
ABSTRACT
Checkpoint blockade therapy is effective against many cancers; however, new targets need to be identified to treat patients who do not respond to current treatment or demonstrate immune escape. Here, we showed that blocking the inhibitory receptor Killer cell lectin-like receptor G1 (KLRG1) enhances anti-tumor immunity mediated by NK cells and CD8+ T cells. We found that loss of KLRG1 signaling alone significantly decreased melanoma and breast cancer tumor growth in the lungs of mice. In addition, we demonstrated that KLRG1 blockade can synergize with PD-1 checkpoint therapy to increase the therapeutic efficacy compared to either treatment alone. This effect was even observed with tumors that do not respond to PD-1 checkpoint therapy. Double blockade therapy led to significantly decreased tumor size, increased frequency and activation of CD8+ T cells, and increased NK cell frequency and maturation in the tumor microenvironment. These findings demonstrate that KLRG1 is a novel checkpoint inhibitor target that affects NK and T cell anti-tumor immunity, both alone and in conjunction with established immunotherapies.
KEYWORDS:
Introduction
Immunotherapy has brought new hope to countless cancer patients with advanced or previously untreatable disease through the generation of novel therapeutics.Citation1–3 One specific area of this field, monoclonal antibodies (mAbs) that block immune inhibitory “checkpoint” receptors, have achieved great success in tumor control and significantly increased survival in patients. The most well-known checkpoint inhibitor targets are programed death-1 (PD-1),Citation4,Citation5 and cytotoxic T lymphocyte-associated molecule-4 (CTLA-4),Citation6 used for treating melanoma, in addition to other cancers. However, a significant subset of patients will either not respond to treatment or exhibit only transient benefits. Due to the poor efficacy in these patients, it is imperative to investigate additional immune cell types and receptor targets for modulation that can be used alone and in conjunction with current immunotherapies.
Recent studies have shown that natural killer (NK) cells are an important cell type for controlling certain types of cancer growth.Citation7,Citation8 NK cells can identify and kill cancer cells through multiple mechanisms, including degranulation and direct killingCitation9 and can recruit various other immune cell populations to the tumor microenvironment via the release of chemokines and cytokines.Citation10 Reduced NK cell function has been associated with a worse disease outcomeCitation11 and in some patient cohorts, the degree of NK cell infiltration in tumor tissues was associated with better prognosis.Citation11,Citation12 These data imply that therapies blocking inhibition of NK cells could be useful. While most current checkpoint blockade inhibitors target CD8+ T cells,Citation13 targeting new receptors on multiple types of immune cells, in both the adaptive and innate immune system, is essential for expanding the effect of checkpoint blockade and limiting the incidence of tumor immune escape.
One such target is killer cell lectin-like receptor G1 (KLRG1), a well-conserved transmembrane C-type lectin receptor that is expressed on NK cells and antigen experienced T cells. KLRG1 contains an immunoreceptor tyrosine inhibitory motif (ITIM) in its cytoplasmic tail and has two main ligands, E-cadherin and N-cadherin.Citation14,Citation15 The cadherins have long been implicated in tumorigenesis. E-cadherin can be expressed on primary tumors and distant metastatic tumorsCitation16 and N-cadherin upregulation enhances the invasiveness of tumor cells.Citation17 Cadherin levels have been observed to be high in several different human cancers, including melanoma, prostate, breast, hepatocellular carcinoma and renal cancer, among others.Citation18–20 Engagement of KLRG1 with either cadherin leads to ITIM phosphorylation and recruitment of the phosphatases SHIP-1/SHP-2Citation15,Citation21 inducing an inhibitory signaling cascade within the cell. High KLRG1 expression has been correlated with low proliferative capacityCitation22 and increased apoptosisCitation22 on both NK cells and T cells. KLRG1 expression is elevated on human CD8+ T cells during viral infection and may contribute to increased morbidity for several infectious diseases.Citation22,Citation23 While KLRG1 has historically been thought of as a marker of T cell senescence, research has shown that engagement of KLRG1 inhibits IFN-γ and TNF-α production.Citation15,Citation24 Experiments with cultured cells have demonstrated that KLRG1+ NK cells had lower cytotoxicity against target cells,Citation21 which could be reversed by KLRG1 knockdown via siRNA treatment.Citation25 Comparably less research has been done on KLRG1 signaling in cancer, however, significantly increased KLRG1 expression has been observed on T cells in a number of different human cancers,Citation18,Citation26 with 16–48% of tumor infiltrating CD8+ T cells being KLRG1+ in renal cell carcinoma, hepatocellular carcinoma, melanoma, ovarian cancer, and others.Citation18 These cells were found to have poor proliferative capacity with decreased effector cytokine production.Citation26 A recent study has suggested that KLRG1 blockade could decrease tumor burden;Citation18 however, the mechanism behind KLRG1 blockade remains to be elucidated.
Here, we show that KLRG1 blockade enhances anti-tumor immunity mediated by multiple types of immune cells in vivo. We developed KLRG1 deficient animals and a novel anti-KLRG1 blocking mAb which we used to determine the efficacy of KLRG1 blockade alone and in conjunction with PD-1 blockade. Loss of KLRG1 alone was found to significantly decrease melanoma and breast cancer tumor growth in the lungs of mice. We also found that KLRG1 synergizes with PD-1 to increase the therapeutic effect of either treatment alone, as combination blockade treatment led to significantly decreased tumor size. Increased NK cell frequency and maturation in the tumor microenvironment was quantified and increased levels of activated CD8+ T cells were also observed within the tumors of combination mAb treated mice. Overall, KLRG1 blockade was found to be effective at slowing tumor growth alone and in conjunction with PD-1 blockade by enhancing NK cell and CD8+ T cell presence within the tumor microenvironment.
Materials & methods
Mice and institutional animal care and use committee approval
C57BL/6NJ, C57BL/6, and Balb/c mice were purchased from Jackson Laboratory. All tumor measurements/tumor counts were performed blind. Both age and sex-matched mice were used for this study. All mouse experiments were carried out in accordance with the recommendations in the Guide for Care and Use of Laboratory Animals, as defined by the National Institutes of Health. Animal protocols were reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) of Brown University. All animals were housed in a centralized and AAALAC-accredited research animal facility that is fully staffed with technical and veterinary personnel.
Generation of KLRG1−/- mice
Two guideRNAs (gRNAs) were generated to target exon 3 and the surrounding intron of klrg1: 5ʹ-ATTGTGGACCATTCAGCTTG-3ʹ and 5ʹ-CTTAACTATGTAGTCCAGAC-3ʹ. gRNAs were selected using the CRISPR Guide RNA design tool Benchling (https://www.benchling.com/crispr/). Cas9/plasmid injection was performed on C57BL/6NJ zygotes. Non-homologous end-joining led to deletion of the sequence between gRNAs (KLRG1−/- mice). Founders were genotyped via sequencing and long-range PCR. Genotyping primers of KLRG1−/- mice: Forward: TGACCTCATGAACTCTGTGAGC, Reverse: CAGCCATCGATAATGAGATCTG. This work was performed in conjunction with the Brown University Mouse Transgenic and Gene Targeting Facility. Littermate controls were used for each mouse line.
Cell lines and culture conditions
B16-F10 and B16-E-cadherin (a gift from Five Prime Therapeutics, San Francisco, CA) were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM)/High Glucose with L-Glutamine (4 mM), Glucose (4500 mg/L), and sodium pyruvate (Thermo Fisher Scientific, Cat# SH30243.01). 4T1 (purchased from ATCC, Cat# CRL-2539) were cultured in Roswell Park Memorial Institute medium (RPMI-1640) with L-glutamine (2.05 mM/L) (Thermo Fisher Scientific, Cat# SH30027.01). In all cases, media contained 8% fetal bovine serum (FBS) (Atlanta Biologics, Cat# S11550, or Gibco, Cat# 10437–028), penicillin and streptomycin and glutamine (100 U/ml; Gibco, Cat# 10378–016), and 48 μM β-mercaptoethanol (Gibco, Cat# 21985–023). Cells were cultured in 5% CO2. B16-F10 and 4T1 cell lines were stimulated for 24 hours with IFN-γ where noted (20 ng/mL, Cat# 14–8311-63). DO11 N-cadherin GFP and DO11 mock GFP transfectant cell lines were generated as described here.Citation15
In vivo tumor growth experiments
Cells were washed and resuspended in sterile PBS. For most intravenous and subcutaneous B16 experiments, 2 × 105 cells in 200 μl were injected. For some intravenous injection experiments (noted in figure legend), 2 × 104 B16 cells in 200 μl were injected. For intravenous and subcutaneous 4T1 experiments, 1 × 105 cells in 200 μl were injected. Subcutaneous tumor growth was measured using calipers, and tumor volume was estimated using the formula V = (L*W2)/2. Tumor growth in the lungs from intravenous injection was quantified via macroscopic examination. For antibody blockade experiments, anti-KLRG1 (clone 1F10, generated as described below, 200 μg/mouse), anti-PD-1 (clone RMP1-14, BioXcell Cat# BE0146, 200 μg/mouse), or control antibody (rat IgG2a, BioXcell Cat# BE0089, 200 μg/mouse, mouse IgG1, BioXcell Cat# BE0083, 200 μg/mouse) were intraperitoneally injected on day 7, 10, 13, and 16 post-tumor cell injection. Animals were monitored daily and tumor growth was measured every third day. Animals were euthanized on day 21 post-intravenous injection and on day 19 or 22 post-subcutaneous injection or when tumor volume reached 2000 mm3.
Lymphocyte isolation
Spleens were dissociated in 150 mM NH4Cl for 10 minutes, filtered through nylon mesh, and washed twice with 1% PBS-serum. Tumors were dissociated in collagenase IV (Sigma-Aldrich Cat# C5138) using GentleMACS program m_impTumor_01.01 and incubated for 30 minutes at 37°C with shaking. Samples were then further dissociated on GentleMACS program m_impTumor_01.02 and incubated again for 30 minutes at 37°C with shaking. In a final dissociation step, tumors were run on GentleMACS m_impTumor_01.01 program and then passed through nylon mesh, washed with 1% PBS-serum and underlayed onto a two-step continuous Lympholyte gradient (GE Healthcare Bio-Sciences). Lymphocytes were harvested and washed once in 1% PBS-serum. Live cell counts were obtained using a hemocytometer/trypan blue exclusion.
Antibodies and flow cytometry
Cells were stained in 1% PBS-serum containing Fc block (2.4G2, produced in house) and cell surface antibodies for 20 minutes on ice in the dark. Staining with KLRG1 tetramer (NIH Tetramer Core Facility) was performed for 15 minutes at room temperature and 15 minutes on ice, both in the dark. Samples were run on a FACSAria III (BD Biosciences) and analyzed using FlowJo (Tree Star Inc.). The mAbs listed below were used for flow cytometry and purchased from Biolegend, eBioscience, BD Biosciences or Thermo Fischer Scientific: APC-KLRG1 tetramer (NIH Tetramer Facility), APC-PD-1 (Cat# 135229), APC-PD-L1 (Cat# 564715), APC-eF780- CD11b (Cat# 47–0112), BV41-KLRG1 tetramer (NIH Tetramer Facility), BV510-TCRβ (Cat# 109234), BV570-CD45 (Cat# 103136), BV605- CD3 (Cat# 100237), BV605- CD8 (Cat# 100744), BV785-NK1.1 (Cat# 108749), eF450 CD69 (Cat# 50–112-4119), FITC- CD27 (Cat# 11–0271), PerCP-Cy5.5-CD69 (Cat# 561931), PE-PD-1 (Cat# 109103), PE-Cy7-KLRG1 (Cat# 25–5893-82), PerCP-Cy5.5-E-cadherin (Cat# 147317). Brilliant Stain Buffer (BD Biosciences, Cat# 563794) was added to the staining cocktail when multiple Brilliant Violet antibodies were used. The KLRG1 tetramer was produced in the labCitation15 or acquired from the NIH tetramer facility.
Western blot
Samples were separated on 4–20% gradient SDS–PAGE gels and transferred to a nitrocellulose membrane (Bio-Rad Laboratories, Hercules, CA, USA). The membrane was blocked in 5% milk for 1 h at room temperature and incubated with the indicated primary antibody overnight at 4°C. After washing, the membrane was incubated with the respective HRP-conjugated secondary antibody for 1 h at room temperature and developed using a ChemiDoc XRS+ System (Bio-Rad).
Fc silent anti-KLRG1 mAb
The variable light-chain and heavy-chain sequences were recovered from the 1F10 hybridoma and cloned into expression vectors in frame with constant regions to obtain a mouse IgG1/κ antibody harboring the N297Q mutation to abrogate binding to Fcγ receptors. The linearized vectors for the light chain and for the heavy chain were co-transfected into a CHO cell line using the Neon device (Invitrogen). The antibody was purified from the supernatant using the MabSelect PrismA resin (Cytiva). The antibody was eluted with citrate 0.1 M pH4.5 buffer, dialyzed overnight against PBS and 0.22 µM filtered. Size exclusion chromatography, SDS-PAGE and endotoxin levels quality controls were performed.
In vitro cell line proliferation
B16-F10 and B16-E-cadherin cell lines were labeled with Cell Trace Violet Proliferation dye (5uM, Thermo Fisher Scientific Cat# C34557) for 20 minutes in the dark at 37°C. Culture medium was then added at five times the original staining volume and samples were incubated for another 5 minutes. Cells were then pelleted by centrifugation and resuspended in pre-warmed complete culture medium. Cells were finally incubated for 10 minutes before flow cytometry analysis to measure Day 0 staining. Samples were then allocated to flasks and flow cytometry was performed on Day 2 to assess proliferation cycles.
Statistical analysis
Analyses for determining statistical significance were performed using Prism 7.0 (Graph-Pad Software, Inc.). Unpaired two-tailed Student’s t tests were used to compare two individual groups. Graphs represent the mean and error bars indicate standard error of the mean (SEM). The number of animals/sample size and total experimental replicates are noted in each figure legend. *p < .05, **p < .01, ***p < .0001, and ****p < .00001.
Results
Loss of KLRG1 signaling decreases tumor burden
While KLRG1 is often thought of as a marker of lymphocyte maturity, its inhibitory properties in the context of viral infection suggest that it plays a significant role in the NK cell and T cell immune response.Citation22,Citation23 This, along with KLRG1 and its ligands high expression profile in human cancer samples, makes it an intriguing target for checkpoint blockade therapy.Citation18,Citation26 To study the effect of loss of KLRG1 in the context of tumor development, we generated KLRG1 deficient mice directly on the C57BL/6 background (Figure S1A-B). Although KLRG1 deficient mice have been previously generated, they were derived from ES cells on the 129 background. This is significant, as it was recently demonstrated that up to 40 megabases of the original 129 genome can be retained, even after 20 backcrosses to C57BL/6. This is significantly more than the predicted 0.01% of the genomeCitation27 and could potentially induce extraneous results. We did not observe any obvious developmental defects in our KLRG1 deficient mice, with KLRG1 deficient mice displaying no changes in NK or CD8+ T cell frequency and NK cell maturation was not affected (Figure S1C-E).
We have previously reported on the cadherin expression on several different murine cell lines, including A20 (B cell lymphoma), RMA-S (T cell lymphoma), RAW264.7 (macrophage leukemia) and L929 (fibroblast cell line) and found that these cell lines do not express cadherin.Citation15 More recently, we have examined cadherin expression on the murine melanoma model, B16-F10. B16-F10 is a well-characterized murine melanoma cell line, and its growth has been shown to be modulated by both NK cells and T cells.Citation28 Although B16-F10 was negative for E-cadherin, it was positive when stained with the KLRG1 tetramer (). Because the KLRG1 tetramer binds to both E-cadherin and N-cadherin,Citation15 we concluded that B16-F10 must endogenously express N-cadherin. We confirmed that B16-F10 expresses N-cadherin via western blot (data not shown), as the available anti-N-cadherin antibodies do not work well for flow cytometry in our hands. In addition to the parental line of B16-F10, we utilized B16-F10 that was transduced to express high, yet physiological, levels of E-cadherin, another KLRG1 ligand (B16-E-cadherin) (). The proliferation rates of B16-F10 and B16-E-cadherin were comparable in vitro (Figure S2A). While only five out of eighteen C57BL/6 mice intravenously injected with B16-F10 developed at least one tumor in the lungs, 16 out of 18 C57BL/6 mice intravenously injected with B16-E-cadherin developed at least one tumor (). This suggests that E-cadherin increases B16-F10 aggressiveness, possibly through modulation of the immune response.
Figure 1. KLRG1 knockout mice display fewer B16-E-cadherin tumors in the lungs
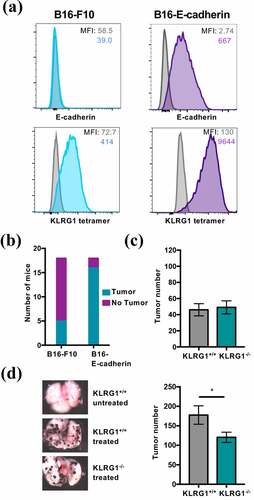
Having shown that E-cadherin expression renders B16-F10 more aggressive, we next investigated whether the KLRG1-cadherin axis was responsible for the observed phenotype. Loss of KLRG1 did not impact the number of parental B16-F10 tumors in the lungs of KLRG1 deficient mice compared to wild-type controls (). This could be due to the relatively low level of N-cadherin expression on B16-F10 (). To examine the effect of loss of KLRG1 signaling in the presence of higher levels of cadherin expression, we performed the same experiment using B16-E-cadherin. Interestingly, there was a significant decrease in B16-E-cadherin tumor number in the lungs of KLRG1 deficient mice compared to control mice (). Together, these data indicate that KLRG1 likely elicits an inhibitory effect on the immune response to cancer and validate KLRG1 as a target for checkpoint blockade therapeutics.
Combined KLRG1 and PD-1 blockade synergize to promote anti-tumor immunity against melanoma tumor growth
We investigated whether KLRG1 blockade could synergize with PD-1 blockade to decrease tumor burden. Combination checkpoint blockade therapies have demonstrated significant efficacy against a number of different types of cancer, often to greater benefit than one therapy alone.Citation29 Since B16 endogenously expresses PD-L1 and this expression is increased approximately 10-fold after IFN-γ stimulation ( and S2B), it is a useful model to examine the effect of combination blockade. To understand if PD-1 checkpoint blockade can provide a synergistic advantage in a mouse model that lacks KLRG1 signaling, we implanted subcutaneous B16-E-cadherin tumors in KLRG1 deficient mice and administered anti-PD-1 mAb or isotype control. We found that KLRG1 deficient mice that received anti-PD-1 mAb had reduced tumor volume compared to KLRG1 deficient mice that received isotype control, and WT mice that received either anti-PD-1 mAb or isotype control (Figure S2C).
Figure 2. Combination KLRG1 and PD-1 therapy synergizes to decrease B16-E-cadherin tumor burden
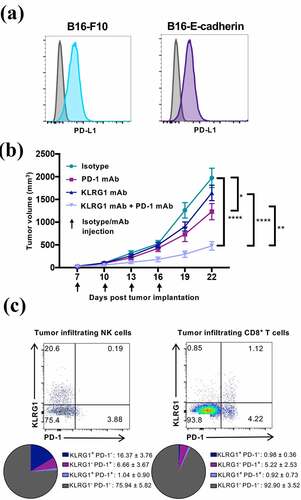
Having validated the potential synergy between KLRG1 and PD-1 pathways in mice that are deficient in KLRG1 signaling, we aimed to test this combination therapy in a wild-type mouse model. To this end, we generated a mouse Fc silent anti-mouse KLRG1 antibody (1F10) derived from a rat anti-mouse KLRG1 mAb (Figure S3A). The original rat IgG2b mAb could not be used in mice due to the interaction between mouse Fc receptors and rat IgG2b antibodies and potential xenogeneic effects. We examined the blocking potential of the newly generated anti-mouse KLRG1 mAb in vitro and showed that the new anti-KLRG1 antibody inhibited the binding of the KLRG1 tetramer to mouse cadherin (Figure S3B). To investigate the effects of KLRG1 and PD-1 combination blockade therapy, B16-E-cadherin tumor-bearing mice were treated with isotype control, anti-KLRG1 mAb, anti-PD-1 mAb, or a combination of both blocking agents. Anti-PD-1 mAb treatment alone demonstrated a very modest effect on B16-E-cadherin growth (). Anti-KLRG1 mAb treatment alone did not impact B16-E-cadherin tumor growth (). Interestingly, combination therapy with anti-KLRG1 and anti-PD-1 mAbs had a synergistic effect, improving the control of tumor growth as compared to either treatment alone ().
We then dissected the immune response to B16-E-cadherin in the tumor bed by analyzing tumor-infiltrating lymphocytes (TILs) (Figure S3C). In the isotype treated mice, ~16% of tumor-infiltrating NK cells and less than 1% of tumor-infiltrating CD8+ T cells expressed solely KLRG1 ( and S3D). Around 6% of tumor-infiltrating NK cells and ~5% of tumor-infiltrating CD8+ T cells were found to be solely PD-1+ ( and S3D). Approximately 1% of infiltrating NK cells and 1% of infiltrating CD8+ T cells were KLRG1+PD-1+ ().
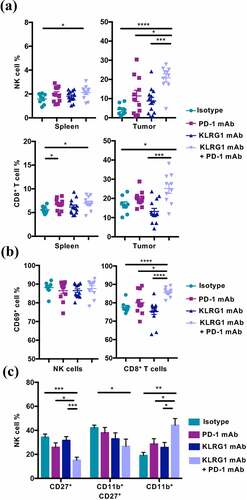
We found that mice that had been treated with KLRG1 and PD-1 double blockade had significantly higher levels of NK cells within the tumor microenvironment compared to all other treatment groups (). There was also an increase in splenic NK cell frequency in the combination therapy treated group (). Our analysis revealed an increase in frequency of CD8+ T cells in mice treated with both mAbs, in both the spleen and tumor microenvironment (). Intriguingly, a higher frequency of TIL CD8+ T cells was positive for the activation marker CD69 in double blockade treated mice compared to all other treatment groups ( and S3E). While the TIL NK cell population in double blockade treated animals did not display enhanced CD69 expression ( and S3E), these NK cells were found to display a more mature (CD27−CD11b+) phenotype (). These data suggest that both NK and CD8+ T cells play a role in the improved tumor control that was observed.
Figure 3. Combination therapy increases NK cell and CD8+ T cell frequency in the tumor microenvironment
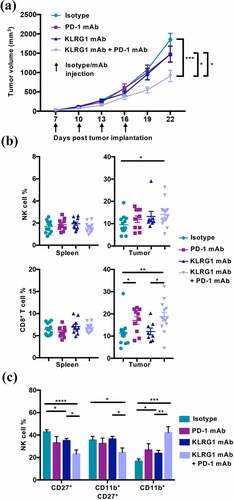
Combined KLRG1 and PD-1 blockade also significantly decreased the growth of parental un-transfected B16-F10 tumors (). This finding is of particular interest because it demonstrates that a tumor that does not respond to anti-PD-1 mAb therapy alone could still benefit from combination therapy with KLRG1 blockade. The effect was less drastic than observed in the B16-E-cadherin model, but this is likely due to the overall lower expression of cadherin in the B16-F10 model (). As observed in the B16-E-cadherin tumors, there was a significantly higher NK cell frequency and CD8+ T cell frequency in the tumors of double mAb treated B16-F10 mice (). These tumor-infiltrating NK cells were significantly more mature in the double blockade treated cohort (), as seen in the B16-E-cadherin model as well. In addition, the KLRG1 and PD-1 expression profiles on tumor infiltrating NK and CD8+ T cells from B16-F10 tumors were similar to what was observed with B16-E-cadherin (Figure S4A-B). Taken together, these data indicate that the combination of anti-KLRG1 mAb and anti-PD-1 mAb have a synergistic therapeutic effect against tumor growth, significantly enhancing CD8+ T cell responses. Similarly, although not significant in some contexts, a reproducible increase of the NK cell frequency is observed. In addition, these findings demonstrate that KLRG1 blockade can be effective against tumor growth in the presence of E-cadherin and N-cadherin, or simply N-cadherin alone.
Figure 4. Combination KLRG1 and PD-1 therapy synergizes to decrease B16-F10 tumor growth
KLRG1 blockade, alone and in combination with PD-1 blockade, increase anti-tumor immunity to 4T1 breast cancer
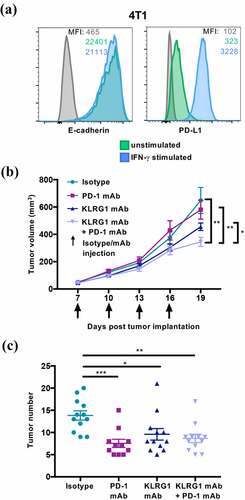
We next investigated whether KLRG1 blockade, both alone and in combination with PD-1 blockade, could decrease tumor growth of a cancer that endogenously expresses high levels of E-cadherin. To examine this question, we utilized the 4T1 model, which is a well-characterized murine mammary carcinoma cell line. We found that 4T1 expresses high levels of E-cadherin and moderate levels of PD-L1 (). We also report that 4T1 does not express N-cadherin (Figure S4C). This allowed us to examine the effect of KLRG1 blockade on a different tumor type and in a different genetic background, as 4T1 is derived from the Balb/c mouse line. Balb/c mice were subcutaneously injected with 4T1 cancer cells. 4T1 tumor-bearing mice were then treated with isotype control, anti-KLRG1 mAb, anti-PD-1 mAb, or a combination of both blocking agents. Anti-PD-1 mAb treatment alone demonstrated no effect on 4T1 tumor growth (). Anti-KLRG1 mAb treatment alone also did not significantly decrease tumor burden, although there was a trend toward lower tumor volume in this group (). Combination therapy with anti-KLRG1 and anti-PD-1 mAbs had a synergistic effect, improving the control of tumor growth as compared to either treatment alone (). There were no significant differences observed in tumor infiltrating lymphocyte frequencies in these mice (data not shown). Importantly, using a second cancer model, we establish that a tumor that does not respond to anti-PD-1 mAb therapy alone could still benefit from combination therapy with KLRG1 blockade.
Figure 5. KLRG1 therapy alone decreases 4T1 tumor burden in the lungs while double blockade therapy decreases subcutaneous 4T1 tumor burden
To examine the possibility of KLRG1 blockade controlling tumor metastasis, we intravenously injected Balb/c mice with 4T1 cancer cells. Animals were then treated with isotype control, anti-KLRG1 mAb, anti-PD-1 mAb, or a combination of both blocking agents. Anti-KLRG1 mAb treatment alone led to significantly fewer tumors in the lungs () compared to isotype treated mice. Anti-PD-1 blockade alone also decreased tumor number compared to isotype (). KLRG1 and PD-1 double blockade treatment decreased tumor number compared to the group treated with isotype, but the combination therapy did not synergize to decrease tumor burden compared to KLRG1 or PD-1 treatment alone (). While PD-1 blockade led to a slightly more significant decrease in tumor burden versus isotype than the other treatments, anti-PD-1 treatment was not significantly different than either anti-KLRG1 treatment or combination treatment. Altogether, we conclude that KLRG1 blockade can be an effective checkpoint inhibitor alone under certain conditions and in combination with PD-1 blockade for multiple types of cancer. It also indicates that KLRG1 blockade can be effective against tumors that solely express E-cadherin.
Discussion
Great strides have been made in T cell immunotherapy research, however, utilizing NK cells to better combat cancer has been studied comparably less. NK cells are intriguing targets of modulation for several reasons, not limited to their cytotoxic abilities and relatively short lifespan.Citation9,Citation11,Citation30 Finally, NK cell tumor infiltration is associated with a better prognosis, while low activity of peripheral blood NK cells is associated with increased cancer risk.Citation9,Citation11,Citation12,Citation30 Therapeutic targets for checkpoint inhibition that are expressed on both NK and CD8+ T cells are of great interest for improving current immunotherapy treatment options. One example of this type of broad-spectrum immune checkpoint is the inhibitory receptor NKG2A. Blockade of NKG2A has been shown to promote NK cell and CD8+ T-cell anti-tumor immunity in mice.Citation31 Monalizumab, a humanized anti-NKG2A antibody, enhanced NK cell activity and rescued CD8+ T cell function in combination with PD-1 blockade and is currently undergoing clinical trials.Citation31
In this study, we focus on KLRG1, a well-conserved transmembrane C-type lectin inhibitory receptor that is expressed on both NK cells and T cells. Engagement of KLRG1 inhibits IFN-γ and TNF-α productionCitation15,Citation24 and high KLRG1 expression correlates with low proliferative capacity and increased apoptosis.Citation22 Importantly, significantly increased KLRG1 expression has been observed on T cells in a number of different human cancers.Citation18,Citation26 These KLRG1+ cells were found to have poor proliferative capacity, with decreased effector cytokine production.Citation26 KLRG1 has two main ligands, E-cadherin and N-cadherin,Citation14,Citation15,Citation21 both of which are known to play a vital role in tumorigenesis and tumor progression.Citation32 While E-cadherin is essential for maintaining epithelial tissue integrity,Citation33 N-cadherin promotes increased cell motility and migration.Citation17 E-cadherin expression is usually lost during the epithelial to mesenchymal transition,Citation34–37 which is a key player in metastasis.Citation38,Citation39 However, it has been shown that E-cadherin can be expressed in primary tumors and again upregulated once cancer cells reach distant sites, including lymph nodes.Citation16 N-cadherin, while absent or expressed at low levels in normal epithelial tissue, has been shown to enhance migratory and invasive capacities of tumor cells when it is upregulated.Citation17,Citation40 Overall, cadherin expression during tumor development is still an area of intense investigation. However, it is clear that it plays an important role. We show here that cadherin allows tumor cells to undergo immune evasion and increases cancer aggressiveness, indicating that blocking cadherin interactions should be an important area of therapeutic inquiry.
We selected B16-F10 melanoma and 4T1 mammary carcinoma, which endogenously express moderate levels of N-cadherin and high levels of E-cadherin, respectively. We also took advantage of B16-F10 that has been transduced with E-cadherin. We found that significantly more mice developed tumor nodule(s) in the lungs after injection of B16-E-cadherin compared to mice injected with the parental B16-F10 line, suggesting that the presence of E-cadherin impaired the immune response to tumor development. While KLRG1 deficient mice did not display any changes in tumor development in the lungs after B16-F10 injection compared to controls, KLRG1 deficient mice did display significantly fewer B16-E-cadherin tumors compared to wild-type controls. A similar effect was observed when we administered our Fc-silent KLRG1 monoclonal antibody to mice as a treatment after 4T1 intravenous injection. These findings, along with the fact that lung NK cells express high levels of KLRG1,Citation41 and B16-F10 growth in the lungs has been shown to be heavily controlled by NK cells,Citation28 suggest that KLRG1 inhibits NK cell functions in these models. Intriguingly, NK cells have been implicated in preventing or controlling metastasis by eliminating circulating tumor cells.Citation42 While intravenous injection is considered “experimental metastasis” not “spontaneous metastasis,” it has long been viewed as a preliminary model to understanding metastatic control,Citation43 raising the possibility that KLRG1 blockade could be useful in a metastatic setting. Minimally, the data demonstrate that targeting KLRG1 is sufficient to control metastasis in two different experimental metastasis tumor models. This area of research warrants further inquiry using “spontaneous metastasis” models.
Combination of checkpoint blockade therapies have been shown to be effective against many different types of cancer.Citation29,Citation44 In support of this, we found that while anti-KLRG1 mAb treatment alone was not sufficient to decrease B16-E-cadherin subcutaneous tumor growth, combination treatment of anti-KLRG1 mAb and anti-PD-1 mAb significantly decreased subcutaneous tumor burden. A similar result was observed for B16-F10 and 4T1 subcutaneous growth, although to a slightly lesser extent. The difference observed could be a consequence of B16-E-cadherin expressing N-cadherin and E-cadherin molecules simultaneously, while B16-F10 and 4T1 only express either N-cadherin or E-cadherin, respectively. Nonetheless, it indicates that KLRG1 blockade and PD-1 blockade can synergize to increase anti-tumor immunity.
A prior report has been published on the effect of KLRG1 blockade during cancer; however, the study focused mainly on identifying KLRG1 and its ligands in human cancer data sets. The report notes that tumors were smaller and survival was increased in a small cohort of miceCitation18 but the mechanism behind this effect was not explored. We sought to assess the role of immune cells within the tumor during KLRG1 antibody treatment. Examination of the TIL population in the isotype control group revealed that very few tumor infiltrating lymphocytes were KLRG1/PD-1 double positive. Instead, expression of these markers was compartmentalized, with a clear KLRG1+ NK cell population and a clear PD-1+ CD8+ T cell population within the tumor microenvironment. The mutually exclusive expression of these two inhibitory receptors is not totally surprising, as it has been shown that the HMG-box transcription factor TOX represses KLRG1 expression and up regulates PD-1 expression, at least on CD8+ T cells.Citation45 Irrespective of the dynamic of PD-1 and KLRG1 expression, the lack of a substantial double positive population indicates that double blockade treatment is unlikely to function at the single cell level. Instead, the success of double antibody therapy reveals a cooperation between these two effector cell subsets to combat tumor development. Toward this end, NK cells have repeatedly been shown to be important in helping T cells mount an anti-tumor response, as NK cell frequency correlates with protective intratumoral dendritic cell recruitment, patient responsiveness to PD-1 therapy, and increased overall survival.Citation46–48 Importantly, this does not rule out that either molecule can regulate/inhibit both NK and CD8+ T cells. For instance, it has been shown that PD-1 blockade can unleash NK cells,Citation9,Citation49 as well as CD8+ T cells.Citation50 Our data and a recent studyCitation51 suggest that in current checkpoint blockade therapies, the effect on NK cell function may have been overlooked.
We also found that combination therapy treated tumors contained significantly higher frequencies of both NK cells and CD8+ T cells. This may be the result of higher levels of infiltration into the tumor microenvironment or increased levels of proliferation. Since KLRG1 engagement has been shown to induce defective Akt (ser473) phosphorylation leading to proliferative dysfunction,Citation22,Citation52 it is tempting to speculate that the observed increase in NK and CD8+ T cell populations is due to increased proliferative capacity when KLRG1 is blocked. Increased proliferation levels of NK and CD8+ T cells within the tumor could also work to further increase immune cell infiltration via increased cumulative levels of cytokine and chemokine release. These increased levels of TILs were complemented by increased expression of CD69, a well-known activation marker, on infiltrating CD8+ T cells, indicating their activation in situ. Tumor-infiltrating NK cells from combination therapy treated mice were significantly more mature than infiltrating NK cells in the isotype treated group or either mAb treatment group alone, as seen by a higher frequency of CD27−CD11b+ NK cells. It is well documented that CD27−CD11b+ NK cells have been heavily educated via exposure to activating ligands.Citation53 These findings indicate that both NK cells and CD8+ T cells play a role in the slower tumor growth observed in the combination blockade treated animals, possibly through a combination of enhanced immune cell recruitment and activation.
In conclusion, these results indicate that KLRG1 checkpoint blockade therapy can be effective alone in some contexts and can synergize with PD-1 blockade to increase anti-tumor immunity against local and disseminated tumor models. Our research elucidated that anti-KLRG1 and anti-PD-1 combination mAb therapy significantly decreased primary tumor growth and led to the accumulation of larger populations of mature NK and activated CD8+ T cells within the tumor microenvironment. The ubiquitous expression of cadherin during solid tumor growth and metastasis progression reinforces the attractiveness of the KLRG1/cadherin axes as a potential treatment for cancers. Altogether, our investigation expands the arsenal of checkpoint inhibitor targets available to oncologists.
Disclosure of potential conflicts of interest
CL, MO, BR, and EV are employees of Innate-Pharma. The other authors declare no competing interests.
Disclaimer statement
The views expressed in this manuscript are the authors’ own and not an official position of the institution or funder.
Author contributions
AT and LB conceptualized the project and designed most experiments. AT performed most experiments. GD and CF performed some experiments. EV shared mutant mouse reagents, advice and provided intellectual contribution. CL, MO and BR provided reagents. AT and LB wrote the manuscript with useful input from all authors.
Sources of Support
This work was supported by the following NIH research grants: R01 AI46709 (LB), R01 AI122217 (LB), Brown Respiratory Research Training Program (NHLBI Grant: T32 HL134625) (AT) and F31 CA243305 (AT). GD was supported by a Research Supplement 3R01AI122217-S1 to promote diversity. The FACSAria was funded by an NCCR equipment grant 1S10RR021051 (LB) and upgraded to a FACSAria III by Provost’s equipment fund. EV is supported by funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (TILC, grant agreement No. 694502), the Agence Nationale de la Recherche including the PIONEER Project (ANR-17-RHUS-0007), Equipe Labellisée “La Ligue” (Ligue Nationale contre le Cancer), MSDAvenir, Innate Pharma and institutional grants to the CIML (INSERM, CNRS, and Aix-Marseille University) and to Marseille Immunopole.
Supplemental Material
Download ()Acknowledgments
We thank Kevin Carlson for cell sorting, Dr. Srinivas Kothakota for providing the B16-F10 and B16-E-cadherin cell lines, and the NIH tetramer facility for providing KLRG1 loaded tetramers. We thank Delia Demers for experimental support. We also thank Dr. Courtney Anderson for scientific discussions and Dr. Lalit Beura for critical reading of the manuscript.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
Additional information
Funding
References
- Baumeister SH, Freeman GJ, Dranoff G, Sharpe AH. Coinhibitory Pathways in Immunotherapy for Cancer. Annu Rev Immunol. 2016;34:539–11. doi:10.1146/annurev-immunol-032414-112049.
- Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science (New York, NY). 2015;348(6230):69–74. doi:10.1126/science.aaa4971.
- Sharma P, Allison JP. The future of immune checkpoint therapy. Science (New York, NY). 2015;348(6230):56–61. doi:10.1126/science.aaa8172.
- Okazaki T, Honjo T. The PD-1-PD-L pathway in immunological tolerance. Trends Immunol. 2006;27(4):195–201. doi:10.1016/j.it.2006.02.001.
- Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515(7528):568–571. doi:10.1038/nature13954.
- Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science (New York, NY). 1996;271(5256):1734–1736. doi:10.1126/science.271.5256.1734.
- Chiossone L, Dumas PY, Vienne M, Vivier E. Natural killer cells and other innate lymphoid cells in cancer. Nat Rev Immunol. 2018;18(11):671–688. doi:10.1038/s41577-018-0061-z.
- Shimasaki N, Jain A, Campana D. NK cells for cancer immunotherapy. Nat Rev Drug Discov. 2020;19(3):200–218. doi:10.1038/s41573-019-0052-1.
- Cózar B, Greppi M, Carpentier S, Narni-Mancinelli E, Chiossone L, Vivier E. Tumor-Infiltrating Natural Killer Cells. Cancer Discov. 2021;11(1):34. doi:10.1158/2159-8290.CD-20-0655.
- Raulet DH. Interplay of natural killer cells and their receptors with the adaptive immune response. Nat Immunol. 2004;5(10):996–1002. doi:10.1038/ni1114.
- Ishigami S, Natsugoe S, Tokuda K, Nakajo A, Che X, Iwashige H, Aridome K, Hokita S, Aikou T. Prognostic value of intratumoral natural killer cells in gastric carcinoma. Cancer. 2000;88(3):577–583. doi:10.1002/(SICI)1097-0142(20000201)88:3<577::AID-CNCR13>3.0.CO;2-V.
- Delahaye NF, Rusakiewicz S, Martins I, Ménard C, Roux S, Lyonnet L, Paul P, Sarabi M, Chaput N, Semeraro M, et al. Alternatively spliced NKp30 isoforms affect the prognosis of gastrointestinal stromal tumors. Nat Med. 2011;17(6):700–707. doi:10.1038/nm.2366.
- Robert C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat Commun. 2020;11(1):3801. doi:10.1038/s41467-020-17670-y.
- Gründemann C, Bauer M, Schweier O, Von Oppen N, Lässing U, Saudan P, Becker K-F, Karp K, Hanke T, Bachmann MF, et al. Cutting edge: identification of E-cadherin as a ligand for the murine killer cell lectin-like receptor G1. J Immunol. 2006;176(3):1311–1315. (Baltimore, Md: 1950). doi:10.4049/jimmunol.176.3.1311.
- Tessmer MS, Fugere C, Stevenaert F, Naidenko OV, Chong HJ, Leclercq G, Brossay L. KLRG1 binds cadherins and preferentially associates with SHIP-1. Int Immunol. 2007;19(4):391–400. doi:10.1093/intimm/dxm004.
- Padmanaban V, Krol I, Suhail Y, Szczerba BM, Aceto N, Bader JS, Ewald AJ. E-cadherin is required for metastasis in multiple models of breast cancer. Nature. 2019;573(7774):439–444. doi:10.1038/s41586-019-1526-3.
- Thiery JP, Sleeman JP. Complex networks orchestrate epithelial–mesenchymal transitions. Nat Rev Mol Cell Biol. 2006;7(2):131–142. doi:10.1038/nrm1835.
- Greenberg SA, Kong SW, Thompson E, Gulla SV. Co-inhibitory T cell receptor KLRG1: human cancer expression and efficacy of neutralization in murine cancer models. Oncotarget. 2019;10(14):1399–1406. doi:10.18632/oncotarget.26659.
- Gruss C, Herlyn M. Role of cadherins and matrixins in melanoma. Curr Opin Oncol. 2001;13(2):117–123. doi:10.1097/00001622-200103000-00006.
- Kaszak I, Witkowska-Piłaszewicz O, Niewiadomska Z, Dworecka-Kaszak B, Ngosa Toka F, Jurka P. Role of cadherins in cancer-a review. Int J Mol Sci. 2020;21(20):7624. doi:10.3390/ijms21207624.
- Ito M, Maruyama T, Saito N, Koganei S, Yamamoto K, Matsumoto N. Killer cell lectin-like receptor G1 binds three members of the classical cadherin family to inhibit NK cell cytotoxicity. J Exp Med. 2006;203(2):289–295. doi:10.1084/jem.20051986.
- Wang JM, Cheng YQ, Shi L, Ying RS, Wu XY, Li GY, Moorman JP, Yao ZQ. KLRG1 negatively regulates natural killer cell functions through the Akt pathway in individuals with chronic hepatitis C virus infection. J Virol. 2013;87(21):11626–11636. doi:10.1128/jvi.01515-13.
- Voehringer D, Koschella M, Pircher H. Lack of proliferative capacity of human effector and memory T cells expressing killer cell lectinlike receptor G1 (KLRG1). Blood. 2002;100(10):3698–3702. doi:10.1182/blood-2002-02-0657.
- Robbins SH, Nguyen KB, Takahashi N, Mikayama T, Biron CA, Brossay L. Cutting edge: inhibitory functions of the killer cell lectin-like receptor G1 molecule during the activation of mouse NK cells. J Immunol. 2002;168(6):2585–2589. (Baltimore, Md: 1950). doi:10.4049/jimmunol.168.6.2585.
- Muller-Durovic B, Lanna A, Polaco Covre L, Mills RS, Henson SM, Akbar AN. Killer cell lectin-like receptor G1 inhibits NK cell function through activation of adenosine 5ʹ-monophosphate-activated protein kinase. J Immunol (Baltimore, Md: 1950). 2016;197(7):2891–2899. doi:10.4049/jimmunol.1600590.
- Li L, Wan S, Tao K, Wang G, Zhao E. KLRG1 restricts memory T cell antitumor immunity. Oncotarget. 2016;7(38):61670–61678. doi:10.18632/oncotarget.11430.
- Chisolm DA, Cheng W, Colburn SA, Silva-Sanchez A, Meza-Perez S, Randall TD, Weinmann AS. Defining genetic variation in widely used congenic and backcrossed mouse models reveals varied regulation of genes important for immune responses. Immunity. 2019;51(1):155–68.e5. doi:10.1016/j.immuni.2019.05.006.
- Grundy MA, Zhang T, Sentman CL. NK cells rapidly remove B16F10 tumor cells in a perforin and interferon-gamma independent manner in vivo. Cancer Immunol Immunother CII. 2007;56(8):1153–1161. doi:10.1007/s00262-006-0264-1.
- Patel SA, Minn AJ. Combination cancer therapy with immune checkpoint blockade: mechanisms and strategies. Immunity. 2018;48(3):417–433. doi:10.1016/j.immuni.2018.03.007.
- Platonova S, Cherfils-Vicini J, Damotte D, Crozet L, Vieillard V, Validire P, André P, Dieu-Nosjean M-C, Alifano M, Régnard J-F, et al. Profound coordinated alterations of intratumoral NK cell phenotype and function in lung carcinoma. Cancer Res. 2011;71(16):5412–5422. doi:10.1158/0008-5472.can-10-4179.
- André P, Denis C, Soulas C, Bourbon-Caillet C, Lopez J, Arnoux T, Bléry M, Bonnafous C, Gauthier L, Morel A, et al. Anti-NKG2A mAb is a checkpoint inhibitor that promotes anti-tumor immunity by unleashing both T and NK cells. Cell. 2018;175(7):1731–43.e13. doi:10.1016/j.cell.2018.10.014.
- Yu W, Yang L, Li T, Zhang Y. Cadherin signaling in cancer: its functions and role as a therapeutic target. Front Oncol. 2019;9(989). doi:10.3389/fonc.2019.00989.
- Stemmler MP. Cadherins in development and cancer. Mol Biosyst. 2008;4(8):835–850. doi:10.1039/b719215k.
- Cano A, Pérez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, Del Barrio MG, Portillo F, Nieto MA. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol. 2000;2(2):76–83. doi:10.1038/35000025.
- Medici D, Hay ED, Olsen BR. Snail and Slug promote epithelial-mesenchymal transition through beta-catenin-T-cell factor-4-dependent expression of transforming growth factor-beta3. Mol Biol Cell. 2008;19(11):4875–4887. doi:10.1091/mbc.e08-05-0506.
- Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2(6):442–454. doi:10.1038/nrc822.
- Perl AK, Wilgenbus P, Dahl U, Semb H, Christofori G. A causal role for E-cadherin in the transition from adenoma to carcinoma. Nature. 1998;392(6672):190–193. doi:10.1038/32433.
- Thiery JP. Epithelial-mesenchymal transitions in development and pathologies. Curr Opin Cell Biol. 2003;15(6):740–746. doi:10.1016/j.ceb.2003.10.006.
- Nieto MA, Huang RY, Jackson RA, Thiery JP. EMT: 2016. Cell. 2016;166(1):21–45. doi:10.1016/j.cell.2016.06.028.
- Hazan RB, Qiao R, Keren R, Badano I, Suyama K. Cadherin switch in tumor progression. Ann N Y Acad Sci. 2004;1014:155–163. doi:10.1196/annals.1294.016.
- Michel T, Poli A, Domingues O, Mauffray M, Thérésine M, Brons NH, Hentges F, Zimmer J. Mouse lung and spleen natural killer cells have phenotypic and functional differences, in part influenced by macrophages. PloS One. 2012;7(12):e51230. doi:10.1371/journal.pone.0051230.
- López-Soto A, Gonzalez S, Smyth MJ, Galluzzi L. Control of metastasis by NK cells. Cancer Cell. 2017;32(2):135–154. doi:10.1016/j.ccell.2017.06.009.
- Clark EA, Golub TR, Lander ES, Hynes RO. Genomic analysis of metastasis reveals an essential role for RhoC. Nature. 2000;406(6795):532–535. doi:10.1038/35020106.
- Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci U S A. 2010;107(9):4275–4280. doi:10.1073/pnas.0915174107.
- Page N, Klimek B, De Roo M, Steinbach K, Soldati H, Lemeille S, Wagner I, Kreutzfeldt M, Di Liberto G, Vincenti I, et al. Expression of the DNA-binding factor TOX promotes the encephalitogenic potential of microbe-induced autoreactive CD8(+) T cells. Immunity. 2018;48(5):937–50.e8. doi:10.1016/j.immuni.2018.04.005.
- Barry KC, Hsu J, Broz ML, Cueto FJ, Binnewies M, Combes AJ, Nelson AE, Loo K, Kumar R, Rosenblum MD, et al. A natural killer–dendritic cell axis defines checkpoint therapy–responsive tumor microenvironments. Nat Med. 2018;24(8):1178–1191. doi:10.1038/s41591-018-0085-8.
- Böttcher JP, Bonavita E, Chakravarty P, Blees H, Cabeza-Cabrerizo M, Sammicheli S, Rogers NC, Sahai E, Zelenay S, Reis E Sousa C, et al. NK cells stimulate recruitment of cDC1 into the tumor microenvironment promoting cancer immune control. Cell. 2018;172(5):1022–37.e14. doi:10.1016/j.cell.2018.01.004.
- Bonavita E, Bromley CP, Jonsson G, Pelly VS, Sahoo S, Walwyn-Brown K, Mensurado S, Moeini A, Flanagan E, Bell CR, et al. Antagonistic inflammatory phenotypes dictate tumor fate and response to immune checkpoint blockade. Immunity. 2020;53(6):1215–29.e8. doi:10.1016/j.immuni.2020.10.020.
- Hsu J, Hodgins JJ, Marathe M, Nicolai CJ, Bourgeois-Daigneault MC, Trevino TN, Azimi CS, Sheer AK, Randolph HE, Thompson TW, Zhang L, et al. Contribution of NK cells to immunotherapy mediated by PD-1/PD-L1 blockade. J Clin Invest. 2018;128(10):4654–4668. doi:10.1172/JCI99317.
- Ahmadzadeh M, Johnson LA, Heemskerk B, Wunderlich JR, Dudley ME, White DE, Rosenberg SA. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood. 2009;114(8):1537–1544. doi:10.1182/blood-2008-12-195792.
- Merino A, Zhang B, Dougherty P, Luo X, Wang J, Blazar BR, Miller JS, Cichocki F. Chronic stimulation drives human NK cell dysfunction and epigenetic reprograming. J Clin Invest. 2019;129(9):3770–3785. doi:10.1172/JCI125916.
- Henson SM, Franzese O, Macaulay R, Libri V, Azevedo RI, Kiani-Alikhan S, Plunkett FJ, Masters JE, Jackson S, Griffiths SJ, et al. KLRG1 signaling induces defective Akt (ser473) phosphorylation and proliferative dysfunction of highly differentiated CD8+ T cells. Blood. 2009;113(26):6619–6628. doi:10.1182/blood-2009-01-199588.
- Hayakawa Y, Smyth MJ. CD27 dissects mature NK cells into two subsets with distinct responsiveness and migratory capacity. J Immunol. 2006;176(3):1517. doi:10.4049/jimmunol.176.3.1517.