
ABSTRACT
Breast cancer subtypes have not shown significant response to current immunomodulatory therapies. Although most subtypes are treatable, triple negative breast cancer (TNBC), an aggressive highly metastatic cancer, comprising 10–20% of breast cancers, remains an unmet medical need. New strategies are needed in order to overcome flaws in the responsiveness to current TNBC therapies. Our aims were: first, to determine the efficacy of a novel immunomodulatory peptide, C24D, on TNBC and second, to elucidate the molecular mechanism by which C24D induces immune-modulating tumor killing. Using mass spectrometry analysis, we identified CD45 as the C24D binding receptor. In vitro and in vivo TNBC models were used to assess the efficacy of C24D in reversing TNBC-induced immunosuppression and in triggering immune-modulated tumor cell killing. The CD45 signal transduction pathway was evaluated by western blot and FACS analyses. We revealed that addition of PBMCs from healthy female donors to TNBC cells results in a cascade of suppressive CD45 intracellular signals. On binding to CD45’s extra-cellular domain on TNBC-suppressed leukocytes, the C24D peptide re-activates the Src family of tyrosine kinases, resulting in specific tumor immune response. In vitro, immune reactivation by C24D results in an increase of CD69+ T and CD69+ NK cells, triggering specific killing of TNBC cells. In vivo, C24D induced CD8+ and activated CD56+ tumor infiltrated cells, resulting in tumor apoptosis. Our results should renew interest in molecules targeting CD45, such as the C24D peptide, as a novel strategy for TNBC immunotherapy.
Introduction
Overcoming tumor-induced immune suppression, immunotherapy has revolutionized the treatment of cancer, offering a more effective alternative for multiple types of tumors.Citation1 Tumor escape mechanisms involve loss of antigenicity, immunosuppression and/or reduction of immunogenicity.Citation2 Malignant cell escape mechanisms include MHC downregulation or upregulation of immunosuppressive checkpoint pathways, affecting the anti-tumor response of T cells.Citation3,Citation4 Monoclonal antibodies directed against these immune checkpoint axes, e.g., the programmed cell death protein 1 or programmed cell death ligand 1 (PD1/PDL-1), in or without combination with cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4), have been successfully used for the treatment of some cancers.
Biologically, triple negative breast cancer (TNBC) is highly heterogeneous. TNBC has more tumor infiltrated lymphocytes,Citation5 higher levels of PD-L1 expression on both tumor and immune cellsCitation6 and a greater number of nonsynonymous mutations.Citation7 These characteristics often correlate with better responses to check point inhibitors and to tumor-specific neoantigens.Citation5–7 However, multiple clinical trials have resulted in disappointing response ratesCitation8 and single-agent efficacy is still low.Citation9–11
Between 10% and 20% of breast cancers are TNBCs. Compared to other breast cancer sub-types, TNBC is more metastatic and has a higher rate of recurrence.Citation12 TNBC tumors are negative for progesterone, estrogen and Her2, thereby explaining the lack of targeted therapies. Highly toxic chemotherapies are the most common clinical option. Ongoing efforts in translational research are needed to broaden current clinical applications and overcome unsolved issues.Citation13–15
In this study, we present the novel molecular mechanism of C24D, an immune modulating therapeutic peptide derived from the placental immunomodulatory ferritin (PLIF), its effect on TNBC-dampened immune cells and the resulting immune-modulated TNBC cell killing. The C24D sequence consists of the last 24 amino acids of PLIF and is synthesized as a homodimer peptide with a disulfide bond.Citation16
We revealed that C24D binds to the CD45 receptor on T and NK cells. CD45 is a transmembrane protein tyrosine phosphatase receptor type C (PTPRC), with double opposing effects on T cell receptor (TCR) activity.Citation17 On the one hand, CD45 plays an inhibitory function involving the dephosphorylation of the tyrosine 394 (Y394) in the lymphocyte-specific protein tyrosine kinase (Lck), preventing its activation.Citation18 On the other hand, CD45 plays the role of activator when it dephosphorylates the tyrosine 505 (Y505), an inhibitory site at the C-terminal end of the non-receptor tyrosine-Src kinases. Activated Lck phosphorylates the immunoreceptor tyrosine-based activation motifs (ITAMs) of the T cell receptor (TCR)/CD3 complex. The phosphorylated ITAMs recruit the Zeta-chain-associated protein kinase 70 (ZAP-70), via its Src homology 2 (SH2) domains. Finally, for TCR activation, CD3-bound ZAP-70 is activated by both Lck and (trans)-autophosphorylation at the ZAP-70 tyrosine 493 (Y493).Citation19 Downstream of protein tyrosine kinases is located the proto-oncogene VAV. Ligation of the activating receptor NKG2D on human NK cells results in phosphorylation of the signaling chain DAP10 by Src family kinases and the recruitment of VAV-1. VAV-1 is then phosphorylated by Src family kinases, as required for NK cell activation.Citation20
In this study, we found that the Src family of tyrosine kinases in PBMCs of TNBC patients is inhibited. We also demonstrated that by binding to the CD45 receptor on the TNBC-suppressed leukocytes, C24D breaks the inhibition and triggers an intracellular cascade of signals which results in immune-cell activation and specific killing of TNBC cells.
Material and methods
Human cell lines and culture conditions
MDA-MB-231 and MDA-MB-468 (obtained from ATCC, Biological Industries, Kibbutz Beit Ha Emek, Israel) is a human triple-negative breast cancer cell line that is Her2-neu, estrogen receptor (ER)- and progesterone receptor (PR)-negative. MCF-10A (obtained from ATCC) are normal breast cells. Cells were grown in DMEM supplemented with 10% FCS, l-glutamine (2 mM), Na-pyruvate (1 nM), penicillin (100 μ/ml), streptomycin sulfate (0.1 mg/ml) and nystatine (12.5 μ/ml) complete culture medium (Biological Industries). Cultures were maintained at 37°C in a humidified 5% CO2 incubator. A large stock of cells was prepared to maintain homogeneity and tumorigenicity of the cell line. Cells were not used beyond passage five and examined for mycoplasma (Mycoplasma detection kit, Biological Industries) at least once every six months.
C24D peptide synthesis: The C24D peptide is a 25 amino acid homodimer peptide with a di-sulphyde bond at CG (CG-HHLLRPRRRKRPHSIPTPILIFRSP). An irrelevant scrambled peptide (CG-PPRIHPLKRISRTRLPRLHRPFHIS (S-C24D) was used as control. Both peptides were synthesized by Synpeptide Co., Ltd. (Shanghai, China). The C24D peptide and the control peptide were also synthesized with the addition of Biotin or FITC C-terminal Ahx, 6-aminohexanoic acid residue to the peptide sequence. HPLC showed purity >97%.
PBMC-tumor cell co-cultures and efficacy of peptide
PBMC isolation:
PBMCs were isolated from blood of healthy female donors or breast cancer patients, obtained from the Blood Bank Mada Tel HaShomer or Davidoff Cancer Center, respectively. The protocol was approved by the Institutional Review Board at Rabin Medical Center, Israel (0667–14-RMC). Samples were isolated by Ficoll–Hypaque density gradient (d = 1,077 g/mL, Ficoll-Paque Plus, GE Healthcare, Upsalla, Sweden) by centrifugation at 650 × g for 30 minutes.
Tumor cells:
MDA-MB-231 and MDAM-MB-468 at 4x105/2 ml, derived from an exponentially growing monolayer were incubated in complete medium overnight in 6 well plates. PBMC in complete RPMI medium, supplemented with 5% AB serum instead of FBS (Biological Industries), were added onto the tumor cells (2x106/ml). The C24D peptide was added immediately at 0.1, 1, 10, 30 and 60 µg/ml and incubated for various times at 37° C, 5% CO2. S-C24D was used as negative control. A dose response analysis revealed that 10 µg/ml was the most effective dose for in vitro experiments, based on IFNγ secretion and tumor cell density (Supplementary Figure 1). MCF-10A normal breast cells were used as control, following the same protocol as described for tumor cells.
At the end of the experiments, lymphocytes were extracted from co-cultures, centrifuged and re-suspended in PBS for FACS analysis. Only samples containing more than 98% CD45+ cells after extraction were selected for the experiments.
The CD45+ cells were identified by FACS analysis with an anti-CD45-PB antibody (Supplementary Figure 2). Supernatant from co-cultures recovered from centrifugation of the lymphocytes was centrifuged at 1200 rpm for 10 minutes and stored at −80°C for cytokines determination.
Cytokines:
Human Interferon gamma (IFNγ), Tumor Necrosis Factor alpha (TNFα), Interleukin 2 (IL-2) and IL-1β were determined by ELISA Ready SET-Go (eBioscience, San Diego, CA, USA).
Tumor cell density:
For the determination of tumor cell density, co-cultures were washed once with PBS and cell density was documented by a light inverted microscope (Olympus IX73). For the determination of tumor cell killing, tumor cells were removed from plates and subjected to FACS analysis. Tumor apoptosis was determined in gated tumor cells using an AnnaexinV/PI kit (MEBCYTO® Apoptosis Kit by MBL, MA, USA). Previous to the addition of Annaexin/PI, an anti-CD45-PB (Monoclonal Antibody IOTest Beckman Coulter, Marseille, France) was added to tubes for identification of leukocytes to be discarded in the FACS analysis.
Tumor xenograft growth in nude mice
The animal experimental procedures used in the present study were approved by the Animal Care and Use Committee of Tel Aviv University (TAU 06–01-20220), in accordance with their guidelines. In total, 30 BALB/C nude mice (female; 5–6 weeks of age; each weighing 20–25 g) were purchased from Envigo (Jerusalem, Israel). Tumor xenografts were generated by subcutaneously injecting 4 × 106 MDA-MB-231 cells into the right nude-Balb/C mice flank. Tumor volumes were measured every other day using micrometer calipers and were calculated according to the following formula: tumor volume (mm3) = 0.5 x D x d2, where d and D represent the shortest and the longest diameters, respectively.
Six days after tumor injection, when the xenograft grew to approximately 100 mm3, the mice were divided randomly into four groups: a negative control group, which was to be treated with PBS (n = 6), a second negative control group, to be treated with 60 μg/mouse S-C24D (scrambled C24D) in 200 μl PBS, and two C24D treated groups (C24D: 60 μg/mouse in 200 μl PBS and C24D: 300 μg/mouse in 200 μl PBS), n = 8 in each of the latter 3 groups.
Fresh human PBMCs from 2 different female donors were incubated with C24D or S-C24D or PBS (60 μg or 300/2 x 106 cells in 0.4 ml PBS) for 5 minutes before the first intravenous injection (IV). The PBMCs from the 2 donors were injected separately, in half of the mice in each group. The subsequent IV injections of C24D, S-C24D or PBS were administered twice a week for 2 weeks.
At the end of the experiment (13 days after the administration of the PBMCs), the mice were bled, for the determination of human IFNγ in serum and sacrificed, for the extraction of tumors. The tumors were processed for immunofluorescence.
Immunofluorescent staining
Tumor tissues underwent 4% formalin fixation overnight at 4°C and paraffin embedding. The specimens were then sliced into 5-µm-thick sections and deparaffinized in xylene and rehydrated in alcohol. After antigen retrieval at pH = 6, sections were incubated for 1 hour with primary antibodies: CD8 (Rabbit Monoclonal, 108 R-14, 1:50, Cell Marque, Rockling, CA, USA), CD45 (Mouse Monoclonal, 145 M-94, 1:50, Cell Marque), activated CD56 (Mouse Monoclonal, 156 M-84, 1:50, Cell Marque), Cleaved Caspase-3, (Rabbit Monoclonal, 9664S, 1:100, Cell Signaling, Danvers, MA, USA), HLA-A (Rabbit Monoclonal, Ab52922, 1:100, Abcam). Slides stained with an IgG irrelevant antibody followed by secondary antibodies served as negative controls. Human PBMCs in paraffin slides served as positive controls. The fluorescence detection was performed with Alexa Fluor 488-AffiniPure Donkey Anti-Mouse IgG (H + L) (715–545-151, 1:200); Cy3-AffiniPure Donkey Anti-Rabbit IgG (H + L) (711–165-152, 1:200); both from Jackson Immunoresearch Laboratories, Westgrove, Pennsylvania, USA). DAPI (4ʹ,6-Diamidino-2-P henylindole, Dilactate) (BLG-422801, 1:400, Biolegend, San Diego, CA, USA) was used as counterstain. Fluorescence observation and image acquisition were performed using a Leica TCS SP5 confocal laser-scanning microscope (Leica Microsystems, Germany). Quantitative Image Analysis was preformed using FIJI (Image J2) image analysis software in 5 fields/slide in 5 different slides.
FACS analysis of C24D binding to cells
Fresh lymphocytes (0.5x106/50 µl PBS) were incubated with the following antibodies for multicolor staining: CD3-PC5.5, CD4-PC7, CD8-KO, CD56-PE, CD16-APC, NKG2D-AF750, CD45RA-APC, CD45RO-PC5.5 and CD14-KO (Beckman Coulter) and with 5 µg/50 µl C24D-FITC peptide for 40 minutes at room temperature. The cells were then washed twice (10 min., 1200 rpm, 4°C). Activated lymphocytes obtained from co-cultures with tumor cells were subjected to the same procedure. To ensure specificity, a Fluorescence Minus One (FMO) control was used to identify and gate cells in the context of data spread due to the presence of multiple fluorochromes in any given panel. In brief, for each sample, two tubes were used: one tube containing cells stained with the antibodies identifying the various PBMC subpopulations and a control peptide-FITC labeled and a second tube containing cells stained with the antibodies identifying the different PBMC subpopulations and with the C24D-FITC peptide. The percentage C24D/CD45 binding on the cell surface in each subpopulation was calculated after subtracting the fluorescence obtained for the control tube. The cells were washed with PBS and suspended in 0.5 ml PBS for FACS (Coulter Navios flow cytometer, Indianapolis, IN, USA). Data were analyzed with the Kaluza software (Beckman Coulter, IN, USA).
Anti-CD45-APC antibody was used for the double staining determination of the C24D+/CD45+ cells.
For the determination of the activated PBMCs sub-populations, PBMCs from the co-cultures with tumor cells were counted and transferred to a FACS tube (0.5x106 cells/50 µl PBS) and incubated with the following antibodies: CD4-PC7, CD8-KO, CD56-PE, NKG2D-AF750, CD14-KO, CD69-PC5, CD57-FITC, CD137-ECD (Beckman Coulter). Gated live cells were analyzed using Coulter Navious FACS, Kaluza software.
Mass Spectrometry analysis for the identification of the specific peptide binding protein
Fresh PBMCs (15x106) from human donors were centrifuged and transferred to a chilled microtube. Lysis buffer (150 mM NaCl, 1% triton X-100, 50 mM Tris HCl pH 8, with the addition of proteases inhibitors) was added to the cells for 20 minutes on ice. After centrifugation for 5 minutes at 1000 rpm, the supernatant was transferred to a new microtube for precipitation. Protein concentration was determined by BCA protein assay kit (Thermo Scientific, Rockford IL, USA) following manufacturer instructions. Cell protein (50 µg/200 µl lysis buffer) from fresh lymphocytes was incubated with 15 µg biotinylated C24D peptide for 1 hour at 4°C at constant shaking. After incubation, the biotinylated peptide which bonded to specific proteins formed a complex. The complex was precipitated using streptavidin magnetic beads (µMACS streptavidin Kit, Miltenyi Biotec GmbH. Cat. Number 130–074-101). The complex was placed in a microcolumn in the magnetic field of a μMACS Separator. After rinsing the column, the target molecules which bonded to the biotinylated probe were eluted. The eluted sample was subjected to SDS PAGE in a 10% gel. The gels were stained with Imperial Protein Stain (Thermo Scientific, Cat. Number 24615) for mass spectrometry analysis.
Mass spectrometry analysis was done by The Smoler Proteomics Center, Technion, Haifa, Israel. Samples were reduced, modified and digested by trypsin, analyzed by LC-MS/MS on Q exactive plus (Thermo). Data was analyzed by Discoverer Software, version 1.4, using the Sequest search engine, vs the human Uniprot database. The data was quantified using the same software. Protein identification was filtered for proteins identified with FDR <0.01 with at least 2 peptides. Proteins which were precipitated without biotinylated peptide served as controls. Proteins which had at least 4-fold more mass-to-charge ratio than the control were considered for further analysis.
Following mass spectrometry analysis, the eluted purified sample obtained from precipitation of the lysed PBMCs with biotinylated C24D was subjected to western blot analysis using and anti-CD45 antibody (Abcam, Cat. Number ab123522).
Proximity ligation assay:
The method was modified for Peptide–Target interaction previously described.Citation21 In brief, a 96 well, high binding plate, was coated with His Tag antibody (Monoclonal IgG1, R&D Systems, USA) for 2 hours at room temperature. After washing with PBS-0.05% Tween, recombinant Human CD45 with a C-terminal 6 His Tag (R&D Systems, USA) was added to coated plates overnight at 4°C. Following 3 plate washes, Biotinylated C24D at various concentrations (0.05, 0.5, 1, 5 and 10 μg/ml) was added for 2 hours at room temperature. Streptavidin-HRP (R&D Systems, USA) was added to plates for 2 hours followed by TMB subtract for proximity Peptide/Target detection. HCl 1 M was used to stop the chemical reaction. Proximity ligation was detected by ELISA reader at 450 nm. A biotinylated scrambled version of C24D was used as negative control and a Biotinylated Anti CD45 antibody (Abcam) was used as positive control.
Blocking of the CD45 receptor
The anti-CD45 antibody 10 μg/ml (Abcam, Cat. Number ab123522) and/or the peptide CD45 inhibitor VI (Calbiochem, Burlington, Massachusetts, USA, Cat. Number 530197) were used for blocking the extracellular domain of CD45 in the PBMCs used in the relevant experiments. The blockers were added before addition of PBMCs to the tumor plates, as per manufacturer’s instructions. The efficacy of the blockers is presented in Supplementary Figure 3.
CD45 signal transduction evaluation
PBMC was added onto the tumor cells (2x106/ml) and 10 µg/ml of C24D was added immediately thereafter. Co-cultures were incubated for 5, 15, 30, 60 minutes and 24 hours at 37° C, 5% CO2, depending on each protein’s phosphorylation.
At the end of each incubation period, lymphocytes were extracted, and a sample of the extracted cells was submitted to FACS analysis to corroborate that the sample for western blot analysis contains more than 98% leukocytes (Supplementary figure 2). The extracted leukocytes were then centrifuged and re-suspended in 0.12 ml of lysis buffer (20 mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM NaF, 2 mM Na3VO4, 1% NP40, 10 mM b-glycerophosphate, 30% glycerol, 1 mM EDTA,0.5% sodium-deoxycholate, 0.5% protease inhibitor cocktail), followed by one freeze-thaw cycle of 20 min. Cells were harvested and centrifuged (14,000 rpm, 15 min, 4°C). The supernatants were collected, and aliquots were separated on 10% SDS PAGE, followed by Western blotting with anti-phospho-Lck Y505 (0.5 µg/ml, ab4901, Abcam, Cambridge, UK), anti-phospho-Lck Y394 (0.25 µg/ml, ab201567, Abcam), anti phospho-VAV-1 Y174 (0.23 µg/ml, ab76225, Abcam) and anti-phospho-ZAP70 Y493 (1 µg/ml, ab194800, Abcam). GAPDH (1 µg/ml, ab9485, Abcam) was added as a control for sample loading. After several washings, the secondary antibody, IRDye 800CW Goat anti-Rabbit or IRDye 680CW Goat anti mouse (1 mg/ml, LI-COR, Nebraska, USA) was added for 1 h.
Quantification methods: The membrane was analyzed by Odyssey 2.1 (Infrared Imaging System) for specific band identification. Quantification of phosphorylation was done by Image J (NIH, USA). Percentage (%) of maximal phosphorylation of phosphorylated proteins were first normalized to the levels obtained with GAPDH respectively, and the activation values were normalized for each time point vs its control, incubated with a control peptide (e.g. C24D + lymphocytes vs. lymphocytes control). The values obtained were then expressed as % of maximal activation that was observed in each experiment, at each time point.
For the corroboration of the results obtained in the western blot analysis, FACS analysis was performed with the same antibodies, followed by incubation with a secondary antibody (anti-rabbit-PE, Jackson ImmunoResearch, cat: 711–116-152). Intracellular staining for the phosphorylation of the tyrosine 505 and 394 in Lck, the tyrosine 493 in ZAP-70 and the tyrosine 174 in VAV-1, was performed using a FIX & PERM Cell Permeabilization kit consisting of matched Fixation and Permeabilization Reagent (R&D Systems, Minneapolis, MN, USA).
Statistical analysis
To determine whether the data from test samples were significantly different from control samples, student’s t-tests were performed using GraphPad Software (GraphPad Software, La Jolla, CA). ANOVA, followed by Tukey-Kramer, was used for multiple comparisons. Significance was defined as p<0.05.
Results
C24D induces re-activation of leukocytes, resulting in TNBC cell killing
The immune-modulating tumor killing effect of C24D was previously demonstrated on MCF-7 (ER+) and T47D (HLA-A2) breast cancer cells, co-cultured with human PBMCs from healthy donors.Citation16 In this study, we first set out to determine if C24D also induces effective immune-modulated tumor killing of TNBC cells (MDA-MB-231 and MDA-MD-468).
Observation by microscope revealed that the addition of 10 μg/ml C24D to co-cultures of human PBMCs from healthy donors with MDA-MB-231 resulted in massive tumor apoptosis (). Similar immune-modulated killing results were achieved with a second TNBC cell line: MDA-MD-468 (Supplementary Figure 4b). Treatment of tumors with C24D or with S-C24D (scrambled C24D), in the absence of PBMCs, both resulted in no effect (supplementary Figure 4a). In co-cultures with PBMC alone, we only noted a minor effect on tumor apoptosis.
Figure 1. C24D induced re-activation of leukocytes, resulting in killing of TNBC cells: a. Representative pictures of tumor cell density in co-cultures of MDA-MB-231 cells with PBMCs from healthy female donors, documented by inverted microscope (Bars = 200 μm) ± C24D (10 μg/ml) or scrambled C24D (S-C24D, 10 μg/ml) as control. b. Tumor cell killing by FACS analysis, using AnnaexinV/PI (N = 14). c. Representative FACS dot blot (experiment #11) of AnnaexinV/PI gated and stained tumor (CD45 negative) cells treated with 10 μg/ml C24D for 4–6 days. d. Percentage of CD4+/CD69+ and CD4+/CD57+ cells in co-cultures of PBMC with MBA-MB-231 cells, ± C24D, by FACS analysis. e. Percentage of CD8+/CD69+, CD8+/CD56+ (T/NK cells) and CD8+/CD57+, in the co-cultures, by FACS analysis. f. Percentage of CD56+/CD69+ and CD56+/CD57+, in the co-cultures, by FACS analysis. g. Percentage of CD14+/CD137+ in the co-cultures, by FACS analysis. Analysis of the results presented as Mean ± SD. h. Representative dot blot FACS analysis showing double staining of CD4, CD8 or CD56 gated cells with either CD69 or CD57, comparing effect of C24D versus control. i. Effect of C24D on IFNγ secretion (pg/ml) in supernatants of PBMCs alone or PBMCs co-cultured with MDA-MB-231 (n = 8). j. Effect of C24D on secretion of IL-2 (pg/ml) (n = 7). k. Effect of C24D on secretion of TNFα (pg/ml) (n = 7). l. Effect of C24D on secretion of IL-1β (pg/ml). Analysis of results presented as Mean ± SD
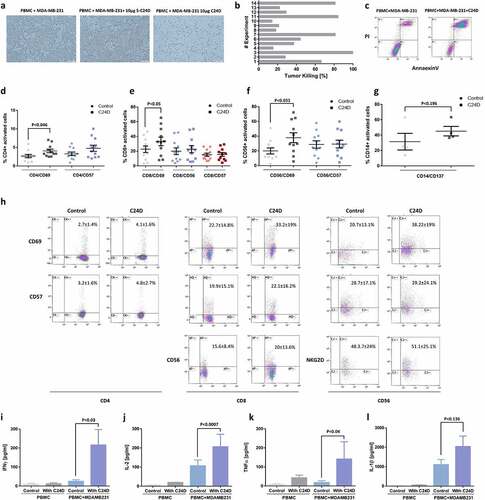
To confirm the microscope observations, we performed FACS analysis using AnnaexinV/PI. With PBMCs from 14 different healthy female donors, the percentage of AnneaxinV/PI positive tumor cells (i.e., MDA-MB-231 cell killing) varied from 15% to 100%, with an average of 45.9 ± 30.6% (). illustrates an example of one such FACS analysis study, showing the results of experiment number 11.
To establish which of the leukocyte sub-populations is activated by C24D and involved in the tumor cell killing, we performed a series of FACS analyses using PBMCs from 11 healthy female donors. Addition of 10 μg/ml C24D to each PBMC/MDA-MB-231 co-culture resulted in a significant increase in the percentage of CD4+/CD69+ cells (from 2.68 ± 1.45% to 4.1 ± 1.58%, p <0.046, ), CD8+/CD69+ cells (from 22.6 ± 14.8% to 33.23%, p <0.05, ) and CD56+/CD69+ (from 19.9 ± 13.61% to 39.1 ± 16.0%, p <0.033, ). CD57 expression was not influenced by the addition of C24D to the co-cultures nor was the percentage of NKG2D+ and CD45RO+ cells. A representative FACS analysis of the activated cells sub-populations is shown in .
In contrast to T and NK activation resulting from addition of C24D, the percentage of activated monocytes (CD14+/CD137+) increased, but non-significantly, from 31.6 ± 9.4% to 45.2 + 5.3%, p <0.19 (). CD14 is a marker for circulating monocytes and CD137 (a member of the tumor necrosis factor receptor family) is expressed on activated monocytes. Addition of C24D to the co-cultures had a non-significant effect on the secretion of IL-1β by monocytes ().
Addition of C24D to the MDA-MB-231/PBMC co-cultures for 4–6 days resulted in a significant increase in secretion of cytokines: 90% of IFN-γ (, p <0.03), 92.2% of IL-2 (, p < 0.0007) and 98% of TNF-α (, p < 0.04). Similar results were observed in co-cultures of PBMCs with MDA-MB-468 (Supplementary Figure 5). No effect was noted when C24D was added to PBMCs alone (, j, k, l)).
Treatment with C24D inhibited tumor growth in vivo
Using a human TNBC mouse model adapted from Zhang et al.Citation22 (), we aimed to determine if the in vitro cytotoxic effect mediated by C24D would also be observed in a meaningful in vivo model. For this purpose, we measured tumor growth over time, IFN-γ in serum and tumor infiltrating human T and activated NK cells in tumor histological sections.
Figure 2. Treatment with C24D reduced TNBC tumor growth in vivo: a. Protocol of the immunocompetent mice in vivo TNBC model. b. Effect of C24D on tumor growth. Tumors were measured twice-weekly in the 4 groups: Control PBS (n = 6), S-C24D (n = 8), C24D (60 μg/ml per mouse, n = 8) and C24D (300 μg/ml per mouse, n = 8). Analysis of results presented as Mean ± SD. c. Photographs of representative tumors, extracted from mice in each group, 13 days after treatment. d. Percentage IFN-γ secretion in serum of mice treated with C24D vs. control (S-C24D). Analysis of results presented as Mean ± SD (p <0.05). e. Immunofluorescence analysis and quantification of tumor (red) infiltrating human CD45+ cells (green) in mice treated with C24D, compared to S-C24D treated mice. f. Immunofluorescence analysis and quantitation of activated human CD56+ tumor (red) infiltrating cells (green). g. Immunofluorescence analysis and quantitation of human CD8+ cells (red). h. Immunofluorescence analysis and quantitation of apoptotic tumor cells (red). Analysis of results presented as Mean ± SD calculated in 5 fields per slide in 5 different slides
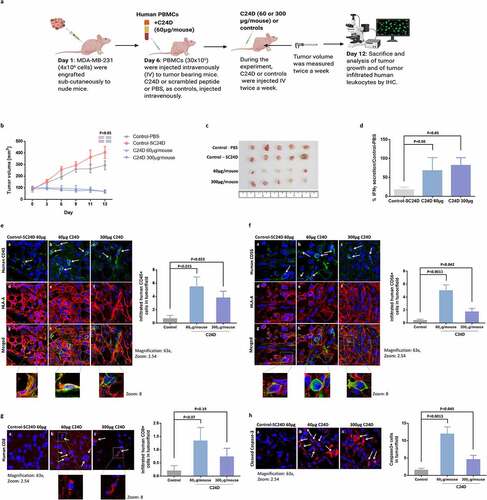
Results showed a significant reduction in tumor growth in mice treated with 60 or 300 μg C24D per mouse, compared to treatment with PBS or S-C24D as controls (p <0.05, ). In , we show the tumors extracted from 5 mice in each of the 4 groups, 13 days after treatment. At the end of the experiment, mice were bled for the determination of human IFN-γ in serum. A significant increase of 68% and 82% (p <0.05) in human IFN-γ secretion was observed in the 60 and 300 μg groups, versus the group with S-C24D treatment ().
Immunofluorescent analysis of tumor sections
The presence of tumor infiltrating and human T and activated NK cells was revealed by an immunofluorescent analysis of tumor sections. A significant number of tumor infiltrating human leukocytes were found in the samples from mice treated with C24D, compared to the S-C24D group (, f, g)). Compared to treatment with S-C24D, treatment with both 60 and 300 μg doses of C24D significantly induced the infiltration of human CD45+ cells (, green) in the human tumor (red) (p <0.015 and p <0.023 respectively).
The following tumor infiltrating human CD45+ sub-populations in mice treated with C24D were found in the immunohistology samples: activated CD56+ (stained with anti-human activated CD56 antibody in green, ) and CD8+ cells (stained with an anti-human CD8 antibody in red, ). Treatment with 60 or 300 μg C24D per mouse resulted in a significant increase in tumor-infiltrating NK (CD56+) cells (p <0.011 and p <0.042, respectively).
A similar pattern was observed for activated human CD8 T cells (p <0.07 and p <0.1, respectively). The non-significant results of human CD8 T cells might be explained by the low number of cells counted.
The presence of infiltrating human T and activated human NK cells supports the significant increase in tumor cell apoptosis (). The lower dose (60 μg) induced a more significant tumor cell apoptosis (p <0.0013) than the higher dose (p <0.049).
Stability of C24D
We performed two C24D stability studies: one in whole blood (ChemPartner Co, Ltd.) and a second, in plasma (in our laboratory). 10 μM (60 μg/ml) of C24D has a T1/2 of 165.03 minutes in human whole blood (Supplementary Figure 3 c). In plasma, target saturation was evaluated by measuring C24D binding to the CD45 receptor, using two different methods: FACS analysis and ELISA. By FACS analysis, we observed that 1.5 μM (10 μg/ml) of C24D bound over 70% of the receptors of the studied PBMC sub-populations (). In the ELISA assay, 3 hours after adding 1.5 μM of C24D in the presence of human plasma, binding to CD45 decreased by 20%.
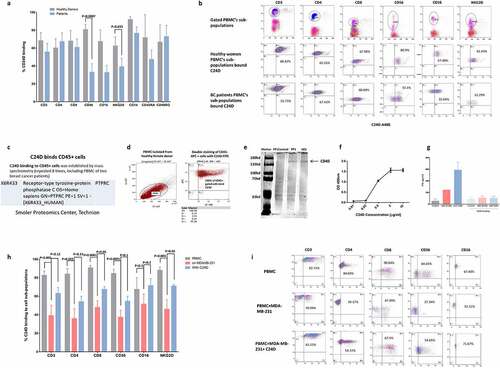
Figure 3. CD45 revealed as the C24D binding receptor: a. Percentage of C24D binding to sub-populations of PBMCs from healthy female donors, compared to C24D binding to sub-populations of PBMC from breast cancer patients, by FACS analysis. b. Representative FACS analysis dot blots showing the gated sub-populations of PBMCs from breast cancer patients (BC) stained with C24D-AF488 peptide. c. The CD45 receptor was identified by mass spectrometry analysis. d. Representative double staining FACS analysis of gated PBMCs stained with CD45-APC antibody and C24D-AF488 peptide. e. Western blot analysis of the precipitated and eluted protein with biotinylated C24D followed by streptavidin (PP). PP control, protein precipitated only with streptavidin. Protein identification in 2 different experiments (PP1 and PP2) was revealed using an anti CD45 antibody. f. Proximity ligation assay by ELISA with different concentrations of C24D bound specifically CD45 recombinant protein. g. Statistical analysis of IFN-γ secretion (in pg/ml) in supernatants of MDA-MB-231/PBMC co-cultures and in supernatants of co-cultures with blocked CD45 receptors (n = 5). h. Percentage of C24D binding to PBMC sub-populations, with or without exposure to tumor cells ± C24D. Analysis of results presented as Mean (%) ± SD. i. Representative FACS dot blot analysis comparing C24D binding to PBMC sub-populations, co-cultured with MDA-MB-231 ± C24D
CD45 revealed as the C24D binding receptor
In an effort to elucidate the molecular mechanism which may explain the above immune-modulated tumor killing, we first proceeded to identify the receptor to which C24D binds. Using PBMCs from healthy female donors and C24D conjugated to Alexa-488 dye, by FACS analysis, we determined the leukocyte sub-populations to which C24D binds (). Surprisingly, we observed that all 9 of the tested T, NK and monocyte sub-populations bound C24D (; a representative dot blot FACS analysis).
Subsequently, we subjected the entire leukocyte population to mass spectrometry analysis. Using PBMCs from 8 different female donors, of which 2 were from breast cancer patients, we precipitated, per donor, the entire leukocyte population with biotinylated C24D. Each isolated complex was subjected to mass spectrometry analysis. The receptor we identified was: CD45 (PTPRC, X6R433 Uniprot), (). The ubiquitous nature of CD45 explained why all the 9 tested T, NK and monocyte sub-populations bound C24D.
To validate the mass spectrometry results, we performed a double-staining FACS analysis with an anti CD45 monoclonal antibody and the C24D-Alexa-488 conjugated peptide (). We observed that 100% of the PBMCs were stained by both antibody and C24D, thereby confirming that CD45 is the binding receptor. This result also suggests that the anti-CD45 antibody and C24D bind to different epitopes of the extra cellular domain of CD45.
Further validation of the mass spectrometry results were obtained by western blot analysis. The protein precipitated with biotinylated C24D, followed by streptavidin magnetic beads and eluted through a magnetic column, was subjected to gel electrophoresis. The membrane was stained with an anti CD45 antibody. Results revealed that the precipitated protein was CD45 ().
To demonstrate direct interaction between C24D and CD45 we performed a Proximity Ligation Assay using a biotinylated C24D peptide. Results showed a specific concentration dependent binding curve of C24D to an immobilized CD45 recombinant protein ().
To support the above results, we blocked the CD45 receptor on PBMCs with a monoclonal antibody and a commercially available blocking peptide and evaluated IFN-γ secretion (). Without blocking, a 2.4-fold increase in IFN-γ was found in the supernatants of C24D treated co-cultures of PBMCs with MDA-MB-231 cells, compared to S-C24D treated cells. In contrast, the results obtained on co-cultures on which the CD45 receptor was blocked, were similar to control (91 ± 35 pg/ml), S-C24D treated cells (108 ± 20 pg/ml) and in C24D treated cells (107 ± 27 pg/ml).
C24D restores the expression of CD45 receptors, previously reduced by exposure to tumor cells
We compared the percentage of C24D which bound to leukocyte sub-populations of PBMCs from healthy female donors versus breast cancer patients. We found that a significantly lower percentage of C24D bonded to the CD56 and NKG2D NK cells sub-populations in breast cancer patients (p <0.0007 and p <0.035 respectively) (, b)).
We also compared C24D binding to PBMC from healthy donors, before versus after exposure to TNBC cells. Exposure of PBMCs to MDA-MB-231 tumor cells resulted in a significant decrease in the percentage of bonded C24D to the following sub-populations: CD3 (p <0.005), CD4 (p <0.002), CD8 (p <0.0001), CD56 (p <0.0001) and NKG2D (p <0.03). Treatment with C24D reversed the decrease of C24D binding to CD8 and NKG2D (p <0.05), and partially reversed the decrease of C24D binding to the CD3, CD4 and CD56 PBMC sub-populations. The percentage of C24D binding to CD16 was not affected by exposure to tumor cells (). A representative dot blot FACS analysis is presented in .
C24D binding to CD45 induces the phosphorylation of Src protein kinases
We then investigated the effect of the C24D/CD45 binding on the CD45 signaling pathway. The phosphorylation of proteins involved in the CD45 signaling pathway was determined by western blot analysis () of co-cultures of MDA-MB-231 cells and PBMC from healthy female donors.
Figure 4. C24D binding to CD45 in dampened PBMCs triggers the CD45 signaling pathway: a. Western blot analysis of PBMCs alone or after exposure to MDA-MB-231 cells, ± C24D, lysed, separated on 10% SDS, and blotted with antibodies for the determination of phosphorylation of Lck, ZAP-70 and VAV-1 proteins. b. Percentage of protein phosphorylation in PBMCs from 13 healthy female donors, alone and after being exposed to MDA-MB-231 cells ± C24D. Statistical analysis of results presented as Mean (%) ± SD. c. Western blot analysis of the phosphorylation of Lck, ZAP-70 and VAV-1 proteins in PBMCs from healthy female donors in which the CD45 receptor was blocked and which were subsequently exposed to MDA-MB-231 cells ± C24D. d. Percentage of protein phosphorylation in PBMCs with blocked CD45 ± C24D (n = 3). Analysis of results presented as Mean ± SE. e. Percentage of protein phosphorylation in live PBMCs co-cultured with MDA-MB-231 for 24hs and treated for 15 minutes with C24D, by FACS analysis (n = 3). Analysis of results presented as Mean ± SD, results normalized/control (S-C24D vs C24D). f. Representative FACS analysis of gated CD45+ cells, stained with Lck, ZAP-70 and VAV-1 protein phosphorylation antibodies
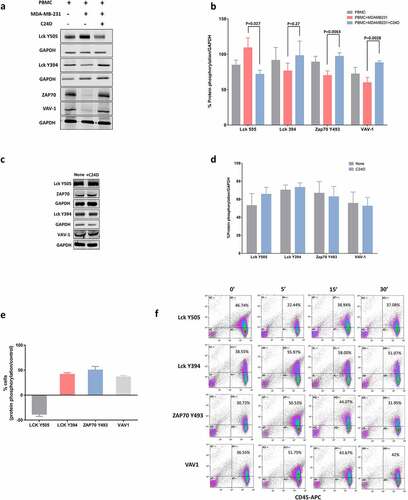
Interestingly, we found that addition of PBMCs to the MDA-MB-231 cells resulted in an increase in the phosphorylation of the tyrosine (Y) 505 in the Lck tyrosine kinase and a decrease in the phosphorylation of the Y394 in the Lck, causing inhibition of Lck. Additionally, we found a decrease in the phosphorylation of the Y493 in the ZAP70 protein kinase and in the proto-oncogene VAV-1, resulting in a deactivation of ZAP70 and VAV-1.
Addition of C24D to the above co-cultures reversed the inhibitory signaling observed in the presence of MDA-MB-231. In 13 separate experiments, we demonstrated that addition of C24D to PBMC/MDA-MB-231 co-cultures significantly reversed immune suppression by de-phosphorylation of Lck Y505 (p <0.027) and phosphorylation ZAP70 Y493 (p <0.0063) and VAV1 (p <0.0028) (). Also, Lck Y394 increased, but not significant statistically (n = 9, p <0.27). Blocking the extracellular domain of CD45 canceled the immune-reactivating effect of treatment with C24D to the PBMC/MDA-MB-231 co-cultures (, d)).
To further confirm the above results, we performed FACS analysis of gated live cells (, f)). PBMCs were identified using a CD45 antibody and the CD45+ cells were gated for further analysis of protein phosphorylation. FACS analysis results demonstrated that addition of C24D to the PBMC/MDA-MB-231 co-cultures resulted in a reduction of 38.1 ± 6.2% versus control of the phosphorylation of Y505 and an increase in the phosphorylation of Y394 in Lck. Upon treatment with C24D, phosphorylation of the Y493 in ZAP70 increased by 50.13 ± 10.2% versus control and by 36.2 ± 4.0% in VAV-1 (). A representative dot blot FACS analysis is depicted in ).
C24D reverses CD45 inhibitory signals in PBMCs from TNBC patients
After evaluating the effect of C24D on the CD45 signaling pathway of healthy-donor-PBMC/MDA-MB-231 co-cultures, we studied the effect of C24D on protein phosphorylation in fresh PBMCs from 5 TNBC patients. Phosphorylation of PBMC proteins was evaluated by western blot analysis (, b)). Addition of C24D induced the de-phosphorylation of Lck Y505 (in average from 118.0 ± 26.3% to 79.2 ± 3.6%), the phosphorylation of the Y394 in Lck (from 87.1 ± 20.4% to 117.4 ± 36.2%), thereby activating Lck. In consequence, ZAP70 Y493 and VAV-1 were phosphorylated (from 92.9 ± 4 to 116.4 ± 10.4% and from 68.9 ± 30.0 to 88.4 ± 26.9%, respectively), activating ZAP70 and VAV-1 (). These results were similar to those from healthy-donor-PBMC/MDA-MB-231 co-cultures with C24D.
Figure 5. C24D reversed CD45 suppressive signals in PBMCs from TNBC patients. a. PBMCs from 5 metastatic breast cancer patients were incubated with the C24D peptide for 5 to 60 minutes. Cells were lysed and subjected to SDS PAGE. Individual western blot analysis of lysed cells blotted with antibodies to detect the protein phosphorylation. b. Statistical analysis, per TNBC patient, of western blots, depicting phosphorylation of proteins, normalized to control. c. Mean (%) ± SD of phosphorylation of the Lck, ZAP-70 and VAV-1 proteins in the 5 TNBC patients, normalized to control. d. Representative western blot analysis of protein phosphorylation in fresh PBMCs from a healthy female donor. C24D was added to the PBMCs for at least 24 hours. e. Percentage of protein phosphorylation of 5 separate experiments (Mean ± SE, no statistical difference in protein phosphorylation was observed). f. Effect of C24D on secretion of IFN-γ, IL-2 and TNF-α in supernatants of PBMCs from healthy female donors, co-cultured with normal breast cell MCF-10A, ± C24D (n = 3). Results presented as percentage of cytokine secretion normalized to control. g. MCF-10A (breast cancer normal cells) killing by FACS analysis, using AnnaexinV/PI (N = 3). Representative FACS dot blot (experiment #1) of AnnaexinV/PI gated and stained breast normal cells (CD45 negative) treated with 10 μg/ml C24D for 4–6 days. Analysis of results presented as Mean ± SD
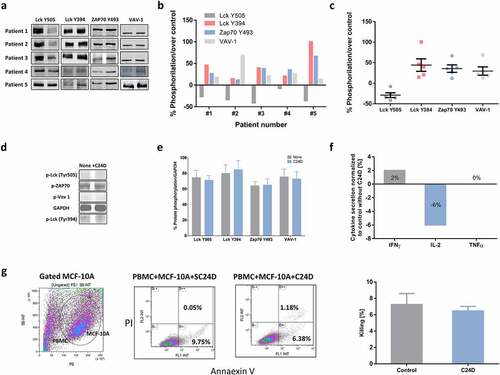
In contrast, no effect was observed on Lck, ZAP70 and VAV-1 activation when fresh PBMCs from healthy donors, in the absence of tumors, were incubated with C24D for 5 to 24 hours, suggesting that C24D has no effect on cells which are not immunosuppressed (, e)). These results were reinforced by the lack of increase in INF-γ, TNF-α and IL-2 secretion after the addition of C24D to a co-culture of MCF-10A normal breast cells and PBMCs from healthy donors (). The cytokine concentrations obtained in control cells vs C24D treatment were INF-γ: 17.7 ± 1.3 pg/ml in control vs 17.9 ± 0.9 pg/ml after C24D, TNF-α: 5.6 ± 0.1 pg/ml in control vs 5.6 ± 0.2 pg/ml in C24D in the treated cells, and IL-2: 43.0 ± 1.2 pg/ml in control vs 40.4 ± 3.8 pg/ml in C24D treated cells. Additionally, treatment with C24D to MCF-10A and PBMCs co-cultures failed to induce killing. Treatment with C24D or S-C24D induced only spontaneous cell apoptosis of 6.47 ± 0.94% and 7.25 ± 2.34% killing, respectively ().
C24D molecular mechanism
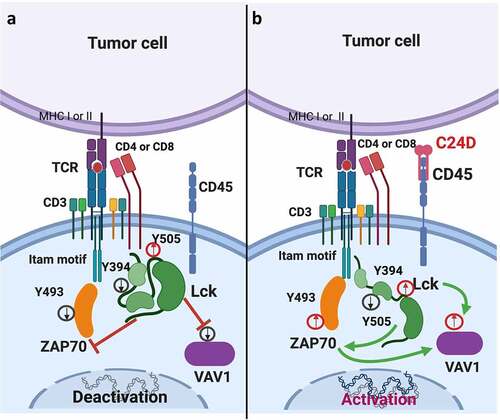
depicts the suggested CD45 molecular mechanism observed when human PBMCs are exposed to TNBC cells. depicts the reversing effect on tumor-immunosuppressed leukocytes resulting from binding of C24D to the CD45 receptor.
Figure 6. Schematic diagram: Suggested model of how C24D reverses tumor-induced immune suppression through the CD45 signaling pathway. a. In PBMCs from healthy donors exposed to TNBC tumors, phosphorylation of the inhibitory tyrosine (y) 505 in Lck (a member of the Src tyrosine kinases) increased, phosphorylation of the tyrosine 394 in Lck decreased, de-activating the Lck protein in the peripheral blood leukocytes. In consequence, tyrosine 493 in ZAP70, and VAV-1 were de-phosphorylated, de-activating ZAP70 and VAV-1. As a result, TCR activity decreased, dampening immune response. b. Binding of the C24D peptide to the CD45 receptor on leukocytes which were previously exposed to TNBC cells, reverses the suppression by de-phosphorylation of the tyrosine 505 and phosphorylation of the tyrosine 394 in Lck. Consequently, the tyrosine 493 in ZAP70 and VAV-1 are phosphorylated, resulting in TCR activation, tumor cell killing and secretion of IFN-γ, IL-2 and TNF-α
Discussion
Despite enormous efforts to find successful therapies, TNBC treatment remains an unmet medical need. The large number of clinical trials currently running on checkpoint inhibitors for the treatment of breast cancersCitation23,Citation24 reflects the great interest in (and need for) finding better therapies. The main challenge for developing effective TNBC immunotherapies is the elucidation of the flaws in the responsiveness of breast cancers to immunotherapy. New strategies are thus needed to overcome these hurdles.Citation15,Citation25
In our study, we revealed a novel role (immunotherapy) for an old target (CD45). The molecular mechanism deployed by this new/old target is fundamentally different from that of currently reported immune-checkpoint strategies and may overcome many of the difficulties encountered so far in breast cancer immunotherapy. The CD45 receptor is a cell surface glycoprotein with tyrosine phosphatase activity. It is expressed on all nucleated hematopoietic cells, except for mature erythrocytes and plateletsCitation17,Citation26,Citation27. CD45 has been identified as a rheostat of T and B cell activation, doing so by positively and negatively regulating Src family protein tyrosine kinases and thereby, the signaling cascade in leukocytes.Citation28–30
We observed that on exposure to TNBC cells, PBMCs from healthy female donors showed no significant immune response against the tumor cells. We revealed that the lack of response against the TNBC cells might be due to the increase in the phosphorylation of the tyrosine 505 and decrease in the phosphorylation of the tyrosine 394 in Lck, thus deactivating Lck (one of the members of the Src family protein tyrosine kinases). In consequence, the tyrosine 493 in ZAP70 was dephosphorylated, inhibiting TCR activation.Citation31–33 Decrease in ZAP70 activation affected VAV-1 phosphorylation, resulting in NK deactivation ().Citation34
Conversely, treatment with C24D reverses the CD45 inhibitory signals in PBMCs which were exposed to tumors. Mass spectrometry results revealed that C24D binds to a short isoform of the human extra cellular domain of CD45, the receptor-type tyrosine-protein phosphatase C (PTPRC). The binding receptor was validated by western blot analysis using an anti-CD45 antibody to confirm CD45 as the protein precipitated from PBMCs with C24D. C24D/CD45 interaction was demonstrated by a Proximity Ligation Assay. Validation of CD45 as C24D binding receptor was also demonstrated when the effect of C24D on IFN-γ secretion was canceled by blocking the extracellular domain of CD45. Upon binding to the CD45 receptor of tamed PBMCs, C24D induces the activation of the CD45 signal transduction pathway.
It has been reported that loss of CD45 results in hyperphosphorylation of the Lck C-terminal tail (Y505), thereby reducing the amount of active Lck. The reduction in Lck impairs T cell development when TCR signaling is required.Citation35–37 We observed a significant decrease in C24D binding to almost all the tested sub-populations (CD3, CD4, CD8 T cells and CD56 and NKG2D NK cells) in PBMCs from healthy donors co-cultured with MDA-MB-231 cells. The significant decrease in the percentage of C24D binding to PBMCs suggests a loss of CD45 (or of a specific CD45 isoform) after exposure to TNBC.
In PBMCs from breast cancer patients, we observed a lower percentage of C24D binding only to NK cells (CD56, CD16 and NKG2D positive cells). The difference in patients compared to co-cultures, is possibly due to the direct contact between the cells in the co-cultures.
The percentage of C24D binding to T and NK cells in PBMCs co-cultured with MDA-MB-231 was restored after treatment with C24D. Concurrently, C24D treatment induced Lck, ZAP70 and VAV-1 phosphorylation, resulting in reactivation of the CD45-signaling cascade () and the triggering of massive tumor cell killing.
Increase in CD69 expression reflects an immunoreactive phenotype associated with enhanced cytotoxicity against various target cells.Citation38 Increase of CD69 expression in NK cells in breast cancer patients after immunotherapy was associated with better survivalCitation39 and longer disease free survivalCitation40 compared to patients with lower receptor expression.
We found that addition of C24D to the MDA-MB-231/PBMC co-cultures induced a significant increase in activated CD4+/CD69+, CD8+/CD69+ T cells and CD56+/CD69+ NK cells and a significant increase in type 1 cytokines which are critical for optimal anti-tumor response: INF-γ, TNF-α and IL-2.Citation41,Citation42 In contrast, C24D had no significant effect on CD14+/CD137L+ activated monocytes.Citation43 Furthermore, IL-1β secretion did not change upon C24D treatment.Citation44
Increase in circulating CD8+ or CD4+ cells positive for CD57 (a marker of terminal differentiation) has been associated with poor prognosis in several solid and hematological tumors.Citation45,Citation46 Additionally, an inverse correlation between circulating CD57+ and tumor-infiltrating NK cells was found in Her2+ breast cancer patients.Citation47 Treatment with C24D to PBMC/MDA-MB-231 co-cultures did not result in an increase of CD57+ clones, enabling effective tumor cell killing.
We also evaluated the effect of treatment with C24D in an in vivo model using TNBC cells in nude mice transplanted with human PBMCs. Analysis of the xenografts from the C24D-treated mice showed that tumor growth was arrested with both the 60 and the 300 μg/mouse doses. However, better results were achieved with 60 μg/mouse. Also in the in vitro studies, higher than optimal doses resulted in a lower response. Such bell-shaped dose responses, resulting from immunological feedback, drug-receptor binding affinity, target saturation and drug stability have been reported.Citation48 Also anti-PD1 antibodies display similar bell-shaped dose-response and receptor saturation characteristics.Citation49The combination of the relatively long stability of C24D and receptor saturation may explain the reduction of the immune response with amounts larger than the optimal dose.
In the serum of the treated mice, we found an increase in human INF-γ. An immuno-fluorescent analysis of the MDA-MB-231 xenografts demonstrated a significant increase in tumor cell apoptosis, explained by a significant increase in infiltrated human CD45+ cells, composed of human CD8 + T cells and activated human CD56+ NK cells. Clinical studies have found a connection between tumor-infiltrated NK and CD8+ cells and anti-cancer immune responses linked to breast cancer patients’ survival.Citation50 Particularly with TNBC, a 10% increase in tumor-infiltrating lymphocytes resulted in improved prognosis.Citation9,Citation46 Moreover, a recent study correlated TNBC patients’ survival (up to 18-year follow-up) with high frequency of CD8+ tumor infiltrated cells.Citation51
Since CD45 is a ubiquitous receptor expressed on all leucocytes,Citation17,Citation26,Citation27 we performed preliminary assays to test C24D’s specificity. We treated PBMCs exposed to normal breast cells (MCF-10A) with C24D and evaluated killing and cytokine secretion. In contrast to PBMCs exposed to TNBC cells, addition of C24D did not induce an immune-stimulating effect, suggesting that C24D only affects immune-suppressed cells.
CD45 has been called a signaling “gatekeeper”, filtering weak or false signaling events and allowing effective signaling.Citation35 Considering the results presented herein, it is possible that breast tumors exploit CD45’s gatekeeper function, suppressing immune response, which is subsequently reversed by treatment with C24D.
Given the central role played by CD45 in the immune system, and given the positive results obtained with C24D treatment, we propose that CD45 can be a promising therapeutic target for the reactivation of immune responses in cancer.Citation28,Citation52 We hope this study rejuvenates the interest in delineating new roles in immunotherapy for the “old” CD45 target.
However, further studies are essential for the development of an effective immunomodulatory compound. Studies for the identification of the C24D/CD45 binding epitope and for a more comprehensive elucidation of the anti-tumor mechanism have been initiated and will undoubtedly shed light on this novel therapeutic strategy.
List of abbreviations:
TNBC triple negative breast cancer
PD1/PDL-1 the programmed cell death protein 1 or programmed cell death ligand 1 CTLA-4: cytotoxic T-lymphocyte-associated antigen 4
PTPRC protein tyrosine phosphatase receptor type C
TCR T cell receptor
Lck lymphocyte-specific protein tyrosine kinase
ITAMs immunoreceptor tyrosine-based activation motifs
ZAP70 Zeta-chain-associated protein kinase 70
PBMC peripheral blood mononuclear cells
IFNγ Interferon gamma
TNFα Tumor Necrosis Factor alpha
IL-2 Interleukin 2
FMO fluorescence minus one
Authors’ contributions:
AR and RY: obtained funding and planned the study. OZ and ES: were involved in the methodology development. JL, SM, SD: conducted in vitro and in vivo experiments, analyzed data and contributed to generating figures and statistical analysis. IL involved in developing methodology and interpretation of FACS data. AR supervised the study and interpreted the results. The manuscript was written by AR and edited and revised by RY and CC.
Ethics approval and consent to participate:
Peripheral blood from healthy female donors or breast cancer patients was isolated from blood, obtained from the Blood Bank Mada Tel HaShomer or Davidoff Cancer Center, respectively. The protocol was approved by the Institutional Review Board at Rabin Medical Center, Israel (0667-14-RMC).
The animal experimental procedures used in the present study were approved by the Animal Care and Use Committee of Tel Aviv University (TAU 06-01-20220), in accordance with their guidelines
Consent for publication:
This manuscript has not been previously published and is not under consideration for publication elsewhere.
Availability of data and material:
The authors confirm that the data supporting the findings of this study are available within the article and its supplementary materials.
Competing interests and funding:
The authors declare no potential conflicts of interest.
“Imanu Immunotherapy, Inc.” (Imanu) has an exclusive license from Ramot at Tel Aviv University, Ltd. for the C24D technology. The technology is protected by patents, some granted, some pending.
Imanu funded the research reported herein but was not involved in the study design or analysis of the results.
Supplemental Material
Download ()Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
References
- Murciano-Goroff YR, Warner AB, Wolchok JD. The future of cancer immunotherapy: microenvironment-targeting combinations. Cell Res. 2020;30(6):507–14. doi:10.1038/s41422-020-0337-2.
- Beatty GL, Gladney WL. Immune escape mechanisms as a guide for cancer immunotherapy. Clinical Cancer Res. 2015;21(4):687–692. doi:10.1158/1078-0432.CCR-14-1860.
- Friedrich M, Jasinski-Bergner S, Lazaridou M-F, Subbarayan K, Massa C, Tretbar S, Mueller A, Handke D, Biehl K, Bukur J, et al. Tumor-induced escape mechanisms and their association with resistance to checkpoint inhibitor therapy. Cancer Immunology, Immunotherapy. 2019;68(10):1689–1700. doi:10.1007/s00262-019-02373-1.
- Töpfer K, Kempe S, Müller N, Schmitz M, Bachmann M, Cartellieri M, Schackert G, Temme A. Tumor evasion from T cell surveillance. J Biomed Biotechnol. 2011;2011:918471. doi:10.1155/2011/918471.
- Denkert C, Von Minckwitz G, Darb-Esfahani S, Lederer B, Heppner BI, Weber KE, Budczies J, Huober J, Klauschen F, Furlanetto J. Tumour-infiltrating lymphocytes and prognosis in different subtypes of breast cancer: a pooled analysis of 3771 patients treated with neoadjuvant therapy. Lancet Oncol. 2018;19(1):40–50. doi:10.1016/S1470-2045(17)30904-X.
- Yuan C, Liu Z, Yu Q, Wang X, Bian M, Yu Z, Yu J. Expression of PD-1/PD-L1 in primary breast tumours and metastatic axillary lymph nodes and its correlation with clinicopathological parameters. Sci Rep. 2019;9(1):14356. doi:10.1038/s41598-019-50898-3.
- Luen S, Virassamy B, Savas P, Salgado R, Loi S. The genomic landscape of breast cancer and its interaction with host immunity. Breast. 2016;29:241–250. doi:10.1016/j.breast.2016.07.015.
- Robert C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat Commun. 2020;11(1):3801. doi:10.1038/s41467-020-17670-y.
- Hartmann FJ, Babdor J, Gherardini PF, Amir EAD, Jones K, Sahaf B, Marquez DM, Krutzik P, O’Donnell E, Sigal N, et al. Comprehensive immune monitoring of clinical trials to advance human immunotherapy. Cell Rep. 2019;28(3):819–831. doi:10.1016/j.celrep.2019.06.049.
- Zhu H, Du C, Yuan M, Fu P, He Q, Yang B, Cao J. PD-1/PD-L1 counterattack alliance: multiple strategies for treating triple-negative breast cancer. Drug Discov Today. 2020;25(9):1762–1771. doi:10.1016/j.drudis.2020.07.006.
- Zhao X, Wangmo D, Robertson M, Subramanian S. Acquired resistance to immune checkpoint blockade therapies. Cancers. 2020;12(5):1161. doi:10.3390/cancers12051161.
- Garrido-Castro AC, Lin NU, Polyak K. Insights into molecular classifications of triple-negative breast cancer: improving patient selection for treatment. Cancer Discov. 2019;9(2):176–198. doi:10.1158/2159-8290.CD-18-1177.
- Keenan TE, Tolaney SM. Role of immunotherapy in triple-negative breast cancer. Journal of the National Comprehensive Cancer Network. 2020;18(4):479–489. doi:10.6004/jnccn.2020.7554.
- D’Abreo N, Adams S. Immune-checkpoint inhibition for metastatic triple-negative breast cancer: safety first? Nat Rev Clin Oncol. 2019;16(7):399–400. doi:10.1038/s41571-019-0216-2.
- Di Sante G, Pagé J, Jiao X, Nawab O, Cristofanilli M, Skordalakes E, Pestell RG. Recent advances with cyclin-dependent kinase inhibitors: therapeutic agents for breast cancer and their role in immuno-oncology. Expert Rev Anticancer Ther. 2019;19(7):569–587. doi:10.1080/14737140.2019.1615889.
- Solodeev I, Zahalka MA, Moroz C. The novel C24D synthetic polypeptide inhibits binding of placenta immunosuppressive ferritin to human t cells and elicits anti–breast cancer immunity. In Vitro and in Vivo Neoplasia. 2014;16(9):741–750. doi:10.1016/j.neo.2014.08.005.
- Huntington ND, Tarlinton DM. CD45: direct and indirect government of immune regulation. Immunol Lett. 2004;94(3):167–174. doi:10.1016/j.imlet.2004.05.011.
- Chang VT, Fernandes RA, Ganzinger KA, Lee SF, Siebold C, McColl J, Jönsson P, Palayret M, Harlos K, Coles CH, et al. Initiation of T cell signaling by CD45 segregation at ‘close contacts’. Nat Immunol. 2016;17(5):574–582. doi:10.1038/ni.3392.
- Filipp D, Ballek O, Manning J. Lck, membrane microdomains, and tcr triggering machinery: defining the new rules of engagement. Front Immunol. 2012;3:155. doi:10.3389/fimmu.2012.00155.
- Barreira M, Rodríguez-Fdez S, Bustelo XR. New insights into the Vav1 activation cycle in lymphocytes. Cell Signal. 2018;45:132–144. doi:10.1016/j.cellsig.2018.01.026.
- Bellucci A, Fiorentini C, Zaltieri M, Missale C, Spano P. The “In Situ” Proximity Ligation Assay to Probe Protein–Protein Interactions in Intact Tissues. In: Ivanov A, editor. Exocytosis and endocytosis. methods in molecular biology (methods and protocols), vol 1174:397-405 New York (NY): Humana Press; 2014. doi:10.1007/978-1-4939-0944-5_27.
- Zhang -Q-Q, Chen J, Zhou D-L, Duan Y-F, Qi C-L, Li J-C, He X-D, Zhang M, Yang Y-X, Wang L. Dipalmitoylphosphatidic acid inhibits tumor growth in triple-negative breast cancer. Int J Biol Sci. 2017;13(4):471–479. doi:10.7150/ijbs.16290.
- Rothlin CV, Ghosh S. Lifting the innate immune barriers to antitumor immunity. J Immuno Therapy Cancer. 2020;8(1):e000695. doi:10.1136/jitc-2020-000695.
- Di Cosimo S. Advancing immunotherapy for early-stage triple-negative breast cancer. The Lancet. 2020;396(10257):1046–1048. doi:10.1016/S0140-6736(20)31962-0.
- Wilson TR, Udyavar AR, Chang C-W, Spoerke JM, Aimi Savage J, Savage HM, Daemen JA, O’Shaughnessy JA, Bourgon MR, Lackner ,MR. Genomic alterations associated with recurrence and tnbc subtype in high-risk early breast cancers. Mol Cancer Res. 2019;17(1):97–108. doi:10.1158/1541-7786.MCR-18-0619.
- Leitenberg D, Novak TJ, Farber D, Smith BR, Bottomly K. The extracellular domain of CD45 controls association with the CD4-T cell receptor complex and the response to antigen-specific stimulation. J Exp Med. 1996;183(1):249–259. doi:10.1084/jem.183.1.249.
- Hermiston ML, Xu Z, Weiss A. CD45: a critical regulator of signaling thresholds in immune cells. Annu Rev Immunol. 2003;21(1):107–137. doi:10.1146/annurev.immunol.21.120601.140946.
- Rheinländer A, Schraven B, Bommhardt U. CD45 in human physiology and clinical medicine. Immunol Lett. 2018;196:22–32. doi:10.1016/j.imlet.2018.01.009.
- Thomas ML, Brown EJ. Positive and negative regulation of Src-family membrane kinases by CD45. Immunol Today. 1999;20(9):406–411. doi:10.1016/S0167-5699(99)01506-6.
- Saunders AE, Johnson P. Modulation of immune cell signalling by the leukocyte common tyrosine phosphatase, CD45. Cell Signal. 2010;22(3):339–348. doi:10.1016/j.cellsig.2009.10.003.
- Gaud G, Lesourne R, Love PE. Regulatory mechanisms in T cell receptor signalling. Nat Rev Immunol. 2018;18(8):485–497. doi:10.1038/s41577-018-0020-8.
- Klammt C, Novotná L, Li DT, Wolf M, Blount A, Zhang K, Fitchett JR, Lillemeier BF. T cell receptor dwell times control the kinase activity of Zap70. Nat Immunol. 2015;16(9):961–969. doi:10.1038/ni.3231.
- Lo W-L, Shah NH, Ahsan N, Horkova V, Stepanek O, Salomon AR, Kuriyan J, Weiss, Weiss A. ALck promotes Zap70-dependent LAT phosphorylation by bridging Zap70 to LAT. Nat Immunol. 2018;19(7):733–741. doi:10.1038/s41590-018-0131-1.
- Bustelo XR. Vav family exchange factors: an integrated regulatory and functional view. Small GTPases. 2014;5(2):e973757. doi:10.4161/21541248.2014.973757.
- Courtney AH, Shvets AA, Lu W, Griffante G, Mollenauer M, Horkova V, Lo W-L, Yu S, Stepanek O, Chakraborty AK, et al. CD45 functions as a signaling gatekeeper in T cells. Sci Signal. 2019;12(604):eaaw8151. doi:10.1126/scisignal.aaw8151.
- Koretzky GA, Picus J, Thomas ML, Weiss A. Tyrosine phosphatase CD45 is essential for coupling T-cell antigen receptor to the phosphatidyl inositol pathway. Nature. 1990;346(6279):66–68. doi:10.1038/346066a0.
- Smith-Garvin JE, Koretzky GA, Jordan MS. T cell activation. Annu Rev Immunol. 2009;27(1):591–619. doi:10.1146/annurev.immunol.021908.132706.
- Clausen J. Functional significance of the activation-associated receptors CD25 and CD69 on human NK-cells and NK-like T-cells. Immunobiology. 2003;207(2):85–93. doi:10.1078/0171-2985-00219.
- Yacyshyn MBB, Poppema S, Berg A, Maclean GD, Reddish MA, Meikle A, Longenecker BM. CD69+ and HLA-DR+ activation antigens on peripheral blood lymphocyte populations in metastatic breast and ovarian cancer patients: correlations with survival following active specific immunotherapy. Int J Cancer. 1995;61(4):470–474. doi:10.1002/ijc.2910610407.
- Mandó P, Rizzo M, Roberti MP, Juliá E, Pampena MB, Pérez De La Puente C, Loza CM, Ponce C, Nadal J, Coló FA, et al. High neutrophil to lymphocyte ratio and decreased CD69+NK cells represent a phenotype of high risk in early-stage breast cancer patients. Onco Targets Ther. 2018;11:2901–2910. doi:10.2147/OTT.S160911.
- Radulovic K, Rossini V, Manta C, Holzmann K, Kestler HA, Niess JH. The early activation marker CD69 regulates the expression of chemokines and CD4 T cell accumulation in intestine. PLoS One. 2013;8(6):e65413. doi:10.1371/journal.pone.0065413.
- Cibrián D, Sánchez-Madrid F. CD69: from activation marker to metabolic gatekeeper. Eur J Immunol. 2017;47(6):946–953. doi:10.1002/eji.201646837.
- Tang X, Yocum DE, DeJonghe D, Nordensson K. Characterizing a soluble survival signal for activated lymphocytes from CD14+ cells. Immunology. 2002;107(1):56–68. doi:10.1046/j.1365-2567.2002.01463.x.
- Hadadi E, Zhang B, Baidžajevas K, Nurhashikin Y, Puan KJ, Ong SM, Yeap WH, Rotzschke O, Kiss-Toth E, Wilson H, et al. Differential IL-1β secretion by monocyte subsets is regulated by Hsp27 through modulating mRNA stability. Sci Rep. 2016;6(39035). doi:10.1038/srep39035
- Kared H, Martelli S, Ng TP, Henriquez S, Lécuroux C, Bitu M, Avettand-Fenoel V, Churaqui F, Catalan P, Chéret A, et al. CD57 in human natural killer cells and T-lymphocytes. Cancer Immunol Immunother. 2016;65(4):441–452. doi:10.1007/s00262-016-1803-z
- Nieto-Velázquez NG, Torres-Ramos YD, Muñoz-Sánchez JL, Espinosa-Godoy L, Gómez-Cortés S, Moreno J, Moreno-Eutimio MA. altered expression of natural cytotoxicity receptors and nkg2d on peripheral blood NK cell subsets in breast cancer patients. Transl Oncol. 2016;9(5):384–391. doi:10.1016/j.tranon.2016.07.003.
- Muntasell A, Servitja S, Cabo M, Bermejo B, Pérez-Buira S, Rojo F, Costa-García M, Arpí O, Moraru M, Serrano L, et al. High numbers of circulating CD57+NK cells associate with resistance to HER2-specific therapeutic antibodies in HER2+primary breast cancer. Cancer Immunol Res. 2019;7(8):1280–1292. doi:10.1158/2326-6066.CIR-18-0896.
- Patil VM, Noronha V, Joshi A, Abhyankar A, Menon N, Banavali S, Gupta S, Prabhash K. Low doses in immunotherapy: are they effective? Cancer Res Stat Treat. 2019;2(1):54–60. doi:10.4103/CRST.CRST_29_19.
- Adams S, Gatti-Mays ME, Kalinsky K, Korde LA, Sharon E, Amiri-Kordestani L, Bear H, McArthur HL, Frank E, Perlmutter J, et al. Current landscape of immunotherapy in breast cancer: a review. JAMA Oncol. 2019;5(8):1205–1214. doi:10.1001/jamaoncol.2018.7147.
- Egelston CA, Avalos C, Tu TY, Rosario A, Wang R, Solomon S, Srinivasan G, Nelson MS, Huang Y, Lim MH, et al. Resident memory CD8+ T cells within cancer islands mediate survival in breast cancer patients. JCI Insight. 2019;4(19):e130000. doi:10.1172/jci.insight.130000.
- Vihervuori H, Autere TA, Repo H, Kurki S, Kallio L, Lintunen MM, Talvinen K, Kronqvist P. Tumor-infiltrating lymphocytes and CD8+ T cells predict survival of triple-negative breast cancer. J Cancer Res Clin Oncol. 2019;145(12):3105–3114. doi:10.1007/s00432-019-03036-5.
- Dahlke MH, Larsen SR, Rasko JE, Schlitt HJ. The biology of CD45 and its use as a therapeutic target. Leuk Lymphoma. 2004;45(2):229–236. doi:10.1080/1042819031000151932.