
ABSTRACT
Chimeric antigen receptor (CAR) T cell is a promising method in cancer immunotherapy but faces many challenges in solid tumors. One of the major problems was immunosuppression caused by PD-1. In our study, the expression of c-Met in GC was analyzed from TCGA datasets, GC tissues, and cell lines. The c-Met CAR was a second-generation CAR with 4–1BB, cMet-PD1/CD28 CAR was c-Met CAR adding PD1/CD28 chimeric-switch receptor (CSR). In vitro, we measured the changes of different subgroups, phenotypes and PD-1 expression in CAR-T cells. We detected the secretion levels of different cytokines and the killing ability of CAR-Ts. In vivo, we established a xenograft GC model and observed the anti-tumor effect and off-target toxicity of different CAR-Ts. We find that the expression of c-Met was increased in GC. CD3+CD8+ T cells and CD62L+CCR7+ central memory T cells (TCM) were increased in two CAR-Ts. The stimulation of target cells could promote the expression of PD-1 in c-Met CAR-T. Compared with Mock T, the secretion of cytokines as IFN-γ, TNF-α, IL-6, IL-10 secreted by two CAR-Ts was increased, and the killing ability to c-Met positive GC cells was enhanced. The PD1/CD28 CSR could further enhance the killing ability, especially the long-term anti-tumor effect of c-Met CAR-T, and reduce the release level of IL-6. CAR-Ts target c-Met had no obvious off-target toxicity to normal organs. Thus, the PD1/CD28 CSR could further enhance the anti-tumor ability of c-Met CAR-T, and provides a promising design strategy to improve the efficacy of CAR-T in GC.
Introduction
Gastric cancer (GC) is a common malignant tumor with a heavy burden all over the world. According to the latest global cancer statistics, there were more than 1 million new cases and about 783,000 deaths of GC in 2018, making it as the third leading cause of cancer-related death globally.Citation1 East Asia is the most serious region of GC in the world.Citation1 Although in recent years, with the development of surgical technology, radiotherapy, neoadjuvant chemotherapy, the prognosis of GC has improved, but in most countries, the 5-year survival rate was only 20–40%.Citation2 Therefore, it is very important to find a new treatment strategy that can prolong the life of patients.
The immune system plays an important role in the occurrence and development of cancer.Citation3 Immunotherapy is a promising tumor treatment method.Citation4 In recent years, a variety of immunotherapies, including adoptive cell therapy (ACT)Citation5,Citation6 and immune checkpoint inhibitors (ICI),Citation7,Citation8 have been deeply studied and widely used. Chimeric antigen receptor (CAR) T cell therapy is a novel kind of ACT. CAR-Ts are T cells transformed with a single-chain variable fragment (scFv) of the antibody of tumor associated antigen (TAA) to recognize tumor cells and activate T cells in a major histocompatibility complex (MHC) independent manner.Citation9,Citation10 CAR-Ts have achieved remarkable clinical effects in blood tumors.Citation11,Citation12 In the basic research of CAR-T in GC, since 2015, researchers have developed CARs targeting NKG2DL,Citation13,Citation14 CEA,Citation15,Citation16 HER2,Citation17,Citation18 FOLR1,Citation19 Claudin 18.2,Citation20 Mesothelin,Citation21,Citation22 Trop2,Citation23 PSCA,Citation24 PD-L1,Citation25 and so on, and optimized the design of CAR, such as CARs secreting cytokines,Citation15 CARs with multi-target tandems.Citation23 In clinical trials, a number of CAR-T targeting digestive system tumors are under investigation but have not yet achieved significant results.Citation26–29 Thus, it is necessary to find a more suitable target for CAR-T of GC.
Mesenchymal-epithelial transition factor (c-Met) is a receptor tyrosine kinase (RTK) encoded by MET proto-oncogene. The natural ligand of c-Met is hepatocyte growth factor (HGF), so c-Met is also called hepatocyte growth factor receptor (HGFR). A large number of studies have shown that the amplification or over-expression of MET can lead to a poor prognosis in GC patients.Citation30,Citation31 A meta-analysis of 14 independent studies showed that increased c-Met expression was significantly associated with decreased overall survival (HR 2.82, 95% CI 1.86–4.27) in GC.Citation32 In view of this, scientists considered the HGF/c-Met signaling pathway as a target for molecular targeted therapy of GC, and have successively developed the monoclonal antibodies of Rilotumumab (AMG102) targeting HGF,Citation33 and Onartuzumab (MetMab) targeting c-MetCitation34 in clinical trials, and achieved better clinical results. Therefore, we consider c-Met as a target for CAR-T of GC.
The occurrence and development of tumors are closely related to immune escape,Citation35 and immune checkpoints play an important role in immune escape.Citation36 Programmed cell death protein 1 (PD-1) and its ligand PD-L1 are important immune checkpoint proteins. Scientists have developed PD-1 monoclonal antibodies nivolumab,Citation37 pembrolizumab,Citation38 toripalimab,Citation39 SHR-1210,Citation40 PD-L1 monoclonal antibody avelumab,Citation41 and conducted a large number of clinical trials on GC.Citation42 Through a meta-analysis study, we found that these antibodies have a better anti-tumor efficacy in GC patients with PD-L1+, MSI-H, or EBV.+Citation7 The presence of the immunosuppressive microenvironment will reduce the efficacy of CAR-T in solid tumors. The PD1/CD28 chimeric-switch receptor (CSR) improves immunosuppression by fusing the extracellular domain of PD-1 with the transmembrane and intracellular domains of CD28, thus transforming the inhibitory signal of PD-1 into the activation signal of CD28.Citation43 CAR-Ts with PD1/CD28 CSR had a better anti-tumor effect than CAR-T combined with PD-1 antibody.Citation44
In this study, we used c-Met, a target for molecular targeted therapy of GC widely expressed on the surface of GC cells, as the target of CAR-T and detected the efficacy and safety of c-Met CAR-T in vitro and in vivo. The structure of PD1/CD28 CSR was optimized in cMet-PD1/CD28 CAR-T in order to enhance the efficacy of c-Met CAR-T by reversing PD-1 immunosuppression.
Materials and methods
Bioinformatics analysis
In order to study the expression of MET gene in GC, we downloaded the gene expression RNAseq data of gastric adenocarcinoma and normal gastric tissue from the TCGA GTEx dataset and the phenotype data from the TCGA STAD dataset in UCSC Xena (https://xenabrowser.net/) at December 16, 2019.Citation45 The expression of MET gene and its relationship with molecular type, clinicopathological stage, and Lauren type in GC patients were analyzed using R and RStudio software.
Immunohistochemistry (IHC)
With the approval of the Ethics Committee of Lanzhou University Second Hospital and the informed consent of patients, paraffin sections of GC and adjacent tissues from 50 patients with GC who were operated in Lanzhou University Second Hospital were obtained from the Department of Pathology. The steps of IHC are as follows: Following deparaffinization, rehydration, and antigen repair, sections were exposed in 3% H2O2 to eliminate endogenous peroxidase activity. Blocked in 3% bovine serum albumin (BSA) for 30 min at room temperature (RT), Rabbit anti-Met monoclonal antibody (Abcam, USA) was incubated as primary antibody overnight at 4 °C, followed by HRP-labeled Goat anti-Rabbit IgG (H + L) (Beyotime, China) as secondary antibody for 1 hour at RT. It is then colored by DAB and counterstained by hematoxylin. The average optical density (AOD) of IHC images was quantitatively analyzed by ImageJ software. The results of IHC were determined by a professional pathologist. The staining intensity was divided into 0 (non), 1+ (weak), 2+ (medium) and 3+ (strong). The staining area was divided into 0 (non), 1+ (<25%), 2+ (25%-50%), 3+ (≥50%). H-score = staining intensity × staining area.
Cell lines
Human GC cell lines (AGS, BGC-823, HGC-27, MGC-803, MKN-45, SGC-7901) and 293 T were purchased from Cell Culture Center of Chinese Academy of Medical Sciences (Beijing, China). GES-1 (human gastric mucosal epithelial cells) was stored in our laboratory. All the cell lines were identified by STR (short tandem repeats). GC cell lines were cultured in RPMI-1640 medium containing 10% fetal bovine serum (FBS). 293 T was cultured in DMEM high glucose medium containing 10% FBS. HGC27-OE was HGC-27 infected with MET over-expression lentivirus and screened by puromycin. MKN45-Luc is MKN-45 infected with Luciferase-Puromycin (Luc-Puro) lentivirus and screened by puromycin.
Chimeric antigen receptor plasmid design
The c-Met overexpression plasmid (MET-OE) used the mRNA coding region (CDS) of human MET gene transcript 2 (NM_000245) as the over-expression region. GV341-Puro (Shanghai Genechem Co., Ltd, China) was used as vector plasmid, digested by AgeI/NheI. The c-Met CAR plasmid is a second-generation CAR composed of c-Met scFv and CD8α signal peptide, hinge region, transmembrane region, and 4–1BB, CD3ζ. The c-Met scFv was designed according to the heavy and light chains of the humanized c-Met monoclonal antibody Onartuzumab (patent number: WO 2013/003680 A1). GV401-eGFP (Shanghai Genechem Co., Ltd., China) was used as vector plasmid, digested by BamHI. The cMet-PD1/CD28 CAR was a PD1/CD28 CSR structure added to c-Met CAR. The PD1/CD28 CSR was the extracellular domain of PD-1 (AA 1–155) combined with the transmembrane and cytoplasmic domains of CD28 (AA 141–220). Blank GV401 vector was used as the plasmid for Mock T as negative control.
Plasmids and lentivirus production
Vector was digested by High-Fidelity Restriction Enzyme (NEB, USA), target gene was amplified by Super-Fidelity DNA Polymerase (Vazyme, China), and then recombined them with DNA ligase (Vazyme, China). The recombined vectors were transformed and amplified in E. coli. The target plasmids were extracted from E. coli (TIANGEN, China).
Lentivirus were produced in 293 T cells via Lipofectamin 2000 Transfection Reagent (Invitrogen, USA). The target plasmids, core plasmids, and envelope plasmids were transfected at the ratio of 4:3:2 followed by concentrating lentivirus from supernatant 3 days after transfection by ultracentrifugation (Beckman) for 2 h at 25 000 rpm.
Generation and culture of CAR-T cells
A 6-well plate was coated with 1 μg/mL of anti-CD3 (clone OKT3) and anti-CD28 (clone CD28.2) antibodies at 4 °C, overnight, 1 day before blood collection. Peripheral blood mononuclear cells (PBMCs) were isolated from the blood of healthy donors. The use of human PBMCs was approved by the Ethics Committee of Lanzhou University Second Hospital, and all donors gave informed consent. The day of blood collection was signed as d0. After 4-hour culture, the suspension cells were transferred to the 6-well plate already coated with CD3 and CD28 antibodies. Then, a new 6-well plate was coated by 50 μg/mL of RetroNectin (Takara, Japan) at 4 °C, overnight, on d0. After 18-hour stimulation by CD3/CD28 antibodies, the suspension cells were transferred into the RetroNectin coated 6-well plate and infected by lentivirus at MOI = 10, 5 μg/mL of polybrene was used as an infection promoting agent. The day of lentivirus infection was marked as d1. CAR-T cells were cultured in X–VIVO 20 Serum-free Medium (Lonza, Switzerland) containing 200 U/mL of Recombinant Human IL-2 (PeproTech, USA). The cell density was kept at 1 × 106/mL, the medium was changed every 3 days.
Real-time PCR (RT-PCR)
The total RNA was extracted from GC cell lines or CAR-Ts by TRIzol (Invitrogen, USA), chloroform, isopropanol, ethanol, and dissolved in RNase-Free water. Then, RNA was reversely transcribed through PrimeScript RT reagent Kit with gDNA Eraser (Takara, Japan) and quantitated by TB Green Premix Ex Taq (Takara, Japan) according to the manufacturer’s instructions. GAPDH was used as internal control and calculated by 2−ΔΔCt. Primers: GAPDH-F: AGGTCGGAGTCAACGGATTT, GAPDH-R: TGACGGTGCCATGGAATTTG; MET-F: ATGAGAGCTGCACCTTGACT, MET-R: CACCAGCCATAGGACCGTAT; PD-L1-F: GAAAGTCAATGCCCCATACAAC, PD-L1-R: GGACTTGATGGTCACTGCTTGT; CD3ζ-F: GAGGAGTACGATGTTTTGGAC, CD3ζ-R: CTGTACTGAGACCCTGGTAAA; c-Met scFv-F: AGCAGTACTACGCCTACCCT, c-Met scFv-R: ATGCAGCCAGTAGCTTGTGA; PD1/CD28-F: GTGACTTCCACATGAGCGTG, PD1/CD28-R: TCCGGGAAATAGGGGACTTG.
Western blot (WB)
GC cell lines were harvested, lysed by RIPA Lysis Buffer (Solarbio, China), then boiled to prepare for protein. After electrophoresis and PVDF membrane transfer, the target PVDF membrane was blocked, incubated with primary antibody Rabbit anti-Met monoclonal antibody (Abcam, USA) or Rabbit anti-GAPDH Polyclonal antibody (proteintech, China) and secondary antibody HRP-conjugated Goat Anti-Rabbit IgG (H + L) (proteintech, China), and then exposed by electrochemiluminescence (ECL) (Solarbio, China).
Flow cytometry (FCM)
c-Met and PD-L1 expressions on the surface of GC cell lines: anti-c-Met FITC (clone eBioclone 97), anti-CD274 PE (clone MIH1) (eBioscience, USA) were added to the GC cells, incubated at 4 °C for 30 min, washed with cold phosphate-buffered saline (PBS), and resuspend in 200 μL PBS followed by the detection with BD FACSCalibur, analyzed by FlowJo software.
Infection efficiency: Firstly, the positive rate of eGFP was detected at d6. Secondly, Biotinylated Recombinant Protein L (AcroBiosystems, China) was used as primary antibody, incubated at 4 °C for 1 hour, washed with PBS, and PE Streptavidin (Biolegend, USA) was used as secondary antibody, incubated at 4 °C for 1 hour, and tested by FCM after washing.
Cell subgroups and phenotype of CAR-Ts: CAR-Ts were collected at d9-12, and anti-CD3 PE (clone OKT3), anti-CD4 APC (clone OKT4), anti-CD8a APC (clone HIT8a) (MultiSciences, China) were used to detect the proportion of CD3+CD4+ and CD3+CD8+ T cells. Anti-CD45RO PE (clone UCHL1), anti-CD62L PE (clone DREG-56), anti-CCR7 APC (clone G043H7) (Biolegend, USA) were used to detect the phenotype of central memory T (TCM) cells in CAR-Ts.
Activation phenotype of CAR-Ts: CAR-Ts were collected at d9-12, co-cultured with target cells MKN-45 at an effector:target ratio of 2:1, anti-CD25 APC (clone BC96), anti-CD69 PE (clone FN50), anti-CD71 APC (clone CY1G4) (Biolegend, USA); anti-HLA-DR PerCP/Cy5.5 (clone LN3) (eBioscience, USA) were used to detect the changes of activation phenotype about CAR-Ts before or after stimulated by target cells.
PD-1 expression on CAR-Ts: CAR-Ts were collected at d9-12, co-cultured with target cells MKN-45 at an effector:target ratio of 2:1, anti-CD3 PerCp/Cy5.5 (clone OKT3) (MultiSciences, China) and anti-PD-1 PE (clone J105) (eBioscience, USA) were used to detect the change of PD-1 expression on CAR-Ts before or after stimulated by target cells.
Cytokine release assays
CAR-Ts (1 × 105) and MKN-45 were co-cultivated for 24 hours at an effector:target ratio of 5:1. The supernatant was collected, and human IFN-γ, TNF-α, IL-6, IL-10 ELISA Kit (MultiSciences, China) were used to detect the concentration of cytokines in the supernatant according to the manufacturer’s instructions.
Cytotoxicity Assay
CAR-Ts were collected at d12-15, co-cultured with target cells (1 × 104) at the effector:target ratios of 1:1 5:1, 10:1, for 24 hours, MKN-45, HGC-27, HGC27-OE were used as different target cells, respectively. The supernatant was collected and the CytoTox96 Non-Radioactive Cytotoxicity Assay (Promega, USA) was used to detect lactate dehydrogenase (LDH) release according to the manufacturer’s instruction.
CD107a degranulation reaction
CAR-Ts and MKN-45 (1 × 105) were co-cultured in a 96-well plate at a ratio of 5:1, and 5 μL/100 μL of anti-CD107a PE (clone H4A3) (eBioscience, USA) was added to the co-culture medium, incubated in the dark for 4 hours. Monensin Solution (eBioscience, USA) was added to the co-culture medium for another 8 hours. Then, cells were collected and anti-CD3 PerCP/Cy5.5 (clone OKT3), anti-CD8a APC (clone HIT8a) (MultiSciences, China) were added at the same time. The CD3+CD8+CD107a+ cells were detected by FCM.
In vivo experiments
Animal experiments were approved by the Institutional Animal Care and Use Committee of Lanzhou University Second Hospital, comply with the animal welfare. Female NOD-SCID mice aged 4–6 weeks were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. and housed in the SPF animal experiment center of Lanzhou University Second Hospital. The maximum diameter of the tumors did not exceed 15–20 mm.
For the MKN45-Luc xenograft models, 1 × 106 MKN5-Luc cells in 100 μL PBS were injected subcutaneously into one side of flank. Two weeks later, when the tumor nodes were palpable, the mice were randomly divided into three groups (Mock T, c-Met CAR-T, cMet PD1/CD28 CAR-T). Then, mice were intratumorally injected with 5 × 106 CAR-Ts, twice, on d0 and d7. Every 7 days, the bioluminescence images were acquired using In-Vivo FX PRO (Bruker), and the tumor volume was calculated by the formula: V = π/6 × L× WCitation2 ≈ 0.52 × L × W.Citation2 Besides, the body weight was analyzed every 7 days.
HE staining
On d28 (CAR-Ts group) or when the tumor diameter was more than 15–20 mm (Mock T group), the mice were sacrificed by cervical dislocation, and the stomach, small intestine, heart, liver, spleen, lung, kidney from each groups were collected, and fixed in 4% paraformaldehyde for 24 hours. The tissue was paraffin-embedded and sliced into 4-µm sections, baked at 65 °C for 1 h, deparaffinized in xylene, rehydrated by graded ethanol, stained with hematoxylin and eosin (HE).
Statistical analysis
Data are displayed as mean ± SD. Statistical significance tests were analyzed by Student’s t-test or variance. p < .05 indicates statistical significance. All statistical analysis and charts were performed by GraphPad Prism V8.3 software.
Results
C-Met expression was increased in GC
By bioinformatics analysis, we found that the transcription level of MET gene in GC tissues was significantly higher than that of normal gastric tissues (p < .0001). In addition, in molecular type, the MET transcription level of microsatellite instability (MSI) was higher than that of microsatellite stability (MSS) (p = .0025). In clinicopathological stage, the MET transcription level of stage III-IV was higher than stage I-II (p = .045). In Lauren type, the MET transcription level of intestinal type was higher than that of diffuse type (p = .019) (). Through the analysis of IHC staining of GC and adjacent normal tissue sections from 50 patients, it was found that the AOD and H-score of c-Met from GC tissue were significantly higher than that of adjacent tissue (p < .0001) (). PCR, WB and FCM were used to detect the MET gene transcription, c-Met protein expression, and c-Met expression on cell surface of 7 GC or gastric mucosal epithelial cells including GES-1, AGS, BGC-823, HGC-27, MGC-803, MKN-45 and SGC-7901. It was found that MKN-45 has the highest c-Met expression and HGC-27 has the lowest in the above 7 cell lines (, D, E). These two cell lines were used in the following experiments. HGC-27 infected with MET-OE lentivirus could over-express c-Met in HGC-27 and construct HGC27-OE cells successfully which was confirmed by PCR, WB and FCM (p < .001) ().
Figure 1. The expression of c-Met was increased in the tissues and cells of gastric cancer. A, Differences in the expression of MET gene in gastric cancer and normal tissues, and the relationship between MET gene expression and microsatellite stability, pathological stage, Lauren classification in gastric cancer tissues. MSS: microsatellite stability, MSI: microsatellite instability. B, Immunohistochemical staining of c-Met in and adjacent tissues (a ~ c) and gastric cancer (d ~ f), and statistical analysis of AOD (g) and H-score (h), n = 50, (a ~ e) 200×, (f) 400 × . AOD: average optical density. C, Transcription levels of MET and PD-L1 genes in different gastric cancer cell lines detected by PCR, n ≥ 3. D, Expression levels of c-Met protein in different gastric cancer cell lines detected by WB, n ≥ 3. E, Expression levels of c-Met on the surface of gastric cancer cell lines detected by FCM, n ≥ 3. Column charts show mean ± SD, compared with GES-1 * p < .05, ** p < .01, *** p < .001, **** p < .0001, p values were determined by two-tailed, unpaired Student’s t test, or one-way ANOVA with Dunnett post hoc test
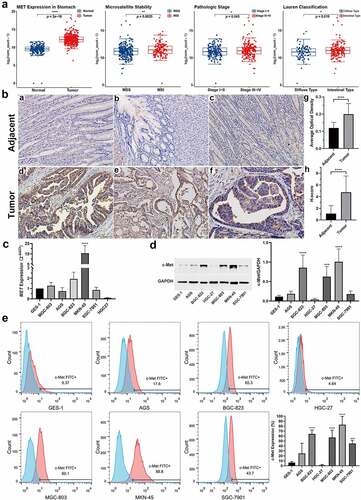
Figure 2. The structure and infection efficiency of CAR-Ts. A, c-Met expression in HGC27-OE cells after the infection of MET-OE lentivirus detected by (a) qRT-PCR, (b) WB, (c) FCM. B, The structures of c-Met CAR, cMet-PD1/CD28 CAR plasmids and PD1/CD28 CSR. C, The infection efficiency of c-Met CAR-T and cMet-PD1/CD28 CAR-T at MOI = 10 determined by eGFP+. D, The infection efficiency of c-Met CAR-T and cMet-PD1/CD28 CAR-T at MOI = 10 determined by Protein L+. E, Gene transcription levels of c-Met scFv and PD1/CD28 CSR after infection detected by PCR. CSR: chimeric-switch receptor, scFv: single-chain variable fragment. F, The proliferation of Mock T, c-Met CAR-T, cMet-PD1/CD28 CAR-T after lentivirus infection. Column charts represent mean ± SD, n ≥ 3, *** p < .001, **** p < .0001, p values were determined by two-tailed, unpaired Student’s t test or one-way ANOVA with Dunnett post hoc test
CAR-T could be successfully constructed by lentivirus
The structure of CAR-Ts is shown in . c-Met CAR and cMet-PD1/CD28 CAR lentivirus have a high infection efficiency at MOI = 10, the infection efficiency of c-Met CAR-T was 59.78 ± 9.83%, and the infection efficiency of cMet-PD1/CD28 CAR-T reached 45.51 ± 6.65% (). Protein L can combine with the κ light chain of scFv in CAR. When MOI = 10, according to the proportion of Protein L positive cells (minus Mock T), the infection efficiency of c-Met CAR-T was 54.81 ± 3.97%, and that of cMet-PD1/CD28 CAR-T was 45.21 ± 6.89% (). qRT-PCR showed that the transcription levels of c-Met scFv and PD1/CD28 CSR were significantly increased (p < .0001) after infection by MOI = 10 (). In addition, c-Met CAR-T and cMet-PD1/CD28 CAR-T made by lentivirus have a good proliferation activity. On the 15th day after infection, the expansion fold reached 20 times ().
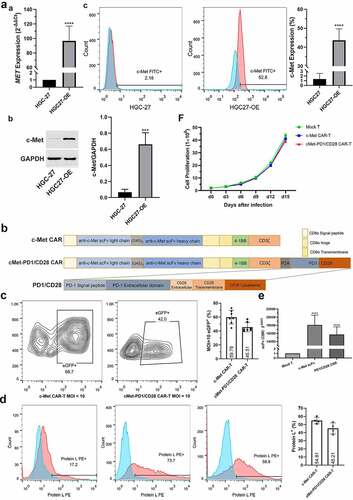
CD8+ T cells and CD62L+CCR7+ central memory T (TCM) cells in CAR-Ts were increased
On the d9 after infection, the proportions of CD3+CD4+, CD3+CD8+ subgroups and TCM phenotype of CD45RO+, CD62L+CCR7+ were detected. Compared with Mock T, the ratio of CD3+CD8+ was increased in c-Met CAR-T (p = .040), but there was no significant change in CD3+CD4+ (p = .155). Compared with Mock T, the proportion of CD3+CD8+ in cMet-PD1/CD28 CAR-T was increased (p = .018), and that of CD3+CD4+ was decreased (p = .045). Compared with c-Met CAR-T, the proportion of CD3+CD4+ and CD3+CD8+ in cMet-PD1/CD28 CAR-T were not significantly changed (). In the central memory phenotype, compared with Mock T, the proportion of CD62L+CCR7+ in c-Met CAR-T was increased (p = .007), but no difference on CD45RO+ (p = .238). Compared with Mock T, the proportion of CD62L+CCR7+ was increased (p < .0001) in cMet-PD1/CD28 CAR-T, and the CD45RO+ also increased (p = .027). Compared with c-Met CAR-T, the proportion of CD62L+CCR7+ in cMet-PD1/CD28 CAR-T was increased (p = .009), but CD45RO+ did not change significantly (p = .059). The proportion of CD62L+CCR7+ TCM in two CAR-Ts was increased, indicating that the PD1/CD28 CSR structure may further increase the proportion of CD62L+CCR7+ TCM cells ().
Figure 3. The changes of subgroups and central memory phenotype of CAR-Ts. A, Changes of CD3+CD4+ and CD3+CD8+ subgroups in two CAR-Ts. B, Changes of CD45RO+ and CD62L+CCR7+ TCM phenotype in two CAR-Ts. TCM: central memory T cell. Column charts represent mean ± SD, n ≥ 3, compared with Mock T * p < .05, ** p < .01, *** p < .001, **** p < .0001, p values were determined by one-way ANOVA with Tukey post hoc test
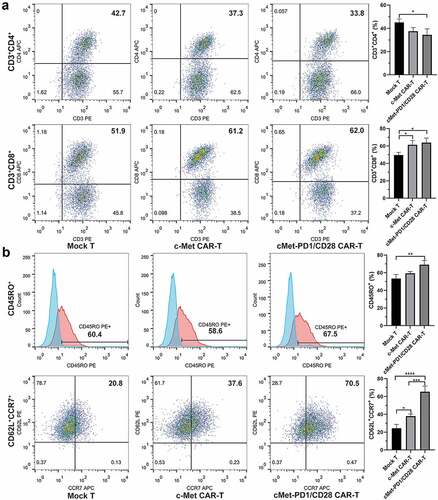
The activated phenotype and PD-1 expression were increased in c-Met CAR-T after being stimulated by target cells
At d9~ d12 after infection, c-Met CAR-T were co-cultivated with the target cell MKN-45 for 24 hours at a ratio of effector:target = 2:1. FCM found that the early activation marker CD69 and mid-term activation marker CD71 in both Mock T and c-Met CAR-T were increased after stimulation by MKN-45 (p < .05), while the expression of activation marker CD25 and HLA-DR did not change significantly (). FCM found that the expression level of PD-1 in cMet-PD1/CD28 CAR-T was significantly higher than that of c-Met CAR-T (p = .0003), indicating that the PD1/CD28 CSR was infected successfully. At d9~ d12, two CAR-Ts were co-cultured with MKN-45 at the 2:1 effector:target ratio for 24 h. After co-cultivation, the expression of PD-1 in Mock T, c-Met CAR-T, cMet-PD1/CD28 CAR-T was increased (p < .05), indicating that the stimulation of target cells could promote the expression of PD-1 in CAR-Ts ().
Figure 4. The changes of activation phenotype and PD-1 expression of CAR-Ts before or after the stimulation by target cells. A, Changes of activation phenotype of CD25, CD69, CD71, HLA-DR in c-Met CAR-T before or after MKN-45 stimulation. B, Changes of PD-1 expression in different CAR-Ts before or after MKN-45 stimulation. CAR-Ts and MKN-45 were co-cultured for 24 hours at a ratio of 2:1. The column chart shows mean ± SD, n ≥ 3, * p < .05, ** p < .01, *** p < .001. Statistical analysis was carried out by one-way ANOVA with Sidak post hoc test
PD1/CD28 stimulated the secretion of IFN-γ and TNF-α, inhibit the secretion of IL-6 in CAR-Ts
At d12-d15 after infection, the effector (CAR-Ts) and target cells (MKN-45, HGC-27, HGC27-OE) were co-cultured at the ratio of 5:1 for 24 hours, then the supernatant was collected. The cytokine secretion in the supernatant was detected by ELISA. We found that compared with Mock T, the secretion of IFN-γ and TNF-α was increased in c-Met CAR-T (p < .0001), and the inflammatory factor IL-6 and immunosuppressive factor IL-10 also increased (p < .0001). Compared with c-Met CAR-T, PD1/CD28 CSR could further increase the secretion level of IFN-γ and TNF-α (p < .001), while reducing the secretion of IL-6 (p < .0001). In addition, the level of cytokine release was related to the c-Met expression on the surface of target cells. In HGC-27 cells, which with very low c-Met expression, stimulate the CAR-T with a low level of cytokine secretion. After stimulation of CAR-Ts with HGC27-OE which over-expressed MET, the secretion levels of the above four cytokines were significantly increased (p < .0001) ().
Figure 5. The cytokine secretion, killing capacity, degranulation level were increased in two CAR-Ts. A, Different secretion levels of IFN-γ, TNF-α, IL-6, IL-10 in c-Met CAR-T, cMet-PD1/CD28 CAR-T after the stimulation by different target cells. The two CAR-Ts were incubated with different target cells at the ratio of 5:1 for 24 hours. B, The killing ability of c-Met CAR-T and cMet-PD1/CD28 CAR-T on different target cells under different effect:target ratios, and the effect of c-Met expression in target cells on the killing ability of CAR-Ts. CAR-Ts were co-cultured with different target cells at the ratios of 10:1, 5:1, and 1:1 for 24 hours. C, CD107a degranulation levels of c-Met CAR-T and cMet-PD1/CD28 CAR-T after stimulation by MKN-45. CAR-Ts and MKN-45 were co-cultured at the ratio of 5:1 for 12 hours. Column charts or line charts represent mean ± SD, n ≥ 3, * p < .05, *** p < .001, **** p < .0001, statistical analysis was conducted by one-way ANOVA with Sidak post hoc test
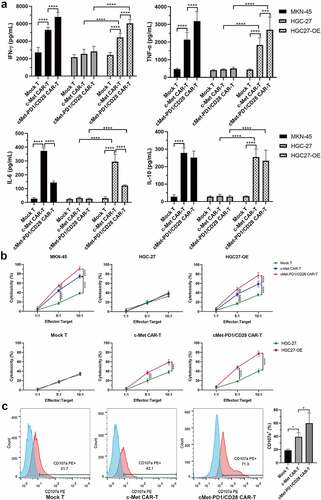
PD1/CD28 further enhanced the killing ability of c-Met CAR-T on target cells and increase the level of CD107a degranulation
At d12-d15 after infection, different effectors (CAR-Ts) and target cells (MKN-45, HGC-27, HGC27-OE) were co-cultured at different effector:target ratios of 10:1, 5:1, 1:1 for 24 hours, the supernatant of the co-culture medium was collected, and the killing activity of CAR-Ts was reflected by the amount of LDH release. In MKN-45 and HGC27-OE cells, the LDH release level of c-Met CAR-T was higher than that of Mock T when the effect target ratio was 5:1 or 10:1 (p < .0001), and the LDH release level of cMet-PD1/CD28 CAR-T was higher than that of c-Met CAR-T at these two ratios (p < .01) (), indicating that the killing activities of CAR-Ts against c-Met positive GC cells (MKN-45, HGC27-OE) were gradually increased with the increase in the effector:target ratio, and PD1/CD28 CSR could further enhance the killing ability of c-Met CAR-T. If the target cell was HGC-27, in which c-Met expression was very low, the LDH release level did not improve significantly when the effector:target ratio increased (). However, if the target cell was HGC27-OE which overexpressed c-Met, when the effector:target ratio was 5:1 or 10:1, the killing abilities of two CAR-Ts on HGC27-OE were significantly improved than HGC-27 (p < .0001) (), indicating that the killing ability of c-Met CAR-T to target cells depends on the expression of c-Met on target cells.
At d12~ d15, effector (CAR-Ts) and target cells (MKN-45) were co-cultured at the ratio of 5:1 for 12 hours. According to FCM, we found the CD107a expression level on the surface of c-Met CAR-T was higher than Mock T (p = .032), and cMet-PD1/CD28 CAR-T could further increase the CD107a degranulation level than c-Met CAR-T (p = .030) ().
c-Met CAR-T and cMet-PD1/CD28 CAR-T had obvious inhibitory effects on c-Met positive subcutaneous GC tumors, and no obvious off-target toxicity to normal organs in vivo
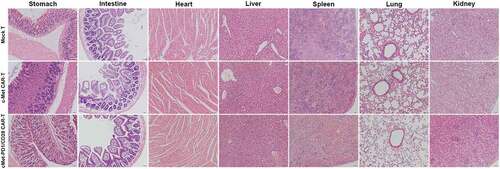
In the subcutaneous GC tumor mouse model made by MKN-45, both c-Met CAR-T and cMet-PD1/CD28 CAR-T treatments could decrease the total bioluminescence intensity and tumor volume than Mock T (p < .01). We find that the bioluminescence intensity and tumor volume in Mock T group gradually increased with time, while the bioluminescence intensity and tumor volume decreased significantly after the c-Met CAR-T and cMet-PD1/CD28 CAR-T treatment. Furthermore, on day 21 after CAR-T treatment, the total bioluminescence intensity and tumor volume of cMet-PD1/CD28 CAR-T treatment group were significantly less/smaller than those of c-Met CAR-T group (p < .01) (, C, D). Besides, in the CAR-Ts treatment groups, mice were sacrificed and normal tissues of important organs were collected at d28. From HE staining of stomach, small intestine, heart, liver, spleen, lung and kidney, we found that neither c-Met CAR-T nor cMet-PD1/CD28 CAR-T caused inflammation or necrosis of normal tissues in these normal tissues of important organs ().
Figure 6. The two CAR-Ts have obvious anti-tumor activity in subcutaneous xenograft model of gastric cancer. A, Flow chart of in vivo experiment. B, Bioluminescence imaging of MKN-45 subcutaneous tumor model at different time points. C, Statistical analysis of total bioluminescence intensity at different time points and on d21. D, Statistical analysis of tumor volumes at different time points and on d21. E, Statistical analysis of body weight at different time points. s.c.: subcutaneously, i.t.: intratumorally, BLI: Bioluminescence Imaging. Column charts and line charts represent mean ± SD, n ≥ 3, ** p < .01, the Multiple t test or Student ’s t test was used for statistical analysis
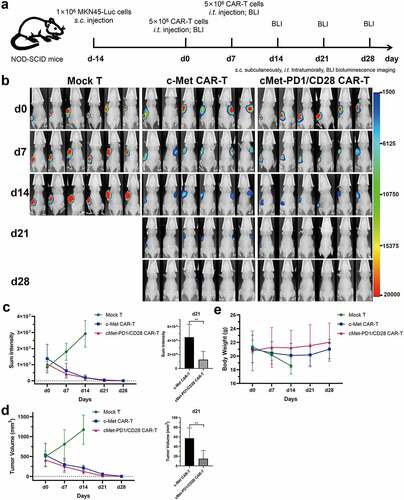
Figure 7. HE staining of normal tissues of stomach, small intestine, heart, liver, spleen, lung and kidney in different CAR-T treatment groups
Discussion
In this study, we designed c-Met CAR-T and verified its efficacy and safety in GC through in vitro and in vivo experiments. Then, we optimized the design of CAR-T by adding PD1/CD28 CSR, constructed cMet-PD1/CD28 CAR-T, to increase the anti-tumor efficacy and improve the security of c-Met CAR-T.
By analyzing the stomach adenocarcinoma data in the TCGA database, we found that the MET gene transcription level of tumor tissue was much higher than that of normal tissue. Specifically, the transcription level was higher in GC patients with microsatellite instability (MSI), pathological stage III-IV, or intestinal type of Lauren type. By IHC of tissues from GC patients, we found that c-Met was expressed in cell membrane and cytoplasm, and the expression of c-Met in GC tissues was higher than that in adjacent normal tissues. These results provided strong support for the selection of c-Met as the target of CAR-T.
Scientists have developed one to four generations of CAR structures, of which the second-generation CAR has the structure of CD3ζ (T cell activation signal I) and costimulatory molecule CD28 or 4–1BB (T cell activation signal II), which is most commonly used in the current research.Citation46 Studies have shown that the 4–1BB co-stimulation structure in CAR-T could inhibit CAR-T cell depletion and led to a longer persistence than CD28.Citation47 Therefore, we used the 4–1BB second-generation CAR as our design. In view of the high expression of c-Met in GC tissues and GC cells, and the application of c-Met in molecular targeted therapy of GC, we chose c-Met as the target of CAR-T for GC. The anti-c-Met scFv was designed according to the gene sequence of heavy chain and light chain of c-Met antibody Onartuzumab. When using lentivirus for infection, the efficiency was more than 50%. Only two studies on c-Met CAR-T have been carried out. In 2017, Carl June’s team constructed a c-Met CAR-T based on c-Met antibody, and studied the efficacy and safety of c-Met CAR-T in breast cancer in basic and clinical trials.Citation48 In the same year, Thayaparan et al. designed a c-Met CAR based on the N and K1 domains of HGF which is the natural receptor of c-Met, and verified its efficacy in mesothelioma.Citation49 The above two c-Met CAR-T studies have shown that c-Met CAR-T has a better anti-tumor effect and higher safety. Our study found that CD8+ T cells, CD62L+CCR7+ TCM cells, CD69+, CD71+ activated phenotypes were increased in c-Met CAR-T, the secretion of cytokines such as TFN-γ, TNF-α, IL-6 were increased, and the killing activity of c-Met-positive GC cells was enhanced. However, the stimulation of target cells significantly increased the expression of PD-1 on c-Met CAR-T, and this may have a negative impact on the anti-tumor efficacy of CAR-T.
The efficacy of CAR-T clinical trials in solid tumors was not as significant as that in hematological tumors, which may be related to the complex immune microenvironment and the widespread existence of immunosuppressive signals in solid tumors.Citation50,Citation51 Immune checkpoint inhibitor (ICI) plays an important role in the immunotherapy of gastric cancer.Citation7 In view of the different anti-tumor principles and respective advantages of ICI and CAR-T, scientists have proposed many strategies for the combined application of ICI and CAR-T and found the combination has a synergistic effect. In 2013, John et al. found that combining the PD-1 antibody pembrolizumab with HER2 CAR-T could enhance the inhibitory effect of CAR-T on tumor growth.Citation52 Gargett et al. found that PD-1 blockade could reduce GD2 CAR-T cell death caused by repeated antigen stimulation and improve CAR-T survival.Citation53 In 2017, researchers from the University of Pennsylvania reported a case report of a patient with diffuse large B-cell lymphoma that failed in CD19 CAR-T treatment; however, after combining with PD-1 monoclonal antibody pembrolizumab, the CD19 CAR-T amplification was enhanced, and an obvious anti-tumor reaction was appeared.Citation54 However, this combination of CAR-T and ICI may directly increase toxic and adverse effects. In addition to combination therapy, some scholars considered knocking out the expression of PD-1 on CAR-T cells, and they used technologies such as CRISPR/Cas9,Citation55,Citation56 TALENCitation57 to generate PD-1 knockout CAR- T cells, thereby eliminating the immunosuppression caused by PD-1/PD-L1 and improving the efficacy of CAR-T. In Ren J’s study, they simultaneously knocked out three genes, TCR, β2 microglobulin, and PD-1 by CRISPR/Cas9 and generated allogeneic universal CAR-Ts.Citation58 However, it has been confirmed that proper PD-1 expression can protect CD8+ T cells from over stimulation, excessive proliferation, and terminal differentiation. The complete deletion of PD-1 may lead to the failure of CD8+ T cells.Citation59 Therefore, completely knocking out PD-1 may adversely affect the activity of CAR-T. In addition to the above two methods, adding a gene sequence which can secrete PD-1 or PD-L1 antibody in CAR structure could make CAR-T secrete PD-1/PD-L1 antibody by paracrine or autocrine while binding with tumor cells. It can not only eliminate immunosuppression but also reduce the adverse effects of systemic application of PD-1 inhibitors. In 2016, Suarez et al. inserted a PD-L1 antibody fragment in the CAR-T targeting carbonic anhydrase IX (CAⅨ) and found that the immunosuppressive molecules PD-1, Tim3, and LAG3 in this CAIX CAR-T were down-regulated and the ability of NK cells recruitment was enhanced.Citation60 Yuan et al. constructed a dual-function CAR-T targeting c-Met and PD-1 and found that it not only blocked PD-L1 but decreased the expression of LAG-3 and TIM-3.Citation61 Similarly, CD19 CAR-T containing anti-PD-1 scFv can also limit the up-regulation of PD-1 in CAR-T cells after antigen stimulation and enhance the proliferation ability of CAR-T.Citation62 In addition, Rafiq et al. found that the PD-1 antibody-secreting CAR-T could survive longer, and because the secreted PD-1 antibody is localized near the tumor, it could avoid the related toxicity caused by systemic application of ICI.Citation63 However, the design of adding the PD-1/PD-L1 antibody gene sequence to the CAR plasmid greatly increases the length of CAR plasmid which will improve the difficulty of CAR-T cell infection. The above strategies have certain advantages, but they have their own disadvantages.
The design of PD1/CD28 CSR could reverse the PD-1 inhibitory signal. The chimeric-switch receptor artificially combined extracellular domain of PD-1 with the transmembrane and cytoplasmic domains of CD28. In 2012, Prosser et al. designed this PD1/CD28 CSR and transferred it into CTL cells. They found that the PD1/CD28 structure could increase the phosphorylation levels of ERK and Akt and enhance the proliferation of CTL cells and secretion ability of cytokines.Citation43 In 2013, Ankri et al. compared the structure of PD1/CD28 and PD1/4-1BB chimeric-switch receptors and found PD1/CD28 secreted higher levels of cytokines such as IFN-γ and IL-2.Citation64 In 2016, the team of Yangbing Zhao and Carl June from the University of Pennsylvania introduced this PD1/CD28 CSR into CAR-T to construct mesothelin (MSLN) and prostate stem cell antigen (PSCA) CAR-Ts. They found that the anti-tumor effects of these PD1/CD28 CAR-Ts were better than the combined therapy of CAR-T and PD-1 antibody pembrolizumab.Citation44 In addition to the PD1/CD28 CSR, which could convert the PD-1 inhibitory signal into the CD28 activation signal, the over-expression of PD-1 extracellular domain on CAR-T can also competitively bind to the PD-L1 on tumor cells, which could hinder the signal transduction of PD-1/PD-L1.Citation65 In view of the advantages and disadvantages of the above schemes, we adopted the design of PD1/CD28 CSR in c-Met CAR-T.
In our study, we found the proportion of CD3+CD8+ T cells and CD62L+CCR7+ central memory T cell (TCM) phenotype in c-Met CAR-T were increased. What’s more, the ratio of CD8+ T cells and CD62L+CCR7+ TCM in cMet-PD1/CD28 CAR-T were further increased than c-Met CAR-T. The stimulation of target cells MKN-45 could increase the expression of activated phenotype CD69 and CD71 in c-Met CAR-T. All these changes in cell subgroups and phenotype may help to increase the anti-tumor activity of CAR-T.
The combination of CAR-T and tumor cells would stimulate the secretion of IFN-γ by CAR-T cells, which can promote the expression of PD-L1 on tumor cells and suppress the anti-tumor immune response through PD-1/PD-L1 signals.Citation66,Citation67 In our research, we found that the expression of PD-1 on c-Met CAR-T cells would increase after the stimulation by target cells (MKN-45). Therefore, we design PD1/CD28 CSR structure in CAR-T, in order to reduce the immunosuppression caused by PD-1 over-expression.
In terms of the cytotoxicity to target cells, our study found that the killing effect of c-Met CAR-T on GC cells depends on the expression of c-Met on the cell surface. The cytotoxicity was weak against the target cells with low expression of c-Met (HGC-27), but was strong against the target cells with high expression of c-Met (MKN-45, HGC27-OE). Besides, the CD107a degranulation reaction of c-Met CAR-T was stronger than blank control (Mock T). Compared with c-Met CAR-T, the killing ability of cMet-PD1/CD28 CAR-T against target GC cells with high expression of c-Met (MKN-45, HGC27-OE) was further enhanced, and CD107a degranulation reaction was also enhanced. In terms of cytokine secretion, we found that after co-culture with c-Met-positive GC cells (MKN-45, HGC27-OE), the secretion of anti-tumor cytokines such as IFN-γ and TNF-α in c-Met CAR-T was increased. But the secretion of the immunosuppressive factor IL-10 and the inflammatory factor IL-6 was also increased. It has been confirmed that the abnormal increase in IL-6 secretion was related to adverse reactions such as cytokine release syndrome (CRS) caused by CAR-T.Citation68 After optimization by PD1/CD28 CSR, the secretion of IFN-γ and TNF-α was further increased in cMet-PD1/CD28 CAR-T compared to c-Met CAR-T. However, the secretion of the inflammatory factor IL-6 was decreased in cMet-PD1/CD28 CAR-T. It indicated that the PD1/CD28 CSR could enhance the anti-tumor effect and reduce the incidence of CRS.
Through in vivo animal experiments, we found that both c-Met CAR-T and PD1/CD28 CAR-T could significantly inhibit the growth of gastric cancer subcutaneous tumor in mouse model and did not cause serious off-target toxicity on normal stomach, small intestine, heart, liver, spleen, lung and kidney. PD1/CD28 CSR structure could further inhibit the growth of subcutaneous tumor model than c-Met CAR-T, especially, significantly improve the long-term anti-tumor effect (d21) which may be related to the increase in central memory phenotype confirmed by cell experiments.
In addition to enhancing the function of CAR-T cells and improving the immunosuppressive microenvironment, homing and infiltration of tumor cells is also important to CAR-T. Chemokine and chemokine receptors play a key role in mediating the directed migration of CAR T cells. In 2018, Adachi K et al. developed a kind of CAR-T that could secrete IL-7 and CCL19. It was found that the secretion of cytokine IL-7 and chemokine CCL19 could promote the infiltration of T cells and dendritic cells (DC) in tumor tissue, reduce T cell depletion, and improve the anti-tumor activity of CAR-T.Citation69 In addition, IL-7 and CCL21 also significantly improve the proliferation and chemotaxis of CAR-T cells, enhancing the survival and infiltration of CAR-T and DC; besides, CCL21 could inhibit tumor angiogenesis.Citation70 The modification of IL-8 receptor CXCR1 or CXCR2 significantly enhanced the migration and persistence of CAR-T cells in tumors.Citation71 CAR-T cells expressing CXCR2 modified by integrin αvβ6-CAR migrated more effectively to tumor cells producing IL-8 receptors.Citation72 CCR4 could promote the migration of CD30 CAR-Ts to Hodgkin’s lymphoma cells that secreting CCL17 and CCL22,Citation73 and CCR2b could improve the tumor homing of Meso CAR-T.Citation74 Chemotherapeutic drugs such as doxorubicin or docetaxel could promote the secretion of CXCR3 ligand CXCL9, 10, 11, thus promoting the transport of T cells to tumor.Citation75,Citation76 In addition to chemokines, some scientists also consider targeting tumor extracellular matrix (ECM), which contain cancer-associated stromal cells (CASCs) and heparan sulfate proteoglycan. Fibroblast activation protein (FAP) is the surface mark of CASCs, and FAP CAR-T could inhibit tumor matrix formation and angiogenesis by targeting FAP+ CASCs.Citation77 CAR-T cells expressing heparanase could degrade heparan sulfate proteoglycan, destroy ECM, and enhance the invasive ability and anti-tumor activity of GD2 CAR-T.Citation78 In our research, we used intratumoral injection of CAR-T cells in vivo experiments, and CAR-T cells were directly located inside the solid tumor. In further research, we need to consider intravenous injection of CAR-T, and ways to enhance the chemotaxis of CAR-T and tumor infiltrating lymphocytes.
In conclusion, c-Met CAR-T has an effective cytotoxicity on c-Met positive GC cells in vitro and can significantly inhibit the growth of GC tumor models in vivo without off-target toxicity. PD1/CD28 CSR can further enhance the anti-tumor activity of c-Met CAR-T, increase the proportion of central memory T cells, prolong the long-term anti-tumor effect, and reduce the secretion of inflammatory factor IL-6, which could reduce the incidence of cytokine release syndrome.
Our study provides a new research direction and design idea for the development of CAR-T for GC. The application of PD1/CD28 CSR plays a positive role in improving the efficacy of CAR-T in solid tumors. The efficacy and safety of c-Met CAR-T and cMet-PD1/CD28 CAR-T for advanced GC patients should be verified by PDX model and clinical trials in the following research.
Authors’ contributions
CC and YL designed the research, wrote the manuscript; YL and HL funded the research; CC and YG performed the experiments, analyzed the data; FZ, ZZ, YZ, YH, LW, NZ assisted in the experiments; FT, HL assisted in the experiments, retouched in writing.
Competing interests
The authors declared they have no conflict of interest.
Acknowledgments
We thank Ms. Lu-xi Yang, Ms. Hui-juan Cheng from Key Laboratory of Digestive System Tumors of Gansu Province and Hai-ning Li from Gansu Provincial Cancer Hospital for the assistance with FCM. We thank Dr. Fu-rong Wang from the Pathology Department of Lanzhou University Second Hospital for the help of IHC.
Data availability statement
All of the data and materials are available from the corresponding author upon reasonable ask.
Additional information
Funding
References
- Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–16. doi:10.3322/caac.21492.
- Allemani C, Matsuda T, Di Carlo V, Harewood R, Matz M, Nikšić M, Bonaventure A, Valkov M, Johnson CJ, Estève J, et al. , CONCORD working group. global surveillance of trends in cancer survival 2000–14 (CONCORD-3): analysis of individual records for 37 513 025 patients diagnosed with one of 18 cancers from 322 population-based registries in 71 countries. Lancet. 2018;391:1023–1075. doi:10.1016/S0140-6736(17)33326-3.
- Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature. 2017;541(7637):321–330. doi:10.1038/nature21349.
- Couzin-Frankel J. Breakthrough of the year 2013. Cancer Immunotherapy Science. 2013;342:1432–1433. doi:10.1126/science.342.6165.1432.
- Chen C, Ma YH, Zhang YT, Zhang F, Zhou N, Wang X, Liu T, Li YM. Effect of dendritic cell-based immunotherapy on hepatocellular carcinoma: a systematic review and meta-analysis. Cytotherapy. 2018;20(8):975–989. doi:10.1016/j.jcyt.2018.06.002.
- Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science. 2015;348(6230):62–68. doi:10.1126/science.aaa4967.
- Chen C, Zhang F, Zhou N, Gu YM, Zhang YT, He YD, Wang L, Yang LX, Zhao Y, Li YM. Efficacy and safety of immune checkpoint inhibitors in advanced gastric or gastroesophageal junction cancer: a systematic review and meta-analysis. Oncoimmunology. 2019;8(5):e1581547. doi:10.1080/2162402X.2019.1581547.
- Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science. 2018;359:1350–1355. doi:10.1126/science.342.6165.1432.
- June CH, Sadelain M. Chimeric antigen receptor therapy. N Engl J Med. 2018;379(1):64–73. doi:10.1056/NEJMra1706169.
- June CH, O’Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science. 2018;359(6382):1361–1365. doi:10.1126/science.aar6711.
- Wang M, Munoz J, Goy A, Locke FL, Jacobson CA, Hill BT, Timmerman JM, Holmes H, Jaglowski S, Flinn IW, et al. X19 CAR T-cell therapy in relapsed or refractory mantle-cell lymphoma. N Engl J Med. 2020;382(14):1331–1342. doi:10.1056/NEJMoa1914347.
- Ghorashian S, Kramer AM, Onuoha S, Wright G, Bartram J, Richardson R, Albon SJ, Casanovas-Company J, Castro F, Popova B, et al. Enhanced CAR T cell expansion and prolonged persistence in pediatric patients with ALL treated with a low-affinity CD19 CAR. Nat Med. 2019;25(9):1408–1414. doi:10.1038/s41591-019-0549-5.
- Liu X, Sun M, Yu S, Liu K, Li X, Shi H. Potential therapeutic strategy for gastric cancer peritoneal metastasis by NKG2D ligands-specific T cells. Onco Targets Ther. 2015;8:3095–3104. doi:10.2147/OTT.S91122.
- Tao K, He M, Tao F, Xu G, Ye M, Zheng Y, Li Y. Development of NKG2D-based chimeric antigen receptor-T cells for gastric cancer treatment. Cancer Chemother Pharmacol. 2018;82(5):815–827. doi:10.1007/s00280-018-3670-0.
- Shibaguchi H, Luo N, Shirasu N, Kuroki M, Kuroki M. Enhancement of antitumor activity by using a fully human gene encoding a single-chain fragmented antibody specific for carcinoembryonic antigen. Onco Targets Ther. 2017;10:3979–3990. doi:10.2147/OTT.S140174.
- Chi X, Yang P, Zhang E, Gu J, Xu H, Li M, Gao X, Li X, Zhang Y, Xu H, et al. Significantly increased anti-tumor activity of carcinoembryonic antigen-specific chimeric antigen receptor T cells in combination with recombinant human IL-12. Cancer Med. 2019;8(10):4753–4765. doi:10.1002/cam4.2361.
- Han Y, Liu C, Li G, Li J, Lv X, Shi H, Liu J, Liu S, Yan P, Wang S, et al. Antitumor effects and persistence of a novel HER2 CAR T cells directed to gastric cancer in preclinical models. Am J Cancer Res. 2018;8:106–119.
- Song Y, Tong C, Wang Y, Gao Y, Dai H, Guo Y, Zhao X, Wang Y, Wang Z, Han W, et al. Effective and persistent antitumor activity of HER2-directed CAR-T cells against gastric cancer cells in vitro and xenotransplanted tumors in vivo. Protein Cell. 2018;9(10):867–878. doi:10.1007/s13238-017-0384-8.
- Kim M, Pyo S, Kang CH, Lee CO, Lee HK, Choi SU, Park CH. Folate receptor 1 (FOLR1) targeted chimeric antigen receptor (CAR) T cells for the treatment of gastric cancer. PLoS One. 2018;13:e0198347. doi:10.1371/journal.pone.0198347.
- Jiang H, Shi Z, Wang P, Wang C, Yang L, Du G, Zhang H, Shi B, Jia J, Li Q, et al. Claudin18.2-specific chimeric antigen receptor engineered t cells for the treatment of gastric cancer. J Natl Cancer Inst. 2019;111:(4):409–418. doi:10.1093/jnci/djy134.
- Lv J, Zhao R, Wu D, Zheng D, Wu Z, Shi J, Wei X, Wu Q, Long Y, Lin S, et al. Mesothelin is a target of chimeric antigen receptor T cells for treating gastric cancer. J Hematol Oncol. 2019;12(1):18. doi:10.1186/s13045-019-0704-y.
- Zhang Z, Jiang D, Yang H, He Z, Liu X, Qin W, Li L, Wang C, Li Y, Li H, et al. Modified CAR T cells targeting membrane-proximal epitope of mesothelin enhances the antitumor function against large solid tumor. Cell Death Dis. 2019;10(7):476. doi:10.1038/s41419-019-1711-1.
- Zhao W, Jia L, Zhang M, Huang X, Qian P, Tang Q, Zhu J, Feng Z. The killing effect of novel bi-specific Trop2/PD-L1 CAR-T cell targeted gastric cancer. Am J Cancer Res. 2019;9:1846–1856.
- Wu D, Lv J, Zhao R, Wu Z, Zheng D, Shi J, Lin S, Wang S, Wu Q, Long Y, et al. PSCA is a target of chimeric antigen receptor T cells in gastric cancer. Biomark Res. 2020;8(1):3. doi:10.1186/s40364-020-0183-x.
- Qin L, Zhao R, Chen D, Wei X, Wu Q, Long Y, Jiang Z, Li Y, Wu H, Zhang X, et al. Chimeric antigen receptor T cells targeting PD-L1 suppress tumor growth. Biomark Res. 2020;8(1):19. doi:10.1186/s40364-020-00198-0.
- Zhang C, Wang Z, Yang Z, Wang M, Li S, Li Y, Zhang R, Xiong Z, Wei Z, Shen J, et al. Phase I escalating-dose trial of CAR-T therapy targeting CEA(+) metastatic colorectal cancers. Mol Ther. 2017;25(5):1248–1258. doi:10.1016/j.ymthe.2017.03.010.
- Feng K, Liu Y, Guo Y, Qiu J, Wu Z, Dai H, Yang Q, Wang Y, Han W. Phase I study of chimeric antigen receptor modified T cells in treating HER2-positive advanced biliary tract cancers and pancreatic cancers. Protein Cell. 2018;9(10):838–847. doi:10.1007/s13238-017-0440-4.
- Beatty GL, O’Hara MH, Lacey SF, Torigian DA, Nazimuddin F, Chen F, Kulikovskaya IM, Soulen MC, McGarvey M, Nelson AM, et al. Activity of mesothelin-specific chimeric antigen receptor t cells against pancreatic carcinoma metastases in a phase 1 trial. Gastroenterology. 2018;155(1):29–32. doi:10.1053/j.gastro.2018.03.029.
- Guo Y, Feng K, Liu Y, Wu Z, Dai H, Yang Q, Wang Y, Jia H, Han W. Phase I study of chimeric antigen receptor-modified T cells in patients with EGFR-positive advanced biliary tract cancers. Clin Cancer Res. 2018;24(6):1277–1286. doi:10.1158/1078-0432.CCR-17-0432.
- Catenacci DV, Ang A, Liao WL, Shen J, O’Day E, Loberg RD, Cecchi F, Hembrough T, Ruzzo A, Graziano F. MET tyrosine kinase receptor expression and amplification as prognostic biomarkers of survival in gastroesophageal adenocarcinoma. Cancer. 2017;123(6):1061–1070. doi:10.1002/cncr.30437.
- Fuse N, Kuboki Y, Kuwata T, Nishina T, Kadowaki S, Shinozaki E, Machida N, Yuki S, Ooki A, Kajiura S, et al. Prognostic impact of HER2, EGFR, and c-MET status on overall survival of advanced gastric cancer patients. Gastric Cancer. 2016;19(1):183–191. doi:10.1007/s10120-015-0471-6.
- Peng Z, Zhu Y, Wang Q, Gao J, Li Y, Li Y, Ge S, Shen L. Prognostic significance of MET amplification and expression in gastric cancer: a systematic review with meta-analysis. PLoS One. 2014;9(1):e84502. doi:10.1371/journal.pone.0084502.
- Catenacci D, Tebbutt NC, Davidenko I, Murad AM, Al-Batran SE, Ilson DH, Tjulandin S, Gotovkin E, Karaszewska B, Bondarenko I, et al. Rilotumumab plus epirubicin, cisplatin, and capecitabine as first-line therapy in advanced MET-positive gastric or gastro-oesophageal junction cancer (RILOMET-1): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2017;18(11):1467–1482. doi:10.1016/S1470-2045(17)30566-1.
- Shah MA, Bang YJ, Lordick F, Alsina M, Chen M, Hack SP, Bruey JM, Smith D, McCaffery I, Shames DS, et al. Effect of fluorouracil, leucovorin, and oxaliplatin with or without onartuzumab in HER2-negative, MET-positive gastroesophageal adenocarcinoma: the METGastric randomized clinical trial. JAMA Oncol. 2017;3(5):620–627. doi:10.1001/jamaoncol.2016.5580.
- Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3(11):991–998. doi:10.1038/ni1102-991.
- Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–264. doi:10.1038/nrc3239.
- Kang YK, Boku N, Satoh T, Ryu MH, Chao Y, Kato K, Chung HC, Chen JS, Muro K, Kang WK, et al. Nivolumab in patients with advanced gastric or gastro-oesophageal junction cancer refractory to, or intolerant of, at least two previous chemotherapy regimens (ONO-4538-12, ATTRACTION-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;390(10111):2461–2471. doi:10.1016/S0140-6736(17)31827-5.
- Shitara K, Özgüroğlu M, Bang YJ, Di Bartolomeo M, Mandalà M, Ryu MH, Fornaro L, Olesiński T, Caglevic C, Chung HC, et al. KEYNOTE-061 investigators. Pembrolizumab versus paclitaxel for previously treated, advanced gastric or gastro-oesophageal junction cancer (KEYNOTE-061): a randomised, open-label, controlled, phase 3 trial. Lancet. 2018;392:(10142):123–133. doi:10.1016/S0140-6736(18)31257-1.
- Wang F, Wei XL, Wang FH, Xu N, Shen L, Dai GH, Yuan XL, Chen Y, Yang SJ, Shi JH, et al. Safety, efficacy and tumor mutational burden as a biomarker of overall survival benefit in chemo-refractory gastric cancer treated with toripalimab, a PD-1 antibody in phase Ib/II clinical trial NCT02915432. Ann Oncol. 2019;30(9):1479–1486. doi:10.1093/annonc/mdz197.
- Xu J, Zhang Y, Jia R, Yue C, Chang L, Liu R, Zhang G, Zhao C, Zhang Y, Chen C, et al. Anti-PD-1 antibody SHR-1210 combined with apatinib for advanced hepatocellular carcinoma, gastric, or esophagogastric junction cancer: an open-label, dose escalation and expansion study. Clin Cancer Res. 2019;25(2):515–523. doi:10.1158/1078-0432.CCR-18-2484.
- Bang YJ, Ruiz EY, Van Cutsem E, Lee KW, Wyrwicz L, Schenker M, Alsina M, Ryu MH, Chung HC, Evesque L, et al. III, randomised trial of avelumab versus physician’s choice of chemotherapy as third-line treatment of patients with advanced gastric or gastro-oesophageal junction cancer: primary analysis of JAVELIN Gastric 300. Ann Oncol. 2018;29(10):2052–2060. doi:10.1093/annonc/mdy264.
- Kono K, Nakajima S, Mimura K. Current status of immune checkpoint inhibitors for gastric cancer. Gastric Cancer. 2020;23(4):565–578. doi:10.1007/s10120-020-01090-4.
- Prosser ME, Brown CE, Shami AF, Forman SJ, Jensen MC. Tumor PD-L1 co-stimulates primary human CD8(+) cytotoxic T cells modified to express a PD1: CD28chimeric receptor. Mol Immunol. 2012;51(3–4):263–272. doi:10.1016/j.molimm.2012.03.023.
- Liu X, Ranganathan R, Jiang S, Fang C, Sun J, Kim S, Newick K, Lo A, June CH, Zhao Y, et al. A chimeric switch-receptor targeting PD1 augments the efficacy of second-generation CAR T cells in advanced solid tumors. Cancer Res. 2016;76(6):1578–1590. doi:10.1158/0008-5472.CAN-15-2524.
- Goldman MJ, Craft B, Hastie M, Repečka K, McDade F, Kamath A, Banerjee A, Luo Y, Rogers D, Brooks AN, et al. Visualizing and interpreting cancer genomics data via the xena platform. Nat Biotechnol. 2020;38(6):675–678. doi:10.1038/s41587-020-0546-8.
- Sadelain M, Brentjens R, Rivière I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013;3(4):388–398. doi:10.1158/2159-8290.CD-12-0548.
- Zhao Z, Condomines M, Van Der Stegen S, Perna F, Kloss CC, Gunset G, Plotkin J, Sadelain M. Structural Design of Engineered Costimulation Determines Tumor Rejection Kinetics and Persistence of CAR T Cells. Cancer Cell. 2015;28(4):415–428. doi:10.1016/j.ccell.2015.09.004.
- Tchou J, Zhao Y, Levine BL, Zhang PJ, Davis MM, Melenhorst JJ, Kulikovskaya I, Brennan AL, Liu X, Lacey SF, et al. Safety and efficacy of intratumoral injections of chimeric antigen receptor (car) t cells in metastatic breast cancer. Cancer Immunol Res. 2017;5(12):1152–1161. doi:10.1158/2326-6066.CIR-17-0189.
- Thayaparan T, Petrovic RM, Achkova DY, Zabinski T, Davies DM, Klampatsa A, Parente-Pereira AC, Whilding LM, Van Der Stegen SJ, Woodman N, et al. T-cell immunotherapy of MET-expressing malignant mesothelioma. Oncoimmunology. 2017;6(12):e1363137. doi:10.1080/2162402X.2017.1363137.
- Pang Y, Hou X, Yang C, Liu Y, Jiang G. Advances on chimeric antigen receptor-modified T-cell therapy for oncotherapy. Mol Cancer. 2018;17(1):91. doi:10.1186/s12943-018-0840-y.
- Zhang E, Gu J, Xu H. Prospects for chimeric antigen receptor-modified T cell therapy for solid tumors. Mol Cancer. 2018;17(1):7. doi:10.1186/s12943-018-0759-3.
- John LB, Devaud C, Duong CP, Yong CS, Beavis PA, Haynes NM, Chow MT, Smyth MJ, Kershaw MH, Darcy PK. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin Cancer Res. 2013;19(20):5636–5646. doi:10.1158/1078-0432.CCR-13-0458.
- Gargett T, Yu W, Dotti G, Yvon ES, Christo SN, Hayball JD, Lewis ID, Brenner MK, Brown MP. GD2-specific CAR T cells undergo potent activation and deletion following antigen encounter but can be protected from activation-induced cell death by PD-1 blockade. Mol Ther. 2016;24(6):1135–1149. doi:10.1038/mt.2016.63.
- Chong EA, Melenhorst JJ, Lacey SF, Ambrose DE, Gonzalez V, Levine BL, June CH, Schuster SJ. PD-1 blockade modulates chimeric antigen receptor (CAR)-modified T cells: refueling the CAR. Blood. 2017;129(8):1039–1041. doi:10.1182/blood-2016-09-738245.
- Hu B, Zou Y, Zhang L, Tang J, Niedermann G, Firat E, Huang X, Zhu X. Nucleofection with Plasmid DNA for CRISPR/Cas9-mediated inactivation of programmed cell death protein 1 in CD133-specific CAR T cells. Hum Gene Ther. 2019;30(4):446–458. doi:10.1089/hum.2017.234.
- Rupp LJ, Schumann K, Roybal KT, Gate RE, Ye CJ, Lim WA, Marson A. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci Rep. 2017;7(1):737. doi:10.1038/s41598-017-00462-8.
- Gautron AS, Juillerat A, Guyot V, Filhol JM, Dessez E, Duclert A, Duchateau P, Poirot L. Fine and Predictable Tuning of TALEN Gene Editing Targeting for Improved T Cell Adoptive Immunotherapy. Mol Ther Nucleic Acids. 2017;9:312–321. doi:10.1016/j.omtn.2017.10.005.
- Ren J, Liu X, Fang C, Jiang S, June CH, Zhao Y. Multiplex Genome Editing to Generate Universal CAR T Cells Resistant to PD1 Inhibition. Clin Cancer Res. 2017;23(9):2255–2266. doi:10.1158/1078-0432.CCR-16-1300.
- Odorizzi PM, Pauken KE, Paley MA, Sharpe A, Wherry EJ. Genetic absence of PD-1 promotes accumulation of terminally differentiated exhausted CD8+ T cells. J Exp Med. 2015;212(7):1125–1137. doi:10.1084/jem.20142237.
- Suarez ER, Chang DK, Sun J, Sui J, Freeman GJ, Signoretti S, Zhu Q, Marasco WA. Chimeric antigen receptor T cells secreting anti-PD-L1 antibodies more effectively regress renal cell carcinoma in a humanized mouse model. Oncotarget. 2016;7(23):34341–34355. doi:10.18632/oncotarget.9114.
- Yuan X, Sun Z, Yuan Q, Hou W, Liang Q, Wang Y, Mo W, Wang H, Yu M. Dual-function chimeric antigen receptor T cells targeting c-Met and PD-1 exhibit potent anti-tumor efficacy in solid tumors. Invest New Drugs. 2021;39:34-51. doi:10.1007/s10637-020-00978-3..
- Li S, Siriwon N, Zhang X, Yang S, Jin T, He F, Kim YJ, Mac J, Lu Z, Wang S, et al. Enhanced Cancer Immunotherapy by chimeric antigen receptor-modified T cells engineered to secrete checkpoint inhibitors. Clin Cancer Res. 2017;23(22):6982–6992. doi:10.1158/1078-0432.CCR-17-0867.
- Rafiq S, Yeku OO, Jackson HJ, Purdon TJ, Van Leeuwen DG, Drakes DJ, Song M, Miele MM, Li Z, Wang P, et al. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nat Biotechnol. 2018;36(9):847–856. doi:10.1038/nbt.4195.
- Ankri C, Shamalov K, Horovitz-Fried M, Mauer S, Cohen CJ. Human T cells engineered to express a programmed death 1/28 costimulatory retargeting molecule display enhanced antitumor activity. J Immunol. 2013;191(8):4121–4129. doi:10.4049/jimmunol.1203085.
- Cherkassky L, Morello A, Villena-Vargas J, Feng Y, Dimitrov DS, Jones DR, Sadelain M, Adusumilli PS. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest. 2016;126(8):3130–3144. doi:10.1172/JCI83092.
- Abiko K, Matsumura N, Hamanishi J, Horikawa N, Murakami R, Yamaguchi K, Yoshioka Y, Baba T, Konishi I, Mandai M. IFN-γ from lymphocytes induces PD-L1 expression and promotes progression of ovarian cancer. Br J Cancer. 2015;112(9):1501–1509. doi:10.1038/bjc.2015.101.
- Qian J, Wang C, Wang B, Yang J, Wang Y, Luo F, Xu J, Zhao C, Liu R, Chu Y. The IFN-γ/PD-L1 axis between T cells and tumor microenvironment: hints for glioma anti-PD-1/PD-L1 therapy. J Neuroinflammation. 2018;15(1):290. doi:10.1186/s12974-018-1330-2.
- Norelli M, Camisa B, Barbiera G, Falcone L, Purevdorj A, Genua M, Sanvito F, Ponzoni M, Doglioni C, Cristofori P, et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat Med. 2018;24(6):739–748. doi:10.1038/s41591-018-0036-4.
- Adachi K, Kano Y, Nagai T, Okuyama N, Sakoda Y, Tamada K. IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor. Nat Biotechnol. 2018;36(4):346–351. doi:10.1038/nbt.4086.
- Luo H, Su J, Sun R, Sun Y, Wang Y, Dong Y, Shi B, Jiang H, Li Z. Coexpression of IL7 and CCL21 increases efficacy of CAR-T cells in solid tumors without requiring preconditioned lymphodepletion. Clin Cancer Res. 2020;26(20):5494–5505. doi:10.1158/1078-0432.CCR-20-0777.
- Jin L, Tao H, Karachi A, Long Y, Hou AY, Na M, Dyson KA, Grippin AJ, Deleyrolle LP, Zhang W, et al. CXCR1- or CXCR2-modified CAR T cells co-opt IL-8 for maximal antitumor efficacy in solid tumors. Nat Commun. 2019;10(1):4016. doi:10.1038/s41467-019-11869-4.
- Whilding LM, Halim L, Draper B, Parente-Pereira AC, Zabinski T, Davies DM, Maher JCART. Cells targeting the integrin αvβ6 and co-expressing the chemokine receptor CXCR2 demonstrate enhanced homing and efficacy against several solid malignancies. Cancers (Basel). 2019;5:11. doi:10.3390/cancers11050674.
- Di Stasi A, De Angelis B, Rooney CM, Zhang L, Mahendravada A, Foster AE, Heslop HE, Brenner MK, Dotti G, Savoldo B. T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model. Blood. 2009;113(25):6392–6402. doi:10.1182/blood-2009-03-209650.
- Moon EK, Carpenito C, Sun J, Wang LC, Kapoor V, Predina J, Powell DJ, Riley JL, June CH, Albelda SM. Expression of a functional CCR2 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a mesothelin-specific chimeric antibody receptor. Clin Cancer Res. 2011;17(14):4719–4730. doi:10.1158/1078-0432.CCR-11-0351.
- Gao Q, Wang S, Chen X, Cheng S, Zhang Z, Li F, Huang L, Yang Y, Zhou B, Yue D, et al. Cancer-cell-secreted CXCL11 promoted CD8+ T cells infiltration through docetaxel-induced-release of HMGB1 in NSCLC. J Immunother Cancer. 2019;7(1):42. doi:10.1186/s40425-019-0511-6.
- Hu J, Sun C, Bernatchez C, Xia X, Hwu P, Dotti G, Li S. T-cell homing therapy for reducing regulatory T cells and preserving effector T-cell function in large solid tumors. Clin Cancer Res. 2018;24(12):2920–2934. doi:10.1158/1078-0432.CCR-17-1365.
- Lo A, Wang LS, Scholler J, Monslow J, Avery D, Newick K, O’Brien S, Evans RA, Bajor DJ, Clendenin C, et al. Is disrupted by depleting FAP-expressing stromal cells. Cancer Res. 2015;75(14):2800–2810. doi:10.1158/0008-5472.CAN-14-3041.
- Caruana I, Savoldo B, Hoyos V, Weber G, Liu H, Kim ES, Ittmann MM, Marchetti D, Dotti G. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat Med. 2015;21(5):524–529. doi:10.1038/nm.3833.