
ABSTRACT
The optimization of adoptive transfer approaches of anti-tumor T cells requires both the functional improvement of the injected T cells and the modulation of the tumor microenvironment, favoring the recruitment of these T cells and their activation. We have recently shown the therapeutic benefit of two approaches tested individually in a melanoma model wich were on one hand the adoptive transfer of specific T cells deficient for the expression of the inhibitory receptor PD-1, and on the other hand PD-L1 targeted alpha therapy (TAT). In this study, we sought to investigate the efficacy of these two therapies combined, compared to each monotherapy, in order to evaluate the synergy between these two approaches, in the same melanoma model. Here we used melanoma-specific T-cell clones, previously validated for the edition of PDCD1 gene and with previously demonstrated superior anti-tumor activity than their wild-type counterparts, after adoptive transfer in NSG mice engrafted with PD-L1 expressing human melanoma tumors. We also used a previously validated TAT approach, using a 213Bi-anti-human-PD-L1 mAb, alone or in combination with adoptive cell transfer, in the same mouse model. We confirmed previous results obtained with each monotherapy and documented the safety and the superior ability of a combination between the adoptive transfer of PD-1 deficient T cells and TAT targeting PD-L1 to control the growth of melanoma tumors in NSG mice. This study provides the first proof-of-concept of the efficacy of a combination therapy using TAT, adoptive cell transfer and genomic editing of IC-coding genes.
Background
In advanced melanoma patients, anti-PD-1 therapy has been approved as first line of treatment.Citation1 However, despite unprecedented clinical results, the therapeutic efficacy of such approaches needs to be further improved, as around 60% of patients do not respond to monotherapy.Citation2 In this context, multiple combination therapies are currently tested in melanoma and other solid tumors, but choosing the most relevant immunotherapy-based combinations depend on the presence and status of a preexisting immune infiltrate.Citation3 Indeed, the combination of several immune checkpoint inhibitors (ICI) or ICIs with costimulatory checkpointsCitation4,Citation5 will be more effective for hot tumors (with an active T infiltrate), while for “cold” tumors without T-cell infiltrate or with an altered T-cell infiltrate, adoptive transfer combined with ICIs would be more appropriate. This combination has been clinically tested with CAR T-cells for the treatment of refractory diffuse large B cell lymphomaCitation6 and refractory neuroblastoma.Citation7 In metastatic melanoma, the combination of adoptive transfer with cytokine-primed CTL (cytotoxic T lymphocytes)Citation8 or TIL (tumor infiltrating T lymphocytes)Citation9 with ICI also offers promising results. However, toxicities associated with the systemic use of ICIs remain a major drawbackCitation10 and in this context, the use of genetically modified therapeutic T lymphocytes, inactivated for IC expression, would allow to circumvent these issues. Enhanced anti-tumor functions have already been reported for CAR-T cells and for tumor-specific CTL inactivated using CRISPR/CAS9 for PD-1 expression.Citation11–13 We recently reported the enhanced anti-tumor efficacy of adoptive transfer of PD-1KO melanoma-specific T-cell clones compared to their wild-type counterpart, in a pre-clinical NSG mouse model engrafted with a human melanoma tumor expressing PD-1 ligand, PD-L1.Citation14 However, the adoptive transfer of such modified lymphocytes delayed tumor growth but did not eradicate the established tumors. Therefore, these results could be further improved by combining this treatment with another therapy, directly targeting the tumor. Among the possible therapeutic combinations, it has been shown that the immunomodulatory effects of radiotherapy could act synergistically with immunotherapy in an immunocompetent host.Citation15
Among radiotherapy treatments, targeted alpha particle therapy (TAT), a targeted radionuclide therapy based on alpha particle emitters delivered specifically to the tumor through a specific target is considered as a relevant anti-cancer treatment. Besides the direct cytotoxicity of ionizing radiation, it has been shown that alpha particle emitter could also modulate the immune system by inducing immunogenic cell death which could potentiate anti-tumor efficacy.Citation16,Citation17 In a preclinical model of melanoma, the use of 213Bi-anti-melanin monoclonal antibody (mAb) demonstrated significant therapeutic efficacy.Citation18 Taking advantage of the same mouse model used to demonstrate the superior efficacy of the adoptive transfer of PD-1KO melanoma-specific T-cell clones, we recently showed that TAT targeting PD-L1 significantly delayed tumor growth and improved survival, without inducing severe toxicities.Citation19
In the present study, we sought to investigate whether the combination of a TAT therapy targeting PD-L1 with the adoptive transfer of PD-1KO melanoma-specific T-cell clones could act synergistically to eradicate established tumors.
As a proof of concept, we use the same model of NSG mice engrafted with PD-L1 expressing melanoma cells, treated with 213Bi-anti-human-PD-L1 mAb in combination with the adoptive transfer of PD-1KO T-cell clones. This combined treatment was compared to each monotherapy and to control mice, and demonstrated its enhanced therapeutic efficacy.
Methods
Cell lines and cell culture
Wild-type and PD-1KO Melan-A specific T-cell clones expressing the same TCR were previously described and compared for their anti-tumor potential in NSG mice.Citation14 T-cell clones were cultured in RPMI-1640 medium supplemented with 8% human serum, 2 mM L-glutamine (Gibco, France), 100 U/mL penicillin (Gibco, France), 0.1 mg/mL streptomycin (Gibco, France) and 150 U/mL recombinant human IL-2 (Proleukin, Novartis Pharma, France), and were regularly (every 3 weeks) amplified on irradiated feeder cells and PHA-L (1 µg/mL, Sigma, France).
M113 melanoma cell line stably expressing PD-L1Citation20 was cultured in RPMI-1640 medium supplemented with 10% Fetal Bovine Serum (Eurobio Scientific, France), 2 mM L-glutamine (Gibco, France), 100 U/mL penicillin (Gibco, France), 0.1 mg/mL streptomycin (Gibco, France) and 0.8 mg/mL of G418 antibiotic.
All the cells were cultured at 37°C in a humidified atmosphere containing 5% CO2 and a weekly test was performed through a HEK-Blue Detection Kit (hb-det3, Invivogen) to check the absence of mycoplasma contamination.
Radiolabeling of anti-human-PD-L1 mAb
The GoInVivo™ purified anti-human PD-L1 mouse mAb (anti-PD-L1 mAb) was purchased from Biolegend (San Diego, CA, USA). Before 213Bi radiolabeling, the anti-PD-L1 mAb was modified using SCN-CHX-A”-DTPA (Macrocyclics, Plano, TX, USA) in carbonate buffer (0.05 M, pH 8.7). After 12 h incubation at 25°C, the modified mAb was purified by HPLC on a Sephadex G200 gel-filtration column (Amersham Biosciences, Little Chalfont, UK). For 213Bi radiolabeling, 100 μg of modified anti-PD-L1 mAb was incubated with 213Bi eluted from an 225Ac/213Bi generator (Institute for Transuranium Elements, Karlsruhe, Germany) for 10 min at 37°C in 0.8 M ammonium acetate (pH 5.3), 1.5% ascorbic acid. The resulting 213Bi-labeled immunoconjugate was separated from unbound 213Bi by size-exclusion chromatography using a PD-10 column (GE Healthcare, Chicago, IL, USA). Radiochemical purity was >98.9 ± 1.03%, as determined by instant thin-layer chromatography silica gel (ITLC-SG).
Mouse xenograft model
Eleven to 13-week-old female NSG mice, purchased from Charles River laboratory, with unrestricted access to food and water were kept under specific pathogen-free conditions in the UTE animal facility (SFR François Bonamy, IRS-UN, University of Nantes, license number: B-44-278). The animal experiments were approved by the local veterinary committee (APAFIS #2468 and APAFIS #7823) and performed in accordance with the recommendations established by the Ethic Committee for Animal Experiments of the University of Nantes and the FELASA (Federation of Laboratory Animal Science Associations). Subcutaneous xenograft tumors were established by injections of 106 of human melanoma cells (M113PD-L1+) in 100 µL DPBS (ThermoFisher Scientific, USA) into the flank of NSG mice. Tumor cells were tested for mycoplasma and PD-L1 expression before each graft.
Seven days later, when tumor volumes reached around 80 mm3, mice were randomly allocated into the following experimental groups (): (1) sterile DPBS, (2) ACT with 5 × 106 WT T-cell clones in sterile DPBS, (3) ACT with 5 × 106 PD-1KO T-cell clones in sterile DPBS, (4) TAT, (5) TAT + ACT with 5 × 106 WT T-cell clones in sterile DPBS and (6) TAT + ACT with 5 × 106 PD-1KO T-cell clones in sterile DPBS. At day 7, mice from groups (4), (5) and (6) were treated with TAT by i.v. injections (in tail vein) of 125 kBq/g 213Bi-anti-hPD-L1 mAb. DPBS control group (1), and mice from groups (2) and (3) received i.v. injections of sterile DPBS (100 µL). At day 8, mice from groups (2), (3), (5) and (6) were treated by i.v. injections (retro-orbital) of 5 × 106 WT (groups (2) and (5)) or PD-1KO (groups (3) and (6)) T-cell clones. DPBS control group (1), and mice from group (4) received i.v. injections of sterile DPBS (100 µL).
Figure 1. Experimental design of adoptive cell transfer, TAT, and combination therapy
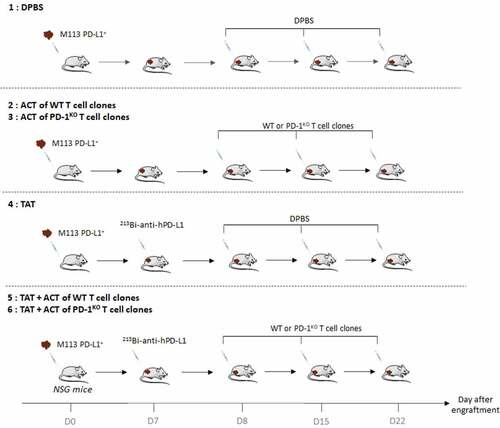
The absence of residual radioactivity on TAT-treated mice was checked before T-cell injection.
Intravenous injections of T-cell clones were repeated twice at days 15 and 22 after engraftment. Tumor burdens were measured by an electronic caliper, and the tumor volume was calculated based on the following formula: volume = (Length x Width2)/2. In addition, animal weight was measured three times a week. Mice were sacrificed taking into account the appearance of tumor necrosis, weight loss (20% of initial weight before tumor transplantation) and tumor size (>2000 mm3), in accordance with national and international policies. Tumors were removed at the time of sacrifice. Statistical analyses were performed using two-way ANOVA multiple comparisons.
Toxicity study
Hematological toxicity was assessed by numeration of red blood cells and platelets on an automated hematology analyzer (Nihon Kohden, France). Bone marrow toxicity was assessed on plasma isolated by centrifugation (10 min at 600 × g). Each sample was assessed in duplicate. Flt3-ligand concentration was quantified by ELISA (Bio-Techne, France) following manufacturer’s protocol. Statistical analysis was performed with two-way ANOVA followed by Sidak’s multiple comparisons test.
Immunohistochemistry
Tumors were collected, formalin-fixed and paraffin-embedded. Immunohistochemistry was performed on 3 µm paraffin sections of each tumor using anti-PD-L1 (10 µg/mL, clone E1L3N, Cell Signaling, USA) or anti-CD3 (6 µg/mL, polyclonal, Agilent, USA) primary antibodies, followed by the Peroxidase/DAB Envision detection system (Agilent, USA) on an automated platform (Dako Autostainer, USA), according to manufacturer’s instructions. The sections were counterstained with Mayer’s hematoxylin. As negative control, isotype antibody was used. CD3+ and PD-L1+cells were quantified on whole tissue sections, with the open-source software Qupath, using automatic classification and positive cell detection workflowsCitation21. Results are expressed as the percentage of positive cells relative to the total number of cells.
Results
Anti-tumor activity of monotherapies and combination therapies
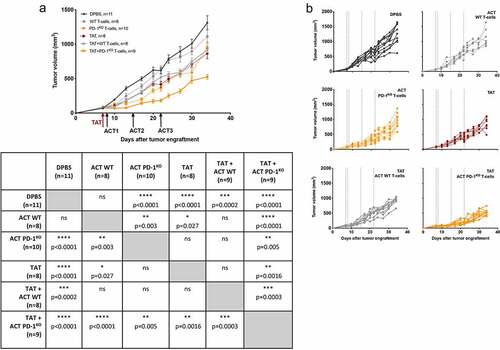
Anti-tumor efficacy of ACT with WT and PD-1KO T-cell clones and TAT alone or in combination was assessed in NSG mice, previously engrafted with a human melanoma cell line expressing PD-L1 (M113PD-L1+). As shown on , and in accordance with our previous reportsCitation14, the adoptive transfer of PD-1KO T-lymphocytes (yellow dotted lines) but not that of WT T-lymphocytes (gray dotted lines), significantly delayed tumor growth compared to the control group (black lines) (p < .0001). In addition, confirming our recently published results,Citation19 anti-PD-L1 TAT also significantly delayed tumor growth (red dotted lines, p < .0001). Monotherapies with either PD-1KO T-cells or TAT both exhibited a better anti-tumor efficacy than monotherapy with WT T-cells (p = .003 and p = .027, respectively). The treatment combining TAT and adoptive transfer of WT T-cells (solid gray lines) also delayed tumor growth compared to control group (p = .0002), in a similar way as monotherapies either with adoptive transfer of PD-1KO T-cells or with TAT. The best result was obtained with the combination of TAT and adoptive transfer of PD-1KO T-cells (yellow solid lines), that delays tumor growth the most, both in comparison to the control group (p < .0001), to groups receiving monotherapies with either WT (p < .0001), PD-1KO T-cells (p = .005) or TAT (p = .0016) and to the group receiving the combination of TAT and WT T-cells (p = .0003). These results showed that the combination of anti-PD-L1 targeted-alpha-therapy and the adoptive transfer of tumor-specific T-lymphocytes with optimized functions resulted in a better anti-tumor efficacy compared to each monotherapy.
Figure 2. Anti-tumor efficacy of mono- and combination therapies. A. M113PD-L1+melanoma tumor growth curves in NSG mice receiving i.v. injection of DPBS (black circles), or monotherapy with either i.v. injections of 5 × 106 of WT (gray circles, dotted line) or PD-1KO (yellow circles, dotted line) melanoma specific T-cells, or TAT (red circles, dotted line), or combination therapies with TAT and WT (gray circles, solid line) or PD-1KO T melanoma-specific T-cells (yellow circles, solid line). Table below the figure illustrates statistical comparisons performed using 2 way ANOVA, followed by Tukey multiple comparisons test. B. Individual curves of M113PD-L1+ melanoma tumor growth, in each group of NSG mice
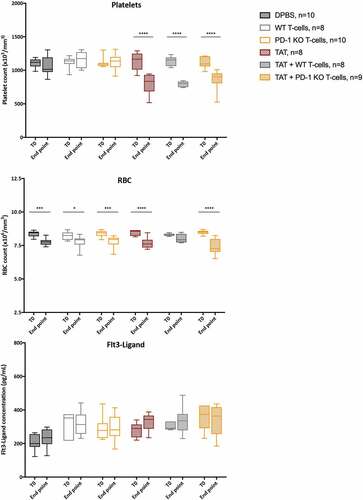
Hematologic and bone marrow toxicity after treatment
Hematologic and bone marrow toxicities were assessed by platelets and RBC counting on one hand and plasma Flt3-Ligand dosing on the other hand. Platelets and RBC counts were evaluated in mice before M113PD-L1+ tumor graft (T0) and at time of sacrifice (end point). As shown in , we observed a significant decrease in platelet counts at end point in all the groups treated with TAT (**** p < .0001). We also observed a significant decrease in RBC counts at end point in all the groups but one. Indeed, RBC drop was observed in control group treated with DPBS (*** p = .0004), in both groups treated with melanoma-specific T-cells either WT (* p = .0398) or PD-1KO (*** p = .0008), in group treated with TAT (**** p < .0001), and in group treated combination therapy consisting in TAT and PD-1KO melanoma-specific T-cells (**** <0.0001). No apparent direct relationship to the type of treatment received was observed, although the decrease appeared more pronounced in 2 of the 3 groups that received TAT. Flt3-Ligand plasma levels at T0 and at end point were not significantly different between the groups, a finding showing the absence of bone marrow toxicity.
Figure 3. Hematologic and bone marrow toxicity after mono- and combination therapies. Platelet counts, RBC counts and plasma Flt3-Ligand concentrations were assessed at T0, before tumor engraftment and at end point, after treatment with DPBS (black boxes) or monotherapy with either i.v. injections of 5 × 106 of WT (gray boxes) or PD-1KO (yellow boxes) melanoma specific T-cells, or TAT (striped red boxes), or combination therapies with TAT and WT (striped gray boxes) or PD-1KO T melanoma-specific T-cells (striped yellow boxes). Box extends from the 25th to 75th percentiles, line represents the median and the whiskers go down to the smallest value and up to the largest. Flt3-Ligand concentrations were assessed in duplicates. Statistical analyses were performed with two-way ANOVA followed by Sidak’s multiple comparisons test
T-cell infiltration and PD-L1 expression
Sixty hours after the last T-cell or DPBS injection, tumors were recovered and paraffin-embedded for immunohistochemical analyses.
CD3 infiltration was assessed on two tumors from groups receiving T-lymphocytes alone or in combination with TAT, 60 hours after the third injection of T-cells. As shown on , we observed T-cell infiltration in tumors from each of these four groups. Nonetheless, at this time point, no significant differences were detected between mice treated with ACT alone or with the combination therapy.
Figure 4. T-cell infiltration of melanoma tumors. A. Example of immunostaining of CD3 of melanoma tumors 60 hr after the third injection of T-cells. B. Example of PD-L1 staining of M113PD-L1+ tumors from mice from each treated group. C. Mean percentages of PD-L1+ cells, in tumors from each group (quantified with the Qupath open-source software). Statistical comparison was performed using multiple T-tests corrected using the Holm–Sidak method (Prism software). * p < .1
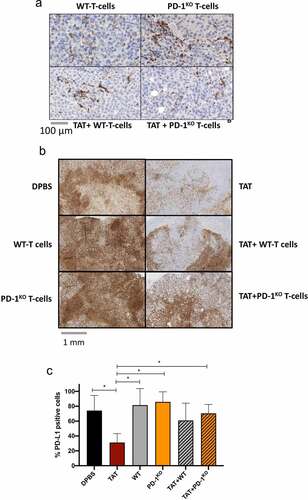
PD-L1 expression was also analyzed on tumors (n = 2) from each group, 60 hours after the third T-cell (or DPBS) injection. As shown on , PD-L1 was homogeneously and strongly expressed on M113PD-L1+ tumor cells from DBPS mice and from mice treated with adoptive cell transfer (74% to 86% of positive cells, ). We observed a significant decrease in PD-L1 expression on tumor cells from TAT-treated mice (31% of positive cells, p = .078), consistent with the therapy targeting PD-L1-expressing cells. Interestingly, the percentage of PD-L1 positive cells increased in the tumors from mice receiving the combination therapies, compared to TAT alone. Indeed, this percentage was around 61% in the tumors from mice treated with TAT + WT T-cells and reached 70% in the tumors from mice treated with TAT + PD-1KO T-cells, significantly higher than that detected in TAT treated mice (p = .08). We confirmed in vitro that this PD-L1 transfected melanoma cell line and its wild-type counterpart were both sensitive to IFN-γ, with a marked increase of PD-L1 expression after 48 h of treatment with recombinant IFN-γ (data not shown).
Discussion
In this study, we demonstrated the better therapeutic efficacy of a treatment combining TAT targeting PD-L1 and ACT of PD-1KO T-cells compared to each monotherapy and to the combination of TAT and ACT of WT T-cells expressing PD-1. The specificity of TAT was assessed by immunohistochemical analyses documenting the decreased PD-L1 expression in TAT-treated tumors compared to control tumors (30% and 75% of PD-L1+cells, respectively). This decrease in PD-L1 expression demonstrates the specific activity of TAT therapy on PD-L1+ tumor cells. Furthermore, we also showed that adoptive transfer of PD-1KO T-cells, and to a lesser extent PD-1+ T-cells (WT), resulted in increased PD-L1 expression in residual tumors after TAT compared to TAT therapy alone. This increased PD-L1 expression could result from the secretion of IFN-γ by T lymphocytes in the tumor microenvironment, as previously described in human melanoma tumorsCitation22, indicating the migration and activation of infused T-cells, especially PD-1KO T-cells, within the tumors.
Since TAT is delivered through i.v. injection, some hematological toxicity related to ionizing radiation is expected. As already observed in our previous studyCitation19, platelet counts were significantly decreased in all groups treated with TAT alone or TAT in combination with melanoma-specific T-cells. More surprisingly, we also observed some toxicity on RBC in all the groups but the one treated with the combination of TAT and WT melanoma-specific T-cells. Although this toxicity does not appear to be directly associated with the treatment, as the same decrease was observed in the control group, it should be considered in future studies. However, this does not appear to be related to erythroblast toxicity since we did not observe any bone marrow toxicity as assessed by Flt3-Ligand levels.
We further sought to formally investigate T-cell infiltration by immunohistochemistry on tumor sections from each group of mice. We confirmed the presence of T-cell infiltration in all tumors that received ACT, but no significant quantitative differences could be shown between mono- and combined therapy conditions. This result resonates with a recent studyCitation23 performed on melanoma-bearing immunocompetent mice with a combination of 177Lutetium-labeled anti-melanin antibody and anti-PD-1 therapy. In this study, the combination therapy showed a superior anti-tumor efficacy compared to each monotherapy but there was no statistical difference in T-cell infiltration between the different groups. This result together with our observations suggest alternative mechanisms beyond increased T-cell infiltration that could explain the better efficacy of combination therapy, although in our setting, T-cell infiltration only depends on injected T-cells and not on the recruitment of T-cells from the endogenous repertoire. It is therefore possible that, in this setting, the superior efficacy of this combination therapy is the result of the addition of cumulative effects of each of the monotherapies alone.
Nonetheless, a methodological issue may also have contributed to the sub-optimal detection of tumor-infiltrating T-cells. Indeed, tumor removal was performed 60 hours after the last T-cell injection, instead of 48 hours as performed in our previous studyCitation14 that could be too late regarding T-cell persistence. In addition, and unlike in this first study, T lymphocytes were injected in the retro-orbital venous sinus, since after TAT injection into mice’s tail vein, we anticipated that subsequent cell diffusion and circulation through this vein could be compromised. Thus, via the retro-orbital route, the kinetics of T-cell arrival at the tumor site, which was not determined, might be faster than when injected into the tail vein.
Nevertheless, PD-L1 expression level and decreased tumor growth upon combination therapy between TAT and PD-1KO T-cells strongly suggest the migration of activated T-cells at the tumor site before tumor removal, and a synergy of this therapeutic combination.
These results should be validated in an immunocompetent mouse model. Indeed, even though the feasibility of anti-PD-L1 TAT is supported by dosimetric studies,Citation24 the use of an immunocompetent model would directly address the safety of targeting PD-L1, which is also expressed by immune cells. In this setting, PD-L1 targeting could result in adverse effects and toxicity that we could not evaluate in this NSG mouse model.
Furthermore, in an immunocompetent host, it has been showed that melanin-targeted radionuclide therapy induced immune cell death, documented by increased cell surface annexin A1 and calreticulin levelsCitation25. In addition, the combination of this therapy with immune checkpoint inhibitors resulted to a better therapeutic efficacy than each monotherapy, by breaking immune tolerance. Thus, the efficacy of our combination therapy could be further improved in immunocompetent hosts, inducing an inflamed tumor microenvironment favoring the recruitment of our PD-1 deficient T-cells in a first step. In a second step, this combination therapy could also favor the recruitment of other tumor-specific T-cells, via the antigen spreading phenomenon. Indeed, epitope spreading, defined by the recruitment of endogenous T-cells specific for antigenic epitopes different from the one initially targeted, is a phenomenon already described after anti-tumor vaccination or ACTCitation26.
To conclude, this study provides the first proof-of-concept of the efficacy of a combination therapy using TAT, adoptive cell transfer and genomic editing of IC-encoding genes. In an immunocompetent host, the antitumor efficacy of this combination therapy should be further enhanced by the induction of immunogenic cell death and recruitment of an endogenous tumor-specific T repertoire.
Acknowledgments
We thank Sylvia Lambot, Sébastien Gouard, Noémie Joalland, Fabienne Haspot, Véronique Daguin, and Séverine Marionnaud-Lambot for expert assistance in in vivo experiments. We thank Patricia Le Saec for expert assistance in radiolabeling. We thank the UTE animal facility (SFR Santé), the Cytometry facility “CytoCell” (SFR Santé), the Radioactivity platform (CRCINA – SFR Santé) for expert technical assistance.
Additional information
Funding
References
- Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, Daud A, Carlino MS, McNeil C, Lotem M, et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N Engl J Med. 2015;372(26):2521–8. doi:10.1056/NEJMoa1503093.
- Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, Schadendorf D, Dummer R, Smylie M, Rutkowski P, et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med. 2015;372(1):23–34. doi:10.1056/NEJMoa1504030.
- Galon J, Bruni D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat Rev Drug Discov. 2019;18:197–218.
- Hellmann MD, Friedman CF, Wolchok JD,Combinatorial Cancer Immunotherapies. Adv Immunol. 2016;130:251–277.
- Buchan SL, Rogel A, Al-Shamkhani A. The immunobiology of CD27 and OX40 and their potential as targets for cancer immunotherapy. Blood. 2018;131(1):39–48. doi:10.1182/blood-2017-07-741025.
- Wang H, Kaur G, Sankin AI, Chen F, Guan F, Zang X. Immune checkpoint blockade and CAR-T cell therapy in hematologic malignancies. J Hematol Oncol. 2019;12(1):20–59. doi:10.1186/s13045-019-0746-1.
- Heczey A, Louis CU, Savoldo B, Dakhova O, Durett A, Grilley B, Liu H, Wu MF, Mei Z, Gee A, et al. CAR T Cells Administered in Combination with Lymphodepletion and PD-1 Inhibition to Patients with Neuroblastoma. Mol Ther. 2017;25(9):2214–2224. doi:10.1016/j.ymthe.2017.05.012.
- Chapuis AG, Lee SM, Thompson JA, Roberts IM, Margolin KA, Bhatia S, Sloan HL, Lai I, Wagener F, Shibuya K, et al. Combined IL-21-primed polyclonal CTL plus CTLA4 blockade controls refractory metastatic melanoma in a patient. J Exp Med. 2016;213(7):1133–1139. doi:10.1084/jem.20152021.
- Mullinax JE, Hall M, Prabhakaran S, Weber J, Khushalani N, Eroglu Z, Brohl AS, Markowitz J, Royster E, Richards A, et al. Combination of Ipilimumab and Adoptive Cell Therapy with Tumor-Infiltrating Lymphocytes for Patients with Metastatic Melanoma. Front Oncol. 2018;8:44. doi:10.3389/fonc.2018.00044.
- Lamichhane P, Deshmukh R, Brown JA, Jakubski S, Parajuli P, Nolan T, Raja D, Badawy M, Yoon T, Zmiyiwsky M, et al. Novel Delivery Systems for Checkpoint Inhibitors. Medicines (Basel). 2019;6(3):74. doi:10.3390/medicines6030074.
- Rupp LJ, Schumann K, Roybal KT, Gate RE, Ye CJ, Lim WA, Marson A. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci Rep. 2017;7(1):737. doi:10.1038/s41598-017-00462-8.
- Guo X, Jiang H, Shi B, Zhou M, Zhang H, Shi Z, Du G, Luo H, Wu X, Wang Y, et al. Disruption of PD-1 Enhanced the Anti-tumor Activity of Chimeric Antigen Receptor T Cells Against Hepatocellular Carcinoma. Front Pharmacol. 2018;9:1118. doi:10.3389/fphar.2018.01118.
- Zhao Z, Shi L, Zhang W, Han J, Zhang S, Fu Z, Cai J. CRISPR knock out of programmed cell death protein 1 enhances anti-tumor activity of cytotoxic T lymphocytes. Oncotarget. 2018;9(4):5208–5215. doi:10.18632/oncotarget.23730.
- Marotte L, Simon S, Vignard V, Dupré E, Gantier M, Cruard J, Alberge J-B, Hussong M, Deleine C, Heslan J-M, et al. Increased antitumor efficacy of PD-1-deficient melanoma-specific human lymphocytes. J Immunother Cancer. 2020;8(1):e000311. doi:10.1136/jitc-2019-000311
- Lhuillier C, Rudqvist N-P, Yamazaki T, Zhang T, Charpentier M, Galluzzi L, Dephoure N, Clement CC, Santambrogio L, Zhou XK, et al. Radiotherapy-exposed CD8+ and CD4+ neoantigens enhance tumor control. J Clin Invest 2021;131.
- Gorin J-B, Ménager J, Gouard S, Maurel C, Guilloux Y, Faivre-Chauvet A, Morgenstern A, Bruchertseifer F, Chérel M, Davodeau F, et al. Antitumor immunity induced after α irradiation. Neoplasia. 2014;16(4):319–328. doi:10.1016/j.neo.2014.04.002
- Gorin J-B, Guilloux Y, Morgenstern A, Chérel M, Davodeau F, Gaschet J. Using α radiation to boost cancer immunity? Oncoimmunology 2014; 3:e954925.
- Allen KJH, Jiao R, Malo ME, Frank C, Fisher DR, Rickles D, Dadachova E. Comparative Radioimmunotherapy of Experimental Melanoma with Novel Humanized Antibody to Melanin Labeled with 213Bismuth and 177Lutetium. Pharmaceutics 2019;11: 348.
- Capitao M, Perrin J, Simon S, Gouard S, Chouin N, Bruchertseifer F, Morgenstern A, Rbah-Vidal L, Chérel M, Scotet E, et al. Anti-Tumor Efficacy of PD-L1 Targeted Alpha-Particle Therapy in a Human Melanoma Xenograft Model. Cancers (Basel) 2021; 13: 1256.
- Simon S, Vignard V, Florenceau L, Dreno B, Khammari A, Lang F, Labarriere N. PD-1 expression conditions T cell avidity within an antigen-specific repertoire. Oncoimmunology 2016;5:e1104448.
- Bankhead P, Loughrey MB, Fernández JA, Dombrowski Y, McArt DG, Dunne PD, McQuaid S, Gray RT, Murray LJ, Coleman HG, et al. QuPath: Open source software for digital pathology image analysis. Sci Rep 2017;7:16878.
- Taube JM, Anders RA, Young GD, Xu H, Sharma R, McMiller TL, Chen S, Klein AP, Pardoll DM, Topalian SL, et al. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci Transl Med. 2012;4(127) 127ra37–7. doi: 10.1126/scitranslmed.3003689.
- Malo ME, Allen KJH, Jiao R, Frank C, Rickles D, Dadachova E. Mechanistic Insights into Synergy between Melanin-Targeting Radioimmunotherapy and Immunotherapy in Experimental Melanoma. Int J Mol Sci. 2020;21(22):8721. doi:10.3390/ijms21228721.
- Josefsson A, Nedrow JR, Park S, Banerjee SR, Rittenbach A, Jammes F, Tsui B, Sgouros SG. Imaging, Biodistribution, and Dosimetry of Radionuclide-Labeled PD-L1 Antibody in an Immunocompetent Mouse Model of Breast Cancer. Cancer Res. 2016;76(2):472–479. doi:10.1158/0008-5472.CAN-15-2141.
- Rouanet J, Benboubker V, Akil H, Hennino A, Auzeloux P, Besse S, Pereira B, Delorme S, Mansard S, D’Incan M, et al. Immune checkpoint inhibitors reverse tolerogenic mechanisms induced by melanoma targeted radionuclide therapy. Cancer Immunol Immunother. 2020;69(10):2075–2088. doi:10.1007/s00262-020-02606-8.
- Brossart P. The Role of Antigen Spreading in the Efficacy of Immunotherapies. Clinical Cancer Research. 2020;26(17):4442–4447. doi:10.1158/1078-0432.CCR-20-0305.