramount for the rational selection and stratification of patients as a means to deliver precision immunotherapy. Immune cell tumor infiltration and tumor metabolic profiles at baseline were assessed as potential response-predictive biomarkers to E-7046 in a first-in-human trial.Citation41 In our preclinical studies, MF-766 did not significantly increase NK or CD8+ T cell infiltration in the EMT6 syngeneic tumor model, which is consistent with its myeloid cell-rich but lymphocyte-low profile (Georgiev et al., in preparation). As a result, a combination of MF-766 and anti-PD-1 in the EMT6 model did not increase overall CR rates despite modulating tumor growth kinetics, so the lack of efficacy likely results from low baseline lymphocyte infiltration in TME. In addition to immune cell infiltration profiles in the TME, concentrations of the EP4 ligand PGE2, overexpression of COX-1/2, and EP4 expression patterns in immune and tumor cells may enable the delivery of EP4 inhibitors as a precision medicine immunotherapy.Citation63 Indeed, in our histoculure studies, MF-766 potentiated Th1 cytokine production only when PGE2 was present. Other possible candidate biomarkers could be derived from a retrospective analysis of EP4/COX-2 pathway interactions. For example, studies have shown that the mechanism by which COX-2/PGE2 might suppress apoptosis leading to increased expression of BCL-2 is via activation of the Ras-MAPK-ERK pathway.Citation64 Under hypoxic conditions, PGE2 signaling via the Ras-MAPK pathway has been shown to promote survival.Citation8 During hypoxia, HIF1 can upregulate COX-2 expression and increase PGE2 production.Citation8 Moreover, PGE2 activates Ras-MAPK, promoting a feedback loop for the COX/PGE2 signal. However, EP4 antagonism may also re-sensitize growing cells to apoptosis. We also observed a correlation between the expression of an EP4 and RAS as well as COX-2 and RAS signaling signature (unpublished data). This may indicate the potential selection of patients based on KRAS and/or BRAF mutation status. Kras is mutated in 45% of CRC cases and 95% of pancreatic adenocarcinomas.Citation65 Association of EP4 with the KRAS signature will likely allow enrichment of patients with COX-2 and/or EP4 overexpression. In addition, previous reports and our unpublished results have demonstrated an association of the STK11/LKB1 loss of function phenotype, which frequently occurs in NSCLC, with EP4 expression.Citation66 In vitro experiments suggest synthetic lethality of EP4 with LKB1 LOF, which has significant overlap with KRAS mutations.Citation66 Taken together, EP4 antagonists could be included in LKB1-deficient NSCLC tumors as part of a triple combination with chemotherapy and pembrolizumab.
The present study largely focused on evaluating immune cell modulation by MF-766 treatment in-vitro and in-vivo. However, targeting EP4 was also reported to control breast tumor growth, angiogenesis, lymphangiogenesis, metastasis,Citation63,Citation67 and to limit breast cancer stem-like cell (SLC) development.Citation68 Therefore, elucidating whether and how MF-766 affects nonimmune cell types, including stromal cells, remains to be determined. In addition, current therapeutics targeting the PGE2/EP signaling axis in the TME are often administered systemically.Citation41 As a result, the global blockade of EP4 signaling is expected to affect other cell types, including still-healthy tissues and cells, in addition to TME components.Citation45 Although E-7046 was well tolerated in a first-in-human clinical study,Citation41 it is necessary to evaluate the on-target effects of E-7046/EP4 antagonists across stromal and immune cells broadly in order to avoid potential adverse effects. In conclusion, we demonstrated that MF-766 can reverse PGE2-mediated immune suppression in several key immune cell populations in vitro and that its combination with anti-PD-1 in vivo drives superior anti-tumor immune responses in syngeneic tumor mouse models, which support the investigation of EP4 antagonists in combination with anti-PD-1 or other therapies in clinic.
Author contributions
Conceptualization: Yun Wang, Brian Haines, Brian Long, Michael Rosenzweig, Andrew Haidle, Yongxin Han, Sheila Ranganath
Investigation: Yun Wang, Long Cui, Peter Georgiev, Latika Singh, Yanyan Zheng, Ying Yu, Jeff Grein, Chunsheng Zhang, Eric Muise, David Sloman, Heidi Ferguson, Hongshi Yu, Cristina St. Pierre, Pranal J Dakle, Doug Wilson, Doug Linn, Vincenzo Pucci, James Baker
Data Curation: Yun Wang, Long Cui, Peter Georgiev, Latika Singh, Yanyan Zheng, Ying Yu, Jeff Grein, Chunsheng Zhang, Eric Muise, David Sloman, Heidi Ferguson, Hongshi Yu, Cristina St. Pierre, Pranal J Dakle, Doug Wilson, Doug Linn, Vincenzo Pucci, James Baker
Methodology: Yun Wang, Long Cui, Peter Georgiev, Latika Singh, Yanyan Zheng, Ying Yu, Jeff Grein, Chunsheng Zhang, Eric Muise, David Sloman, Heidi Ferguson, Hongshi Yu, Doug Wilson, Doug Linn, Vincenzo Pucci, Christopher Brynczka, James Baker
Formal analysis: Yun Wang, Long Cui, Latika Singh, Yanyan Zheng, Jeff Grein, Chunsheng Zhang, Eric Muise
Software: Jeff Grein, Chunsheng Zhang, Eric Muise, Andrey Loboda
Writing-original draft: Yun Wang, Long Cui, Latika Singh, Yanyan Zheng, Jeff Grein, Eric Muise
Writing-review & editing: Yun Wang, Peter Georgiev, Vincenzo Pucci, Andrey Loboda, Brian Long, Richard Wnek, Svetlana Sadekova, Michael Rosenzweig, Andrew Haidle, Yongxin Han, Sheila Ranganath
Conflict of interest:
This work was supported by Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, United States of America. All authors were employees and stockholders in Merck & Co., Inc., Kenilworth, NJ, United States of America, during the period this study was conducted.
The funder provided support in the form of salaries for all authors [YW, LC, LS, YZ, YY, JG, CZ, PG, EM, DS, HF, HY, CP, PD, VP, JB, AL, DL, CB, DW, BH, BL, RW, SS, MR, AH, YH, SR] and research materials but did not have any additional role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript. The specific roles of these authors are articulated in the ‘author contributions’ section.
No authors have competing interests and the commercial affiliation with Merck & Co., Inc., Kenilworth, NJ, USA does not alter our adherence to Oncoimmunology policies on data sharing.
Supplemental Material
Download ()Acknowledgments
We thank Dr. Nick Haining and Dr. Jonathan Bennett for the careful reading and review of the manuscript.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- Koury J, Lucero M, Cato C, Chang L, Geiger J, Henry D, Hernandez J, Hung F, Kaur P, Teskey G, et al. Immunotherapies: exploiting the immune system for cancer treatment. J Immunol Res. 2018;2018:9585614. doi:10.1155/2018/9585614.
- Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39(1):1–16. doi:10.1016/j.immuni.2013.07.012.
- Galon J, Bruni D. Tumor immunology and tumor evolution: intertwined histories. Immunity. 2020;52:55–81.
- Seidel JA, Otsuka A, Kabashima K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: mechanisms of action, efficacy, and limitations. Front Oncol. 2018;8:86. doi:10.3389/fonc.2018.00086.
- Beatty GL, Gladney WL. Immune escape mechanisms as a guide for cancer immunotherapy. Clin Cancer Res. 2015;21(4):687–692. doi:10.1158/1078-0432.CCR-14-1860.
- Cassim S, Pouyssegur J. Tumor microenvironment: a metabolic player that shapes the immune response. Int J Mol Sci. 2019;21(1):157. doi: 10.3390/ijms21010157.
- Iorgulescu JB, Braun D, Oliveira G, Keskin DB, Wu CJ. Acquired mechanisms of immune escape in cancer following immunotherapy. Genome Med. 2018;10(1):87. doi:10.1186/s13073-018-0598-2.
- Greenhough A, Smartt HJ, Moore AE, Roberts HR, Williams AC, Paraskeva C, Kaidi A. The COX-2/PGE2 pathway: key roles in the hallmarks of cancer and adaptation to the tumour microenvironment. Carcinogenesis. 2009;30(3):377–386. doi:10.1093/carcin/bgp014.
- Ricciotti E, FitzGerald GA. Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol. 2011;31(5):986–1000. doi:10.1161/ATVBAHA.110.207449.
- Nakanishi M, Rosenberg DW. Multifaceted roles of PGE2 in inflammation and cancer. Semin Immunopathol. 2013; 35(2):123–137. doi:10.1007/s00281-012-0342-8.
- Kalinski P. Regulation of immune responses by prostaglandin E2. J Immunol. 2012;188(1):21–28. doi:10.4049/jimmunol.1101029.
- Eberhart CE, Coffey RJ, Radhika A, Giardiello FM, Ferrenbach S, DuBois RN. Up-regulation of cyclooxygenase 2 gene expression in human colorectal adenomas and adenocarcinomas. Gastroenterology. 1994;107(4):1183–1188. doi:10.1016/0016-5085(94)90246-1.
- Matsubayashi H, Infante JR, Winter J, Klein AP, Schulick R, Hruban R, Visvanathan K, Goggins M. Tumor COX-2 expression and prognosis of patients with resectable pancreatic cancer. Cancer Biol Ther. 2007;6(10):1569–1575. doi:10.4161/cbt.6.10.4711.
- Khuri FR, Wu H, Lee JJ, Kemp BL, Lotan R, Lippman SM, Feng L, Hong WK, Xu XC. Cyclooxygenase-2 overexpression is a marker of poor prognosis in stage I non-small cell lung cancer. Clin Cancer Res. 2001;7(4):861–867.
- Petkova DK, Clelland C, Ronan J, Pang L, Coulson JM, Lewis S, Knox AJ. Overexpression of cyclooxygenase-2 in non-small cell lung cancer. Respir Med. 2004;98(2):164–172. doi:10.1016/j.rmed.2003.09.006.
- Hwang D, Scollard D, Byrne J, Levine E. Expression of cyclooxygenase-1 and cyclooxygenase-2 in human breast cancer. J Natl Cancer Inst. 1998;90(6):455–460. doi:10.1093/jnci/90.6.455.
- Gupta S, Srivastava M, Ahmad N, Bostwick DG, Mukhtar H. Over-expression of cyclooxygenase-2 in human prostate adenocarcinoma. Prostate. 2000;42(1):73–78. doi:10.1002/(SICI)1097-0045(20000101)42:1<73::AID-PROS9>3.0.CO;2-G.
- Koga H, Sakisaka S, Ohishi M, Kawaguchi T, Taniguchi E, Sasatomi K, Harada M, Kusaba T, Tanaka M, Kimura R, et al. Expression of cyclooxygenase-2 in human hepatocellular carcinoma: relevance to tumor dedifferentiation. Hepatology. 1999;29(3):688–696. doi:10.1002/hep.510290355.
- Czachorowski MJ, Amaral AF, Montes-Moreno S, Lloreta J, Carrato A, Tardón A, Morente MM, Kogevinas M, Real FX, Malats N; SBC/EPICURO investigators. Cyclooxygenase-2 expression in bladder cancer and patient prognosis: results from a large clinical cohort and meta-analysis. PLoS One. 2012;7(9):e45025. doi:10.1371/journal.pone.0045025.
- Rich TA, Shepard R. COX-2 inhibitors as radiation sensitizers for upper GI tract cancers: esophagus, stomach, and pancreas. Am J Clin Oncol. 2003. 26(4):S110–3. doi:10.1097/01.COC.0000074148.37768.3E.
- Albu DI, Wang Z, Huang KC, Wu J, Twine N, Leacu S, Ingersoll C, Parent L, Lee W, Liu D, et al. EP4 Antagonism by E7046 diminishes Myeloid immunosuppression and synergizes with Treg-reducing IL-2-Diphtheria toxin fusion protein in restoring anti-tumor immunity. Oncoimmunology. 2017;6(8):e1338239. doi:10.1080/2162402X.2017.1338239.
- Holt DM, Ma X, Kundu N, Collin PD, Fulton AM. Modulation of host natural killer cell functions in breast cancer via prostaglandin E2 receptors EP2 and EP4. J Immunother. 2012;35(2):179–188. doi:10.1097/CJI.0b013e318247a5e9.
- Sreeramkumar V, Fresno M, Cuesta N. Prostaglandin E2 and T cells: friends or foes? Immunol Cell Biol. 2012. 90(6):579–586. doi:10.1038/icb.2011.75.
- Ching MM, Reader J, Fulton AM. Eicosanoids in cancer: prostaglandin E2 receptor 4 in cancer therapeutics and immunotherapy. Front Pharmacol. 2020;11:819. doi:10.3389/fphar.2020.00819.
- Okuyama T, Ishihara S, Sato H, Rumi MA, Kawashima K, Miyaoka Y, Suetsugu H, Kazumori H, Cava CF, Kadowaki Y, et al. Activation of prostaglandin E2-receptor EP2 and EP4 pathways induces growth inhibition in human gastric carcinoma cell lines. J Lab Clin Med. 2002;140(2):92–102. doi:10.1016/S0022-2143(02)00023-9.
- Mizuno R, Kawada K, Sakai Y. Prostaglandin E2/EP signaling in the tumor microenvironment of colorectal cancer. Int J Mol Sci. 2019;20(24):6254. doi: 10.3390/ijms20246254.
- Xu XC. COX-2 inhibitors in cancer treatment and prevention, a recent development. Anticancer Drugs. 2002;13(2):127–137. doi:10.1097/00001813-200202000-00003.
- Stratton MS, Alberts DS. Current application of selective COX-2 inhibitors in cancer prevention and treatment. Oncology (Williston Park). 2002;16:37–51.
- Bertagnolli MM, Eagle CJ, Zauber AG, Redston M, Solomon SD, Kim K, Tang J, Rosenstein RB, Wittes J, Corle D, et al. Celecoxib for the prevention of sporadic colorectal adenomas. N Engl J Med. 2006;355(9):873–884. doi:10.1056/NEJMoa061355.
- Edelman MJ, Wang X, Hodgson L, Cheney RT, Baggstrom MQ, Thomas SP, Gajra A, Bertino E, Reckamp KL, Molina J, et al. Phase III randomized, placebo-controlled, double-blind trial of celecoxib in addition to standard chemotherapy for advanced non-small-cell lung cancer with cyclooxygenase-2 overexpression: CALGB 30801 (alliance). J Clin Oncol. 2017; 35(19):2184–2192. doi:10.1200/JCO.2016.71.3743.
- Huang H, Aladelokun O, Ideta T, Giardina C, Ellis LM, Rosenberg DW, et al. Inhibition of PGE2/EP4 receptor signaling enhances oxaliplatin efficacy in resistant colon cancer cells through modulation of oxidative stress. Sci Rep. 2019;9(1):4954. doi:10.1038/s41598-019-40848-4.
- Li B, Li X, Xiong H, Zhou P, Ni Z, Yang T, Zhang Y, Zeng Y, He J, Yang F, et al. Inhibition of COX2 enhances the chemosensitivity of dichloroacetate in cervical cancer cells. Oncotarget. 2017;8(31):51748–51757. doi:10.18632/oncotarget.18518.
- Hutchinson L. Immunotherapy: evading immune escape: synergy of COX and immune-checkpoint inhibitors. Nat Rev Clin Oncol. 2015;12:622.
- Li Y, Fang M, Zhang J, Wang J, Song Y, Shi J, Li W, Wu G, Ren J, Wang Z, et al. Hydrogel dual delivered celecoxib and anti-PD-1 synergistically improve antitumor immunity. Oncoimmunology. 2016;5(2):e1074374.
- Mattia C, Coluzzi F. COX-2 inhibitors: pharmacological data and adverse effects. Minerva Anestesiol. 2005;71:461–470.
- Sharma JN, Jawad NM. Adverse effects of COX-2 inhibitors. ScientificWorldJournal. 2005;5:629–645. doi:10.1100/tsw.2005.82.
- Brænne I, Willenborg C, Tragante V, Kessler T, Zeng L, Reiz B, Kleinecke M, von Ameln S, Willer CJ, Laakso M, et al. A genomic exploration identifies mechanisms that may explain adverse cardiovascular effects of COX-2 inhibitors. Sci Rep. 2017;7(1):10252. doi:10.1038/s41598-017-10928-4.
- Majumder M, Nandi P, Omar A, Ugwuagbo KC, Lala PK. EP4 as a therapeutic target for aggressive human breast cancer. Int J Mol Sci. 2018;19(4):1019.
- .Miao J, Lu X, Hu Y, Piao C, Wu X, Liu X, Huang C, Wang Y, Li D, Liu J. Prostaglandin E2 and PD-1 mediated inhibition of antitumor CTL responses in the human tumor microenvironment. Oncotarget. 2017;8(52):89802–89810. doi:10.18632/oncotarget.21155.
- Murciano-Goroff YR, Warner AB, Wolchok JD. The future of cancer immunotherapy: microenvironment-targeting combinations. Cell Res. 2020;30(6):507–519. doi:10.1038/s41422-020-0337-2.
- Hong DS, Parikh A, Shapiro G, Varga A, Naing A, Meric-Bernstam F, Ataman O, Reyderman L, Binder T, Ren M, et al. First-in-human phase I study of immunomodulatory E7046, an antagonist of PGE2-receptor E-type 4 (EP4), in patients with advanced cancers. J Immunother Cancer. 2020;8(1.
- Colucci J, Boyd M, Berthelette C, Chiasson JF, Wang Z, Ducharme Y, Friesen R, Wrona M, Levesque JF, Denis D, et al. Discovery of 4-[1-[([1-[4-(trifluoromethyl)benzyl]-1H-indol-7-yl]carbonyl)amino]cyclopropyl]be nzoic acid (MF-766), a highly potent and selective EP4 antagonist for treating inflammatory pain. Bioorg Med Chem Lett. 2010;20(12):3760–3763. doi:10.1016/j.bmcl.2010.04.065.
- PClark P, Rowland SE, Denis D, Mathieu MC, Stocco R, Poirier H, Burch J, Han Y, Audoly L, Therien AG, et al. MF498 [N-{[4-(5,9-Diethoxy-6-oxo-6,8-dihydro-7H-pyrrolo[3,4-g]quinolin-7-yl)-3-methylbe nzyl]sulfonyl}-2-(2-methoxyphenyl)acetamide], a selective E prostanoid receptor 4 antagonist, relieves joint inflammation and pain in rodent models of rheumatoid and osteoarthritis. J Pharmacol Exp Ther. 2008;325(2):425–434. doi:10.1124/jpet.107.134510.
- Burch JD, Belley M, Fortin R, Deschênes D, Girard M, Colucci J, Farand J, Therien AG, Mathieu MC, Denis D, Vigneault E, et al. Structure-activity relationships and pharmacokinetic parameters of quinoline acylsulfonamides as potent and selective antagonists of the EP(4) receptor. Bioorg Med Chem Lett. 2008;18(6):2048–2054. doi:10.1016/j.bmcl.2008.01.103.
- Take Y, Koizumi S, Nagahisa A. Prostaglandin E receptor 4 antagonist in cancer immunotherapy: mechanisms of action. Front Immunol. 2020;11:324. doi:10.3389/fimmu.2020.00324.
- Liu D, Jenkins RW, Sullivan RJ. Mechanisms of resistance to immune checkpoint blockade. Am J Clin Dermatol. 2019;20(1):41–54. doi:10.1007/s40257-018-0389-y.
- Pitt JM, Vétizou M, Daillère R, Roberti MP, Yamazaki T, Routy B, Lepage P, Boneca IG, Chamaillard M, Kroemer G, et al. Resistance mechanisms to immune-checkpoint blockade in cancer: tumor-intrinsic and -extrinsic factors. Immunity. 2016;44(6):1255–1269. doi:10.1016/j.immuni.2016.06.001.
- Zarghi A, Arfaei S. Selective COX-2 inhibitors: a review of their structure-activity relationships. Iran J Pharm Res. 2011;10:655–683.
- Bonavita E, Bromley CP, Jonsson G, Pelly VS, Sahoo S, Walwyn-Brown K, Mensurado S, Moeini A, Flanagan E, Bell CR, et al. Antagonistic inflammatory phenotypes dictate tumor fate and response to immune checkpoint blockade. Immunity. 2020;53(6):1215–1229. e8. doi:10.1016/j.immuni.2020.10.020.
- Ma X, Holt D, Kundu N, Reader J, Goloubeva O, Take Y, Fulton AM. A prostaglandin E (PGE) receptor EP4 antagonist protects natural killer cells from PGE2-mediated immunosuppression and inhibits breast cancer metastasis. Oncoimmunology. 2013;2(1):e22647. doi:10.4161/onci.22647.
- Harris SG, Padilla J, Koumas L, Ray D, Phipps RP. Prostaglandins as modulators of immunity. Trends Immunol. 2002;23(3):144–150. doi:10.1016/S1471-4906(01)02154-8.
- Jin Y, Smith C, Hu L, Coutant DE, Whitehurst K, Phipps K, McNearney TA, Yang X, Ackermann B, Pottanat T, et al. LY3127760, a selective prostaglandin E4 (EP4) receptor antagonist, and celecoxib: a comparison of pharmacological profiles. Clin Transl Sci. 2018;11(1):46–53. doi:10.1111/cts.12497.
- Vassiliou E, Jing H, Ganea D. Prostaglandin E2 inhibits TNF production in murine bone marrow-derived dendritic cells. Cell Immunol. 2003;223(2):120–132. doi:10.1016/S0008-8749(03)00158-8.
- Böttcher JP, Bonavita E, Chakravarty P, Blees H, Cabeza-Cabrerizo M, Sammicheli S, Rogers NC, Sahai E, Zelenay S, Reis E, Sousa C. NK cells stimulate recruitment of cDC1 into the tumor microenvironment promoting cancer immune control. Cell. 2018;172(5):1022–1037. e14. doi:10.1016/j.cell.2018.01.004.
- Gonzalez H, Hagerling C, Werb Z. Roles of the immune system in cancer: from tumor initiation to metastatic progression. Genes Dev. 2018;32(19–20):1267–1284. doi:10.1101/gad.314617.118.
- Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. 2017;168(4):707–723. doi:10.1016/j.cell.2017.01.017.
- Dohadwala M, Batra RK, Luo J, Lin Y, Krysan K, Pold M, Sharma S, Dubinett SM. Autocrine/paracrine prostaglandin E2 production by non-small cell lung cancer cells regulates matrix metalloproteinase-2 and CD44 in cyclooxygenase-2-dependent invasion. J Biol Chem. 2002;277(52):50828–50833. doi:10.1074/jbc.M210707200.
- Wang J, Zhang L, Kang D, Yang D, Tang Y. Activation of PGE2/EP2 and PGE2/EP4 signaling pathways positively regulate the level of PD-1 in infiltrating CD8(+) T cells in patients with lung cancer. Oncol Lett. 2018;15(1):552–558. doi:10.3892/ol.2017.7279.
- Sun X, Li Q. Prostaglandin EP2 receptor: novel therapeutic target for human cancers (Review). Int J Mol Med. 2018;42(3):1203–1214. doi:10.3892/ijmm.2018.3744.
- Harizi H, Grosset C, Gualde N. Prostaglandin E2 modulates dendritic cell function via EP2 and EP4 receptor subtypes. J Leukoc Biol. 2003;73(6):756–763. doi:10.1189/jlb.1002483.
- Huang L, Li Y, Du Y, Zhang Y, Wang X, Ding Y, Yang X, Meng F, Tu J, Luo L, Sun C. Mild photothermal therapy potentiates anti-PD-L1 treatment for immunologically cold tumors via an all-in-one and all-in-control strategy. Nat Commun. 2019;10(1):4871. doi:10.1038/s41467-019-12771-9.
- Kune GA. Colorectal cancer chemoprevention: aspirin, other NSAID and COX-2 inhibitors. Aust N Z J Surg. 2000;70(6):452–455. doi:10.1046/j.1440-1622.2000.01844.x.
- Majumder M, Xin X, Liu L, Girish GV, Lala PK. Prostaglandin E2 receptor EP4 as the common target on cancer cells and macrophages to abolish angiogenesis, lymphangiogenesis, metastasis, and stem-like cell functions. Cancer Sci. 2014;105(9):1142–1151. doi:10.1111/cas.12475.
- Sheng H, Shao J, Morrow JD, Beauchamp RD, DuBois RN. Modulation of apoptosis and Bcl-2 expression by prostaglandin E2 in human colon cancer cells. Cancer Res. 1998. 58(2):362–366.
- Zeitouni D, Pylayeva-Gupta Y, Der CJ, Bryant KL. KRAS mutant pancreatic cancer: no lone path to an effective treatment. Cancers (Basel). 2016;8(4):45. doi: 10.3390/cancers8040045.
- Skoulidis F, Goldberg ME, Greenawalt DM, Hellmann MD, Awad MM, Gainor JF, Schrock AB, Hartmaier RJ, Trabucco SE, Gay L, et al. STK11/LKB1 mutations and pd-1 inhibitor resistance in kras-mutant lung adenocarcinoma. Cancer Discov. 2018;8(7):822–835. doi:10.1158/2159-8290.CD-18-0099.
- Xin X, Majumder M, Girish GV, Mohindra V, Maruyama T, Lala PK. Targeting COX-2 and EP4 to control tumor growth, angiogenesis, lymphangiogenesis and metastasis to the lungs and lymph nodes in a breast cancer model. Lab Invest. 2012;92(8):1115–1128. doi:10.1038/labinvest.2012.90.
- Majumder M, Xin X, Liu L, Tutunea-Fatan E, Rodriguez-Torres M, Vincent K, Postovit LM, Hess D, Lala PK. COX-2 induces breast cancer stem cells via EP4/PI3K/AKT/NOTCH/WNT axis. Stem Cells. 2016;34(9):2290–2305. doi:10.1002/stem.2426.