
ABSTRACT
Chimeric antigen receptor T cells (CAR-T) targeting CD19 have achieved significant success in patients with B cell malignancies. To date, implementation of CAR-T in other indications remains challenging due to the lack of truly tumor-specific antigens as well as control of CAR-T activity in patients. CD123 is highly expressed in acute myeloid leukemia (AML) blasts including leukemia-initiating cells making it an attractive immunotherapeutic target. However, CD123 expression in normal hematopoietic progenitor cells and endothelia bears the risk of severe toxicities and may limit CAR-T applications lacking fine-tuned control mechanisms. Therefore, we recently developed a rapidly switchable universal CAR-T platform (UniCAR), in which CAR-T activity depends on the presence of a soluble adapter called targeting module (TM), and confirmed clinical proof-of-concept for targeting CD123 in AML with improved safety. As costimulation via 4–1BB ligand (4–1BBL) can enhance CAR-T expansion, persistence, and effector functions, a novel CD123-specific TM variant (TM123-4-1BBL) comprising trimeric single-chain 4–1BBL was developed for transient costimulation of UniCAR-T cells (UniCAR-T) at the leukemic site in trans. TM123-4-1BBL-directed UniCAR-T efficiently eradicated CD123-positive AML cells in vitro and in a CDX in vivo model. Moreover, additional costimulation via TM123-4-1BBL enabled enhanced expansion and persistence with a modulated UniCAR-T phenotype. In addition, the increased hydrodynamic volume of TM123-4-1BBL prolonged terminal plasma half-life and ensured a high total drug exposure in vivo. In conclusion, expanding the soluble adapter optionality for CD123-directed UniCAR-T maintains the platforms high anti-leukemic efficacy and immediate control mechanism for a flexible, safe, and individualized CAR-T therapy of AML patients.
Introduction
Acute myeloid leukemia (AML) is a very heterogeneous disease characterized by relapses attributed to leukemia-initiating cells, also referred to as leukemic stem cells (LSCs), and a generally high mortality.Citation1 So far, allogeneic hematopoietic cell transplantation (allo-HCT) remains the only curative treatment option for transplant-eligible relapsed/refractory AML patients.Citation2,Citation3 However, relapses after allo-HCT occur, and it is often associated with transplant-related mortality and morbidity.Citation4,Citation5 Thus, alternative targeted immunotherapeutic strategies have been developed over the last years.Citation6,Citation7 In B cell malignancies, chimeric antigen receptor (CAR) expressing T cells (CAR-T) targeting CD19 and BCMA showed encouraging clinical results with impressive response rates.Citation8–13 Consequently, the first two autologous CD19-targeting CAR-T therapies, tisagenlecleucel and axicabtagene ciloleucel, were approved by U.S. Food and Drug Administration (FDA) and European Medicines Agency (EMA).Citation8,Citation14 Identifying the optimal signaling domain for second-generation CAR-T between CD28 and 4–1BB, however, remains largely undetermined and context-dependent.Citation15 For conventional CAR-T in the context of B cell malignancies, studies credited the CD28 costimulatory domain enhanced initial effector functions and early tumor clearance, while 4–1BB was linked to slower kinetics, but a higher in vivo persistence and decreased exhaustion.Citation16–18 Third-generation CARs, combining the signaling potential of two costimulatory domains such as CD28 and 4–1BBCitation19 showed even greater T cell expansion and tumor regression linked to a more narrow therapeutic window.Citation18,Citation20–23 The antitumor activity of CAR-T can further be enhanced by genetic modification with transgenes for cytokine secretion or additional costimulatory ligands.Citation24 Here, CD28-based CAR-T comprising an additional coexpression of 4–1BBL exhibited the highest in vivo anti-tumor function.Citation18,Citation25 However, toxicity profiles of conventional CAR-T therapy including treatment-related severe cytokine release syndrome (CRS) and neurotoxicity, as well as the development of refractory escape variants still limits their therapeutic efficacy.Citation26–28 Moreover, broader clinical application of conventional CAR-T in particular for targeting less differentially expressed antigens remains a challenge due to lack of tumor-exclusive antigens and CAR-T controllability. CD123, the IL-3 receptor alpha chain, is highly expressed in AML as well as acute lymphoblastic leukemia (ALL) and has been associated with the LSC compartment.Citation1,Citation29–31 However, CD123 expression was also detected on normal tissues including hematopoietic progenitorsCitation32–35 and endothelia.Citation36–39 Therefore, CD123-specific CAR-T applications are limited to strategies incorporating fine-tuned control mechanisms or may only be used for bridging to allo-HCT. With the aim to safely activate CAR-T in a controlled and dose-dependent manner, our group recently introduced a rapidly switchable universal CAR-T platform (UniCAR).Citation40 The efficacy of the UniCAR platform was successfully evaluated for a series of targets expressed on several hematopoieticCitation40,Citation41 and solid tumorsCitation42–45 in vitro and in vivo. Regarding AML, we recently confirmed pre-clinical proof-of-concept for UniCAR-T cells (UniCAR-T) targeting CD123 demonstrating limited toxicity toward hematopoietic progenitors due to the rapid on/off switch.Citation46 Subsequently, proof-of-concept for safety and efficacy of UniCAR-T in combination with a CD123-specific targeting module (TM), TM123, was demonstrated in a phase I clinical trial including relapsed/refractory AML patients (NCT04230265).Citation47 Rapid clearance of TM123 due to its small size and short terminal plasma half-life offers a fast safety switch during the onset of therapy, where high tumor burden and elevated toxicity risks require a particularly tight controllability of UniCAR-T. However, during consolidation cycles, patients might benefit from TMs with extended terminal plasma half-life and enhanced potential of UniCAR-T stimulation. Here, we provide pre-clinical data for UniCAR-T in combination with a novel CD123-specific TM variant (TM123-4-1BBL) harboring a covalently bound trimeric single-chain 4–1BB ligand (sc4-1BBL) for treatment of AML. In this study, characteristics of TM123-4-1BBL-directed UniCAR-T were evaluated against CD123-positive AML cells in vitro and in a cell line-derived xenograft (CDX) in vivo model. While the increased plasma half-life is expected to slow down the speed of UniCAR-T switch-off, the novel TM could further enhance CAR-T expansion, persistence and effector functions and may be ideal for consolidation cycles due to the additional 4–1BBL costimulus.Citation18,Citation25
Materials and methods
Cell culture
OCI-AML3 (DMSZ, ACC 582), HEK293T (ATCC® CRL-11268), and HT-1080 (ATCC® CCL-121) cells were cultured in DMEM high glucose, Glutamax™ (Thermo Fisher Scientific) supplemented with 10% fetal calf serum (FCS). MOLM-13 (DMSZ, ACC 554), CHO-K1 (DMSZ, ACC 110), and YTS-E13 (Retroviral modified human NK tumor cell line YT (DMSZ ACC 434) with stable expression of CD28/CD3ζ (28/ζ) UniCAR) cells were cultured in RPMI 1640 “complete” medium (supplemented with 10% to 15% fetal calf serum (FCS), 1 mM sodium pyruvate, 2 mM L-Alanyl-L-glutamine, and 1% non-essential amino acids, Merck KGaA, Biochrom KG). Both the adherent and suspension cell lines were mycoplasma tested and grown in a humidified atmosphere containing 5% CO2 at 37°C. Deposited master cell banks were authenticated by German Collection of Microorganisms and Cell Cultures (DSMZ) and stored in liquid nitrogen. Cells were passaged for a maximum of 6 weeks and cell-line identity was regularly ascertained by microscopic morphology checks and surface antigen determination by flow cytometry. Cell lines were handled separately under sterile workbench to avoid cross-contamination.
Lentivirus production and manufacturing of the CAR construct
The design of the 28/ζ UniCAR construct was recently described.Citation40 Transfection of HEK293T cells with polyethylenimine (Polysciences Europe GMbH) was performed using a transgene carrying vector, a packing plasmid pCD/NL-BH encoding Gag and Pol, and a pMD-GM vector bearing the genetic information for the viral envelope (Vesicular Stomatitis Glycoprotein). Lentivirus containing supernatants were concentrated by low-speed centrifugation and stored at −80°C until further use. Lentiviral titers were determined by limiting dilution transduction on human HT1080 cells as described before.Citation48 To generate UniCAR-T, human T cells were isolated from buffy coats (German Red Cross, Dresden, Germany). Therefore, PBMCs were isolated by density gradient centrifugation using Biocoll (Merck KGaA, Biochrom KG) followed by magnetic separation of T cells over CD4/CD8 beads (Miltenyi Biotec) using the AutoMACS® Pro Separator (Miltenyi Biotec). To activate T cells via CD3 and CD28, TransAct™ reagent was used according to the manufacturer’s instructions (Miltenyi Biotec). Subsequently, T cells were incubated for 24 h in TexMACS culture medium (Miltenyi Biotec) supplemented with 100 U/ml IL-2 (Novartis AG), 30 U/ml IL-7 (Miltenyi Biotec) and 30 U/ml IL-15 (Miltenyi Biotec). Consequently, activated T cells were transduced with lentiviral supernatant and spinoculation was performed at 600 × g for 2 h and 36°C. After 24 h on a rocking shaker, culture medium was exchanged (TexMACS supplemented with 200 U/ml IL-2, 600 U/ml IL-7, and 100 U/ml IL-15) and T cell concentration set to 2 × 106 cells/ml. Thereafter, T cells were expanded from 12 to 14 days prior to use in experiments and culture medium was changed every other day. For detection of UniCAR surface expression, T cells were incubated with mouse monoclonal antibody (mAb) anti-UniCAR-tag (UC-tag, anti-La 7B6) followed by staining with secondary goat-anti-mouse AlexaFluor 647 (Dianova).
Design, expression, and purification of targeting modules
The CD123-specific single chain variable fragment (scFv) derived from a murine mAb was modified by de-immunization (TM123) as recently described.Citation46 For the novel TM123-4-1BBL, a single-chain format of the costimulatory ligand 4–1BBL was fused to TM123. Therefore, three 4–1BBL domains (aa 71–254 each, UniProtKB-P41273), linked together by four flexible glycine-serine (G4S)4 linkers, were genetically fused to the C-terminus of TM123. Stable recombinant TM producing CHO-K1 cell lines were established by lentiviral transduction and TMs were purified using Ni-NTA affinity chromatography.Citation49 TM batches were characterized regarding UV-absorption, sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), and Western Blot for content, purity, and identity. For Western Blot analysis, His-tagged recombinant TMs were detected with a primary mouse anti-Penta-His IgG1 antibody (Ab) (Qiagen) and an anti-mouse-AP-conjugated secondary antibody (Jackson ImmunoResearch).
Surface plasmon resonance-based binding assay
Binding kinetics of TM123-4-1BBL and TM123 on human CD123 were analyzed by surface plasmon resonance (SRP) using a Biacore® X100 system (Cytiva). Three different and independent batches of purified TM123-4-1BBL and TM123 were included. As mobile phase 20 mM Sodium phosphate, 2.7 mM KCl, 137 mM NaCl (all Carl Roth GmbH + Co. KG), 0.05% (v/v) Polysorbate 20 (AppliChem) in 1000 ml ddH2O pH 7.4 was used. Therefore, the recombinant fragment crystallizable (Fc)-tagged soluble human CD123 was transiently immobilized on a CM5-sensor (Cytiva) containing a covalently bound anti-human capture antibody (Cytiva, Human antibody capture kit). Consequently, decreasing concentrations of TMs were injected in duplicates for 180 seconds allowing analyte association, followed by a 1200 second dissociation phase. With independent cycles, different TM concentrations ranging from 50 nM to 0.62 nM were tested; in between, the biosensor was regenerated using 3 M Magnesium chloride (Cytiva, Human antibody capture kit). The measured data is displayed in response units (RU) and analyzed using the Biacore® X100 software (Cytiva). The raw data were fitted using a monovalent 1:1 kinetic binding model, and the bulk signal was set constant to zero (RI = 0).
Immunofluorescence staining
To identify target antigens, cell marker expression, and effector cell as well as target cell populations, immunofluorescence staining was performed. For standardized immunofluorescence, staining cells were stained, washed, and analyzed in AutoMACS® running buffer (Miltenyi Biotec) using MACSQuant Analyzer®10 or 16 (Miltenyi Biotec). Target cells OCI-AML3 and MOLM-13 cells were stained with fluorochrome-labeled mAbs directed against human CD123/BV421 (clone 9F5, BD Bioscience). T cells were stained with 4–1BB/BV421 (clone 4B4-1, BD Bioscience) mAb. In the presented in vivo study, organ samples were stained with different panels containing mAbs against human CD3/APC-eFluor780 (clone UCHT1, eBioscience), CD4/PE-Cy7/BV510 (clone SK3, BD Bioscience), CD8/PE (clone RPA-T8, BD Bioscience), CD25/PE (clone M-A251, BD Bioscience) CD28/PE (clone CD28.2, BD Bioscience), CD33/APC (clone WM53, BD Bioscience), CD45/BV510 (clone HI30, BD Bioscience), CD45RO/PE-Cy7 (clone UCHL1, BD Bioscience), CD69/APC (clone FN50, BD Bioscience), CD95/APC (clone DX2, BD Bioscience), CD123/BV421 (clone 9F5, BD Bioscience), CD197/BV421 (clone 150503, BD Bioscience), CD274/BV421 (clone MIH1, BD Bioscience), and murine CD45/BV510 (clone 30-F11, BD Bioscience). The compensation of individual fluorochrome channels was performed using the MACS Comp Bead Kit (Miltenyi Biotec) according to the manufacturer’s protocol. All immunofluorescence staining data were analyzed with FlowJo™v10 (BD Life Sciences).
Flow cytometry-based binding studies and cytotoxic assays
To evaluate TM-binding properties in a cellular context, cells were stained with indicated serial TM dilutions. For detection, His-tagged TMs were labeled with a secondary anti-His-PE Ab (clone GG11-8F3.5.1, Miltenyi Biotec) and analyzed using MACSQuant Analyzer® 10, while the respective half maximal effective concentration (EC50) values were determined using GraphPad Prism 6.0 (GraphPad Software, Inc.). For analysis of cytotoxic potential, UniCAR-T cells were cocultured with antigen-positive tumor cells in the presence or absence of TMs at the indicated concentrations and effector to target cell ratio (E:T ratio). Therefore, 24 h before the use in cytotoxicity assays, target cells were labeled with live cell-dye eFluor 670™ (Thermo Fisher Scientific) to better distinguish them from UniCAR-T cells. On the same day, T cell cultivation medium was exchanged to RPMI 1640 “complete” (supplemented with 10% FCS) to minimize basic T cell activation. The following day, 2 × 104 target cells per well were seeded in a 96-well plate together with transduced UniCAR-T cells at different E:T ratios dependent on the respective experiment. Moreover, TM was added in decreasing concentrations and target cells only or a cell mix without TM served as controls in a total reaction volume of 200 μl RPMI 1640 “complete” with 10% FCS per well. After indicated time points, cytotoxic responses against target cells were determined by flow cytometry utilizing the MACSQuant Analyzer® 10. Ultimately, cell counts of viable eFluor670™-positive target cells were determined and normalized to cell numbers from the control wells containing target cells for only a percentage of lysis. In the case of a long-term rechallenged cytotoxic assay, 5 × 103 OCI-AML3 target cells were seeded at an initial E:T ratio of 1:20 with UniCAR-T cells and 10 nM CD123-specific TMs in a total reaction volume of 200 μl RPMI 1640 “complete” with 10% FCS per well. After 48 h, cells were resuspended and 50 µl per well were transferred for measuring using the MACSQuant Analyzer® 10. Cell counts of OCI-AML3 target cells were normalized to control wells containing target cells for only a percentage of living target cells. In addition, 5 × 103 OCI-AML3 target cells together with fresh TM at a final concentration of 10 nM were added in 50 μl medium to each well. This procedure was repeated for every rechallenge at day 4, day 6, day 9, and day 12. All flow cytometry-based binding study and cytotoxic assay data were analyzed with the FlowJo™v10 software and EC50 values were determined with GraphPad Prism 6.0.
Determination of cytokine-release
After cultivation, cell-free supernatants were analyzed by Enzyme-linked Immunosorbent Assay (ELISA) using BD OptEIA™ kits (BD Biosciences) according to the manufacturer’s protocols.
In vivo pharmacokinetic studies
Animal experiments were performed with 8–12 week old male and female immunodeficient NOD scid gamma (NSG™, The Jackson Laboratory) mice according to the German animal protection law (Landesdirektion Sachsen, TVV 87/2017). TM doses of 1 μg/gbodyweight (BW) or 2.5 μg/gBW in phosphate-buffered saline (PBS, Merck KGaA, Biochrom KG) were applied either intravenously (IV) via the tail vein or intraperitoneal (IP). At defined time points, peripheral blood was collected via sampling from the retro-orbital (RO) venous plexus. TM concentrations in peripheral blood samples were determined with an in-house ELISA as described before.Citation40
NSG CDX mouse model
Animal experiments were performed with 8–12 week old male and female NSG mice according to the German animal protection law (Landesdirektion Sachsen, TVV 61/2017). On day zero, 1 × 105 MOLM-13 AML cells in 100 µl PBS were administered IV via the tail vein into NSG mice. Three days later, 5 × 106 UniCAR-T (82% CAR+) were applied IV via the tail vein in 100 µl PBS. Right away, the first treatment cycle of TM therapy started with a total of two TM therapy cycles, each lasting 10 days with a short break of five days in-between. Both TM123-41BBL and TM123 were administered by IP injections of 1 μg/gBW twice a day. Mice were sacrificed either at a pre-defined endpoint after the 1st TM therapy cycle (n = 3), or if stop criteria according to the German Animal Welfare Act were met (n = 10), including paralysis of the hind extremities, weight loss >20%, weakness or morbid state. Sacrificed mice were analyzed for human cells in peripheral blood, bone marrow, spleen, or metastatic tumor samples. For organ sample preparation, RO obtained blood samples and single-cell suspensions from spleen, metastases, as well as bone marrow obtained from femur and tibia of both hind legs were prepared. Erythrocytes were removed by lysis and cells were stained with indicated mAbs. Human T cell chimerism was defined as the percentage of CD3-positive cells over the entire populations of both human tumor cells and murine CD45 leukocytes together. For MOLM-13, chimerism was defined as the percentage of target cells over the populations of CD3-positive cells and murine CD45 leukocytes. Here, MOLM-13 tumor cells were characterized via expression of CD33, human CD45 and CD4. Stained organ samples were measured on the MACSQuant® Analyzer 10 and analyzed using FlowJo™v10.
Statistics
Tests indicated in figure legends were conducted utilizing GraphPad Prism 6.0. P values < .05 were considered significant.
Results
Novel targeting module TM123-4-1BBL specifically binds UniCAR, CD123-positive AML cell lines, and 4-1BB receptor on T cells
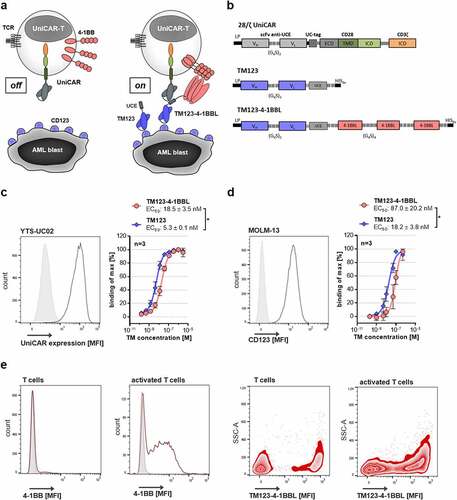
The modular UniCAR platform technology splits tumor antigen recognition and receptor signaling properties into two autonomous components (). The first component is a UniCAR-engineered T cell (UniCAR-T) transduced with an inert fully humanized second-generation CAR bearing a CD28/CD3ζ signaling domain (). In contrast to conventional CARs, the UniCAR is not recognizing tumor-associated antigens (TAAs) directly, but rather a small non-immunogenic peptide with 10 amino acids derived from the human nuclear La/SS-B protein (UniCAR epitope, UCE), which is not accessible on the surface of intact cells under physiological conditions. The second component, soluble adapters termed targeting modules (TMs) consist of the UCE linked to a TAA–specific binding moiety, and thus enable retargeting and activation of UniCAR-T to the respective target cells. For a next-generation TM targeting CD123-positive AML, a sc4-1BBL was fused to CD123-specific TM123 resulting in novel TM123-4-1BBL (). The TM123-4-1BBL production and purification process yielded 2.4 ± 0.5 mg/L (n = 7, ± SD) of protein and SDS-PAGE analysis of purified TM123-4-1BBL revealed a dominant band running at around 95 kDa, which matched the TMs predicted molecular weight of 91.6 kDa (Figure S1a). Moreover, Western Blot analysis with anti-His-tag detection confirmed the identity of the novel protein (Figure S1a). In order to cross-link UniCAR-T and target cells, TMs need to bind their respective target antigen, while simultaneously interacting with the UniCAR via their UCE-tag. Accessibility of TM123-4-1BBL’s UCE-tag was investigated on UniCAR-expressing YTS-E13 cells, revealing a significantly lower UCE-tag accessibility than TM123 (). The binding capabilities of TM123-4-1BBL toward CD123 on target cells were analyzed using AML cell lines MOLM-13 () and OCI-AML3 (Figure S1b). TM-specific half-maximal binding efficiencies (EC50) were comparable for both target cell lines and no significant differences between TM123 and TM123-4-1BBL were observed on OCI-AML3 cells. However, TM123-4-1BBL demonstrated a 5-fold lower binding efficiency for CD123 on MOLM-13 cells compared to TM123. Moreover, SPR-based data that analyzed the binding of CD123-specific TMs to recombinant human CD123 confirmed 8-fold lower affinity for TM123-4-1BBL compared to TM123 (Figure S1d+e). In order to investigate the binding of trimeric 4–1BBL to its natural receptor 4–1BB, activated T cells expressing high levels of 4–1BB were stained with TM123-4-1BBL (). T cells expressing 4–1BB specifically bound to TM123-4-1BBL, whereas TM123 showed no binding (Figure S1c). The differences in the mean fluorescence intensity (MFI) values measured for TM123-4-1BBL binding to activated T cells or native T cells correlate with their respective 4–1BB expression, and thus validate a specific binding of the recombinant 4–1BBL portion to its receptor. Altogether, the binding kinetics of TM123-4-1BBL confirmed its ability for targeting CD123, binding to UniCAR as well as to the costimulatory receptor 4–1BB.
Figure 1. Novel costimulatory TM123-4-1BBL specifically binds UniCAR, target antigen and costimulatory receptor. (a) Schematic representation of the inducible modular UniCAR-T platform. The UniCAR recognizes a small UniCAR epitope (UCE). Therefore, UniCAR-T alone remain in a non-reactive off-mode. In the on-mode, soluble adapters termed targeting modules (TMs) consisting of the UCE linked to a CD123-specific scFv enable precise control over CD123-specific UniCAR-T activation. (b) Schematic representation of the 28/ζ UniCAR construct as well as CD123-specific TM variants TM123 (30.4 kDa) and TM123-4-1BBL (91.6 kDa). TCR: T cell receptor, LP: leader peptide, UC-tag: UniCAR-tag (anti-La 7B6), VH: variable heavy chain, VL: variable light chain, G4S: glycine-serine linker, HIS6x: Hexa-Histidine-tag, ECD: extracellular domain, TMD: transmembrane domain, ICD: intracellular domain. (c) UCE accessibility of CD123-specific TMs was evaluated on UniCAR-expressing YTS-E13 cells. Expression of UniCAR on YTS-E13 cells was determined by immunofluorescence staining with mouse mAb anti-UC-tag followed by staining with secondary goat-anti-mouse AlexaFluor 647. Binding of TM123 and TM123-4-1BBL was analyzed via flow cytometry using a PE-labeled α-His Ab. TM binding data are represented as % binding of max by normalizing the obtained MFI to the highest MFI measured. Data is shown as mean ± SD of three independent repeats measured in biological triplicates. Statistical significance was determined by parametric paired t test, p < .05 = *. (d) Target antigen binding of CD123-specific TMs was evaluated on the CD123-positive target cell line MOLM-13. Expression of CD123 on MOLM-13 was determined by immunofluorescence staining with an α-CD123-APC Ab by flow cytometry. Binding of TM123 and TM123-4-1BBL was analyzed via flow cytometry using a PE-labeled α-His Ab. TM binding data are represented as % binding of max by normalizing the obtained MFI to the highest MFI measured. Data is shown as mean ± SD of three independent repeats measured in biological triplicates. Statistical significance was determined by parametric paired t test, p < .05 = *. (e) T cells were activated with PMA/Ionomycin (Biolegend) for 12 h prior to this experiment. 4–1BB receptor expression was determined by immunofluorescence staining with an α-4-1BB-BV421 Ab by flow cytometry. Binding of 500 nM TM123-4-1BBL to the receptor 4–1BB was determined on T cells and activated T cells by flow cytometry using a PE-labeled α-His Ab. TM binding data are represented as a zebra blot with the side scatter area (SSC-A) against the MFI for one representative donor
Redirection of UniCAR-T using TM123-4-1BBL mediates efficient elimination of AML cell lines in vitro
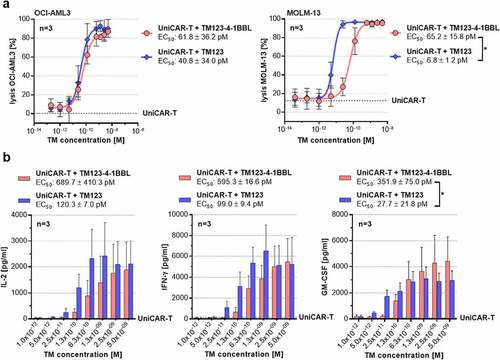
The TM123-4-1BBL-mediated cytotoxic potential of UniCAR-T against CD123-positive AML cell lines was analyzed on OCI-AML3 and MOLM-13 cells (). Therefore, UniCAR-T were incubated with AML target cells and TM in varying concentrations. Consequently, both TM123-4-1BBL and TM123 mediated a dose-dependent tumor cell lysis of OCI-AML3 and MOLM-13 target cells. Compared to TM123, however, TM123-4-1BBL required 10-fold higher doses to reach the half-maximal killing efficiency (EC50) in MOLM-13, while no significant difference in EC50 was observed in OCI-AML3 target cells. To characterize the cytokine release of UniCAR-T activated by TM123-4-1BBL, culture supernatant of UniCAR-T cocultured with MOLM-13 target cells was collected and cytokine levels of interleukin-2 (IL-2), interferon gamma (IFN-γ), and granulocyte-macrophage colony-stimulating factor (GM-CSF) were determined using ELISA (). In line with the cytotoxic potency studies, cross-linkage of UniCAR-T by TM123-4-1BBL and TM123 induced a dose-dependent cytokine secretion of IL-2, IFN-γ and GM-CSF. However, cytokine response for IL-2, IFN-γ and GM-CSF required 5- to 10-fold higher TM doses compared to the TM concentration required for cytotoxic response mediated by UniCAR-T. Hence, cytokine secretion that can be controlled by means of TM dosing would represent an advantageous differentiator from conventional CAR-T and enable a potential therapeutic window. This therapeutic window could allow for a safe treatment of patients with high tumor burden, as tumor cells can be effectively eliminated, while a strong cytokine release due to a high tumor burden is prevented. Taken together, in vitro cytotoxic potency and cytokine secretion data of TM123-4-1BBL suggest a potential therapeutic window regarding CAR-T side effects like CRS.
Figure 2. CD123 redirected UniCAR-T are highly efficient targeting AML. (a) CD123-positive leukemic OCI-AML3 and MOLM-13 cells (2x104) were incubated with UniCAR-T at an E:T ratio of 1:1 and 1:5, respectively. TM123-4-1BBL and TM123 were titrated at indicated concentrations. EC50 concentrations were determined for both TMs after 48 h. Target cell lysis was normalized to control samples without UniCAR-T cells and TM. Coculture of UniCAR-T and target cells without TM (UniCAR-T, dotted line) served as negative control. Data is shown as mean ± SD of three independent donors measured in biological triplicates. Statistical significance was determined by parametric paired t test, p < .05 = * (b) Cytokine levels of UniCAR-T after CD123-specific TM-mediated target cell lysis. UniCAR-T were cocultured with MOLM-13 target cells in a total E:T ratio of 1:5. After 48 h, cell culture supernatant of biological triplicates was collected, pooled and cytokine levels of IL-2, IFN-γ and GM-CSF were determined using ELISA. Coculture of UniCAR-T and MOLM-13 cells without TM (UniCAR-T, dotted line) served as negative control. Data is shown as mean ± SD of three independent donors measured in technical triplicates. Statistical significance was determined by parametric paired t test, p < .05 = *
Costimulation in trans promotes enhanced long-term cytotoxic potency and persistence of UniCAR-T in vitro
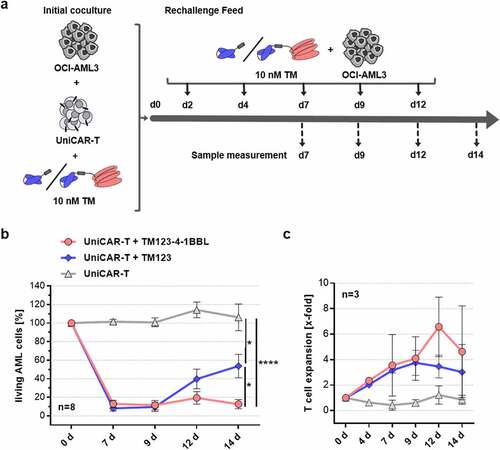
Since 4–1BB costimulation promotes T cell proliferation and persistence,Citation16,Citation50 we aim to simulate in vitro conditions of T cell exhaustion in a long-term rechallenge assay (). UniCAR-T were cocultured in the presence of AML cell line OCI-AML3 at 1:20 E:T ratio and activated with either of the CD123-specific TMs. At the indicated time points, fresh AML cells and respective TM were added to the culture after lysis was monitored. Over the first cycles, both TM123- and TM123-4-1BBL-redirected UniCAR-T were able to induce a complete lysis of target cells with every rechallenge cycle (). After 12 days of coculture, however, TM123-4-1BBL mediated beneficial UniCAR-T properties resulting in enhanced target cell killing capability compared to TM123. Regarding UniCAR-T expansion, no significant differences were observed between TM123- and TM123-4-1BBL-redirected UniCAR-T after 12 days of coculture (). Altogether, this long-term rechallenge assay displayed enhanced effector functions and persistence of TM123-4-1BBL-redirected UniCAR-T most likely mediated by the 4–1BBL costimulation in trans.
Figure 3. Costimulatory TM123-4-1BBL mediates long-term cytotoxic potency and persistence of UniCAR-T in vitro. (a) Experimental schema of a long-term rechallenge cytotoxic assay. UniCAR-T were cocultured with 5 × 103 OCI-AML3 target cells at an initial E:T ratio of 1:20 and 10 nM CD123-specific TMs. At indicated time points (d2 until d12), 5 × 103 OCI-AML3 cells together with 10 nM of respective TM were added and remaining living target cells were determined by flow cytometry. (b) Long-term rechallenge cytotoxic assay data. Cell counts of OCI-AML3 target cells were normalized to control wells containing target cells only for percentage living target cells. Data is shown as mean ± standard error of mean (SEM) of eight independent donors measured in biological triplicates. Statistical significance was determined by parametric unpaired one-way analysis of variance (ANOVA) with Tukey’s multiple comparison test, p < .05 = *, p < .0001 = ****. (c) At indicated time points, living T cell counts were determined by flow cytometry and T cell expansion was calculated by normalization to the initial T cell count. Data is shown as mean ± SEM of three independent donors measured in biological triplicates
Increased hydrodynamic volume of TM123-4-1BBL prolongs terminal plasma half-life and ensures high total drug exposure in vivo
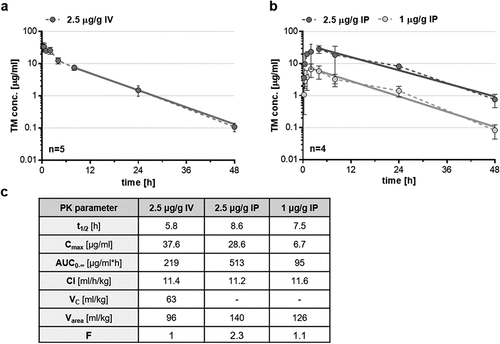
Therapeutic proteins with a small hydrodynamic volume under 70 kDa are rapidly eliminated from circulation by renal filtration.Citation51,Citation52 Consequently, increasing the hydrodynamic volume of TM123 (30.4 kDa) over the filtration threshold by fusion to sc4-1BBL (TM123-4-1BBL: 93.5 kDa) resulted in a prolonged terminal plasma half-life (t1/2) (). The t1/2 of 5.8 h after IV injection (+c) implied a 13-times longer t1/2 as for the short-lived TM123 of about 0.45 h.Citation46 Furthermore, the overall plasma clearance (Cl) of TM123-4-1BBL was 11.4 ml/h/kg after IV injection, which was confirmed by similar Cl values calculated for IP application (). The initial central volume of distribution (VC), the apparent volume of the central compartment from which TM123-4-1BBL elimination occurs, was determined as 63 ml/kg. Moreover, the maximal value of Varea, used to describe TM123-4-1BBL distribution in the total body, was calculated at 96 ml/kg. Comparisons with an extravascular application route revealed that IP injections followed a flip-flop kinetic, where the rate of absorption through the abdominal cavity is determined as the rate limiting step ultimately resulting in a longer terminal plasma half-life of up to 8.6 h (+c). This phenomenon was also observed in higher values of Varea for both IP applications, as TM123-4-1BBL exploited a much higher volume of distribution that served as a reservoir. Furthermore, the high bioavailability (F) of TM123-4-1BBL after IP application excluded great TM loss at the site of injection and observed AUC0-∞ values ensured a high total drug exposure in NSG mice. In conclusion, increasing the hydrodynamic volume of TM123-4-1BBL resulted in an expected terminal plasma half-life extension that could be prolonged even further by IP application.
Figure 4. Pharmacokinetic studies of TM123-4-1BBL in NSG mice. (a) TM123-4-1BBL was administered via intravenous (IV) tail vein injection of 2.5 μg/gBW TM or (b) via intraperitoneal (IP) injections in concentrations of 1 μg/gBW and 2.5 μg/gBW of NSG mice, respectively. Peripheral blood was taken via RO puncture at indicated time points. Plasma concentrations of TM123-4-1BBL were determined performing ELISA. Data is shown as four to five mice per time point analyzed. (c) Indicated pharmacokinetic parameters were defined utilizing PK solver 2.0 and its non-compartmental analysis (NCA). Slope of the terminal phase was calculated from the last three (IV) or four (IP) time points (straight line)
Switch-mediated costimulation in trans enhances UniCAR-T functionality in vivo
To determine the in vivo efficacy of TM123-4-1BBL-redirected UniCAR-T, a systemic xenotransplantation NSG model with CD123-expressing AML cell line MOLM-13 was utilized (). One treatment cycle of TM therapy included IP injections with 1 μg/gBW CD123-specific TM twice a day for 10 consecutive days. At the end of the first treatment cycle, three mice per group were sacrificed and analyzed for AML, T cell chimerism, and UniCAR-T phenotype. In contrast to the AML control, all groups transplanted with UniCAR-T indicated a reduced tumor load in peripheral blood, spleen, and bone marrow, likely due to alloreactivity of donor T cells against the AML cell line. However, only the TM123-4-1BBL group displayed a significant difference (Figure S2a). Alloreactivity potential of UniCAR-T was also observed against other cell lines in xenograft models and appeared to be T cell donor dependent (data not shown). TM123-4-1BBL-redirected UniCAR-T indicated initial costimulatory effects in trans, as they demonstrated higher T cell numbers at the main leukemic site in the bone marrow, though they were not significant (Figure S2b). Compared to the initial CD4/CD8 ratio of UniCAR-T on day of injection, the CD4/CD8 ratio after the 1st therapy cycle suggested a shift toward CD4+ T cells for all groups except TM123-4-1BBL-redirected UniCAR-T, which indicated non-significant tendencies toward CD8+ T cells in the peripheral blood and spleen (Figure S2c). Moreover, the fraction of UniCAR-positive T cells gradually increased from peripheral blood to spleen and bone marrow (Figure S3a). In addition, CD4+ and CD8+ UniCAR-T were characterized for their respective phenotype, which is displayed exemplarily for bone marrow samples in Figure S3b. Here, initially applied UniCAR-T showed a phenotype dominated by central memory and transitional memory T cells. Upon target antigen contact in the bone marrow, the initial composition of UniCAR-T shifted further toward an effector T cell phenotype. Especially CD8+ T cells of TM123- and TM123-4-1BBL-redirected UniCAR-T displayed increased proportions of effector memory and reduced proportions of transitional memory T cells.
Figure 5. Switch-mediated costimulation in trans enhances the functionality of UniCAR-T in a CDX model. (a) Experimental schema of the in vivo study design. (b) Overall survival of MOLM-13 transplanted NSG mice depicted in a Kaplan-Meier diagram. Statistical significance was determined by a Log-Rank (Mantel Cox) test; p < .0001 = ****. Flow cytometric endpoint analysis of (c) the absolute CD33+ MOLM-13 cell line and (d) CD3+ T cell chimerism in peripheral blood, spleen and bone marrow. (e) Endpoint organ samples were analyzed for CD4/CD8 ratio of CAR+ T cells by flow cytometry. Statistical significance for Figure 5c-e was analyzed using a non-parametric one-way ANOVA (Kruskal–Wallis test) combined with Dunn’s multiple comparison test; p < .05 = *, p < .01 = **, p < .001 = ***, p < .0001 = ****. Data is shown as mean ± SD of ten individual animals per group
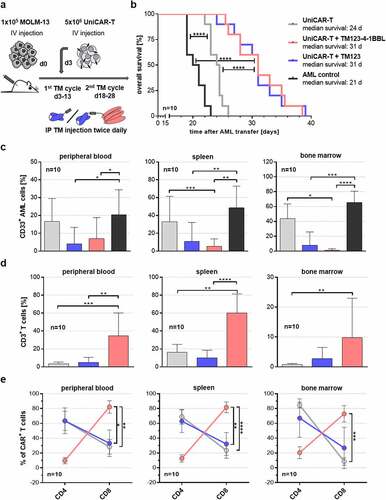
After the first therapy cycle and a resting period of 5 days, the second TM therapy cycle was given, followed by survival and endpoint analysis. Rapid death of the untreated AML control group (median survival: 21 d) was observed, confirming the aggressiveness of the chosen AML model (). Animals receiving UniCAR-T without TM displayed a slightly longer median survival of 24 days very likely due to alloreactive effects of UniCAR-T against MOLM-13. Application of both TM123-4-1BBL (median survival: 31 d) and TM123 (median survival: 31 d) successfully extended the median survival of AML-bearing NSG mice by 10 or 7 days compared to the control groups. Final analysis of animals at the endpoint demonstrated a high reduction of tumor burden in peripheral blood, spleen, and bone marrow mediated by TM-redirected UniCAR-T (). Moreover, analyzing the endpoint T cell chimerism demonstrated the beneficial effects of TM123-4-1BBL on UniCAR-T compared to TM123 and UniCAR-T control. In all three organs, T cell expansion was significantly increased at least threefold after costimulation in trans with TM123-4-1BBL (). Interestingly, costimulatory signaling in trans by TM123-4-1BBL shifted the CD4/CD8 ratio of UniCAR-T nearly completely toward CD8+ effector T cells (). TM123-redirected UniCAR-T and UniCAR-T control, however, rather promoted a CD4+ helper T cell phenotype.
Despite a low tumor burden in all analyzed organs of TM-redirected UniCAR-T treated animals, transferred leukemic cells caused extramedullary AML disease in the form of subcutaneous metastases (Figure S4a). Euthanasia of AML and UniCAR-T control groups had to be performed due to paralysis of the hind legs in 90–100% of animals. In contrast, a large proportion (70%) of CD123-specific TM-treated animals were sacrificed due to extramedullary AML disease that manifested as numerous subcutaneous and organ metastases. All analyzed metastases showed a high CD33+ AML chimerism and were still CD123 positive (Figure S4b).
Altogether, TM123-4-1BBL mediated a highly efficient tumor response of UniCAR-T against AML in a systemic xenotransplantation model. Anti-leukemic efficacy was demonstrated for both TM123-4-1BBL and TM123; however, costimulatory TM123-4-1BBL endowed UniCAR-T with additional properties such as an elevated T cell expansion as well as promoting an effector UniCAR-T phenotype.
Discussion
Despite ongoing improvements in treatment conditions, relapsed or refractory AML is still associated with a poor prognosis. Over the last 25 years, however, emerging targeted immunotherapy strategies have shifted the paradigm, providing a potent option to specifically eliminate chemo-resistant LSCs, which are reported as the main cause of relapse.Citation33 CD19-specific CAR-T has shown excellent clinical responses in the treatment of B cell malignancies; however, conventional CAR-T therapy has inherent limitations challenging its broad clinical application for different types of cancer apart from CD19- and B-cell maturation antigen (BCMA)-expressing indications. Severe on-target/off-tumor toxicities due to normal tissue expression for most cell surface targets prevent the development of effective and tolerable conventional CAR-T therapies. CD123-specific CAR-T needs to have fine-tuned control mechanisms, as CD123 is expressed in terms of hematopoietic progenitorsCitation32,Citation35 as well as endothelia.Citation37,Citation38 Preclinical data indicated that severe toxicities against normal hematopoietic progenitors and AML eradication by CD123-specific CAR-T go hand-in-hand.Citation53,54 To date, second-generation CD123-specific CAR-T constructs CD123CAR-41BB-CD3ζ (NCT03766126), CD123CAR-CD28-CD3ζ-EGFRt (NCT02159495), and UCART123 (NCT03190278) are examined in phase I clinical trials. However, current CD123-specific CAR-T is infused with the intention of being a bridging therapy to allo-HCT to mitigate anticipated long-term hematotoxic effects.Citation54 We are currently exploring our switch-controllable UniCAR platform targeting CD123Citation40,Citation46 in an ongoing clinical phase I study (NCT04230265), which already provided clinical proof-of-concept.Citation47,Citation55 Rapid functional interruption of UniCAR-T-CD123 therapy could abrogate on-target/on-tumor toxicities (e.g. CRS) and should prevent severe long-term toxicity due to CD123 expression in hematopoietic progenitors and mature myelocytes. Importantly, clinical proof-of-function and a successful reactivation of functional persistent UniCAR-T was shown. To extend the soluble adapter options for CD123-directed UniCAR-T therapy, we developed novel TM123-4-1BBL incorporating additional costimulatory signals in trans.
In general, UniCAR-T functionality is determined by the successful establishment of an immunological synapse between target and effector cell mediated by TMs. Compared to TM123, the rather large trimeric 4–1BBL domain exerted a steric hindrance on UCE-tag accessibility as well as a lower binding affinity toward CD123. This effect was also reported for an scFv-sc4-1BBL fusion protein that exhibited strongly impaired target antigen-binding capacities after fusion to the costimulatory ligand in the single-chain format.Citation56 Nevertheless, TM123-4-1BBL displayed specific binding for all three binding partners, with moderate-binding efficiencies for UniCAR and CD123. This could be especially important, because high-binding affinities might negatively influence the toxicity profile, CAR-T persistence as well as the ability to sequentially target multiple tumor cells.Citation57–60
First proof-of-concept studies successfully confirmed a TM123-4-1BBL concentration-dependent lysis of CD123-positive AML cell lines. The cytotoxic potency of TM123-4-1BBL, however, was up to 10-fold lower compared to TM123. This could be due to a steric hindrance of functional immunological synapse formation by trimeric 4–1BBL, as the fusion of 4–1BBL already lead to a comparably lower target binding affinity as well as a reduced accessibility of TM123-4-1BBL´s UCE-tag. Nevertheless, in comparison with alternative constructs that address CD123 as a target structure such as flotetuzumab (MGD006),Citation61 two novel CD123xCD3 Bispecific T cell Engager (BiTE)Citation62 and APVO436,Citation63 TM123-4-1BBL-redirected UniCAR-T still displayed comparable in vitro efficacy in the picomolar range. In addition to target cell lysis, EC50 values for cytokine secretion of IL-2, IFN-γ and GM-CSF revealed higher TM123-4-1BBL doses for reaching half-maximal cytokine secretion levels compared to attaining half-maximal lysis. The same effect was observed for the short-lived TM123 and also recently described by our group for IFN-γ release by TM123-redirected UniCAR-T.Citation46 Hence, cytokine secretion that can be controlled by means of TM dosing would represent an advantageous differentiator from conventional CAR-T and enable a broader therapeutic window. Moreover, the first clinical data revealed that CRS-related side effects can be rapidly abrogated by withdrawal of TM123 (NCT04230265).Citation47
Costimulation via 4–1BB was reported to enhance survival, cytokine release, effector functions, and clonal expansion of preferably CD8+ T cells, thus leading to enhanced antitumor effects.Citation64 Similarly, TM123-4-1BBL mediated costimulatory properties including increased effector functions and persistence of UniCAR-T in a long-term in vitro cytotoxic rechallenge assay resulting in enhanced killing capability of UniCAR-T. The observed effects are in line with data published by Hornig et al. showing an enhanced T cell expansion and cytokine secretion as well as development of cytotoxic T cells with strong effector potential by providing a 4–1BB signal in trans together with a second non-tumor-specific bispecific antibody.Citation65 In the context of a modular targeting platform that consists of a universal effector module and individual targeting modules (TMs) similar to the UniCAR technology, Arndt et al. generated a CD33-specific TM fused into a single 4–1BBL domain. Here, they observed that killing of tumor cells expressing low levels of CD33 was enhanced by costimulation in trans via 4–1BB.Citation66
Short-lived TMs such as TM123 with a molecular weight below 70 kDa are rapidly eliminated from circulation by renal filtration. Hence, terminal plasma half-life of TM123 in NSG mice after IV bolus was reported expectedly short (t1/2: 0.45 h).Citation46 The fusion of a half-life extension moiety in form of sc4-1BBL was described to increase the hydrodynamic volume and extend terminal plasma half-life of an epithelial cell adhesion molecule (EpCAM)-specific scFv-sc4-1BBL fusion protein to 13.3 h in CD1 mice after IV injection.Citation56 In our case, TM123-4-1BBL displayed a terminal plasma half-life of 5.8 h in NSG mice after IV injection extending terminal plasma half-life of short-lived TM123 by 13-fold in NSG mice. Nevertheless, terminal plasma half-life of TM123-4-1BBL was not extended sufficiently to achieve constantly high levels over longer time periods, and thus continuous infusion of TM123-4-1BBL may be required in a clinical setting.
Based on previous reports, which show that provision of 4–1BB signaling in trans is superior to 3rd generation CAR signaling incorporating CD28 and 4–1BB,Citation18,Citation25 we explored the potential of delivering the 4–1BB signaling in trans via an inducible ligand. In our in vivo model costimulation in trans positively affected T cell expansion until study endpoint demonstrating significantly higher T cell numbers for TM123-4-1BBL-redirected UniCAR-T in all organ sites. Here, observed differences in vivo may also be related to the increased terminal plasma half-life of TM123-4-1BBL. However, applied TM doses of both CD123-specific TMs corresponded to concentrations multiple times the effective in vitro EC90 (concentrations to induce 90% lysis) for the complete period of TM therapy. Hence, effects due to higher TM dosing should be unlikely, because both TMs were applied at saturation and no evidence for increased T cell expansion at excessively high TM doses was observed in vitro or in vivo (data not shown). Besides an enhanced T cell expansion, 4–1BB activation was described to preferentially expand CD8+ effector T cells.Citation64,Citation67 Accordingly, TM123-4-1BBL costimulation in trans shifted the CD4/CD8 ratio nearly completely toward a CD8+ effector UniCAR-T phenotype, whereas all other groups displayed a CD4+-predominant phenotype. The findings are directly in line with previous findings by Zhao et al. describing that CAR-T receiving integrated CD28 and 4–1BB signals via coexpression of 4–1BBL display increased T cell persistence accompanied by an elevated CD8+ fraction and reduced exhaustion.Citation18 Similarly, Drent et al. demonstrated that additional 4–1BB signaling mediated by 4–1BBL coexpression provided CD28-based CAR-T with superior proliferative capacity.Citation25 Although TM123-4-1BBL mediated a superior T cell expansion and persistence, both CD123-specific TMs demonstrated a similar median survival benefit. Despite the high anti-tumor activity of TM-redirected UniCAR-T in spleen and bone marrow, the overall response in this mouse model was incomplete. As reported before, the aggressive MOLM-13 model led to metastasis in anatomical niches and sanctuary lesions, in which CAR-T could not be detected.Citation68 Suboptimal IP bolus dosing might have supported tumor evasion toward an immune suppressive environment. Here, future clinical therapy of AML could benefit from the flexibility of the UniCAR platform, as dual targeting of CD123 and programmed death-ligand 1 (PD-L1) with a recently developed anti-PD-L1 targeting module could be capable of inducing an improved anti-leukemic response in anatomical niches.Citation69 In such an optimized experimental design, TM123-4-1BBL therapy could demonstrate superior therapeutic efficacy, since TM123-4-1BBL-redirected UniCAR-T demonstrated a tendency for an enhanced anti-leukemic activity as well as increased numbers of effector UniCAR-T. In line with this, CD28-based CAR-T comprising an additional coexpression of 4–1BBL exhibited the highest in vivo antitumor function.Citation18,Citation25
In summary, our study provides a proof-of-concept that 4–1BB costimulatory signaling can be provided in trans by fusing trimeric 4–1BBL to the TM, which provides the antigen-specific signal for UniCAR-T. The newly developed CD123-specific TM incorporating 4–1BBL demonstrated promising functional features expanding the soluble adapter options for CD123-directed UniCAR-T, while maintaining the platform's high anti-leukemic efficacy and control mechanism for a safe and customizable CAR-T therapy of AML patients. TM123-4-1BBL could offer interesting features during consolidation cycles by further enhancing UniCAR-T reactivation, expansion, persistence, and effector functions due to the additional 4–1BBL costimulus,Citation18,Citation22,Citation25 even though its switch-off response is prolonged. In such a post-debulking set-up, rapid switchability becomes less important compared to persistent functionality as functional superiority of a 4–1BB costimulated 3rd generation CD19 CAR-T was revealed in patients with low disease burden.Citation22
Supplemental Material
Download ()Acknowledgments
We thank Christin Eger, Glenn Gröbe, Rebecca Christiana, Katrin Zimmermann, Juliane Schneider, Susann Gerber, and Robert Kuhnert for technical assistance. We also acknowledge Cordula Gründer, Julia Riewaldt, Kristin Franke, Maria Schreiber, as well as all employees of GEMoaB GmbH and CPT Cellex Patient Treatment GmbH. This work was supported by GEMoaB GmbH and CPT Cellex Patient Treatment GmbH.
Disclosure statement
Conflict-of-interest-disclosure: GE and AE are shareholders of GEMoaB GmbH and CPT Cellex Patient Treatment GmbH. JEM, SL, JD, JS, GJJ, MC, and AE are employees at GEMoaB GmbH and/or its subsidiary company CPT Cellex Patient Treatment GmbH.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
References
- Ehninger A, Kramer M, Röllig C, Thiede C, Bornhäuser M, von Bonin M, Wermke M, Feldmann A, Bachmann M, Ehninger G, et al. Distribution and levels of cell surface expression of CD33 and CD123 in acute myeloid leukemia. Blood Cancer J. 2014;4(6):e218. doi:10.1038/bcj.2014.39.
- Cornelissen JJ, Gratwohl A, Schlenk RF, Sierra J, Bornhäuser M, Juliusson G, Råcil Z, Rowe JM, Russell N, Mohty M, et al. The European LeukemiaNet AML working party consensus statement on allogeneic HSCT for patients with AML in remission: an integrated-risk adapted approach. Nature Reviews Clinical Oncology. 2012;9(10):579. doi:10.1038/nrclinonc.2012.150.
- Lichtenegger FS, Krupka C, Haubner S, Köhnke T, Subklewe M. Recent developments in immunotherapy of acute myeloid leukemia. J Hematol Oncol. 2017;10(1):142. doi:10.1186/s13045-017-0505-0.
- Forman SJ, Rowe JM. The myth of the second remission of acute leukemia in the adult. Blood. 2013;121(7):1077–14. doi:10.1182/blood-2012-08-234492.
- Gill S, Porter DL. Reduced-intensity hematopoietic stem cell transplants for malignancies: harnessing the graft-versus-tumor effect. Annu Rev Med. 2013;64(1):101–117. doi:10.1146/annurev-med-121411-103452.
- Hofmann S, Schubert M-L, Wang L, He B, Neuber B, Dreger P, Müller-Tidow C, Schmitt M. Chimeric antigen receptor (CAR) T cell therapy in acute myeloid leukemia (AML). J Clin Med. 2019;8(2):200. doi:10.3390/jcm8020200.
- Winer ES, Stone RM. Novel therapy in Acute myeloid leukemia (AML): moving toward targeted approaches. Ther Adv Hematol. 2019;10:204062071986064. doi:10.1177/2040620719860645.
- Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, Braunschweig I, Oluwole OO, Siddiqi T, Lin Y, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med. 2017;377(26):2531–2544. doi:10.1056/NEJMoa1707447.
- Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, Jäger U, Jaglowski S, Andreadis C, Westin JR, et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N Engl J Med. 2019;380(1):45–56. doi:10.1056/NEJMoa1804980.
- Schuster SJ, Svoboda J, Chong EA, Nasta SD, Mato AR, Anak Ö, Brogdon JL, Pruteanu-Malinici I, Bhoj V, Landsburg D, et al. Chimeric antigen receptor T cells in refractory B-cell lymphomas. N Engl J Med. 2017;377(26):2545–2554. doi:10.1056/NEJMoa1708566.
- Park JH, Rivière I, Gonen M, Wang X, Sénéchal B, Curran KJ, Sauter C, Wang Y, Santomasso B, Mead E, et al. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. N Engl J Med. 2018;378(5):449–459. doi:10.1056/NEJMoa1709919.
- Berdeja JG, Madduri D, Usmani SZ, Singh I, Zudaire E, Yeh T-M, Allred AJ, Olyslager Y, Banerjee A, Goldberg JD, et al. Update of CARTITUDE-1: a phase Ib/II study of JNJ-4528, a B-cell maturation antigen (BCMA)-directed CAR-T-cell therapy, in relapsed/refractory multiple myeloma. J Clin Oncol. 2020;38(15_suppl): 8505–8505. doi:10.1200/JCO.2020.38.15_suppl.8505.
- Munshi NC, Anderson, Jr LD, Shah N, Jagannath S, Berdeja JG, Lonial S, Raje NS, Siegel DSD, Lin Y, Oriol A, et al. Idecabtagene vicleucel (ide-cel; bb2121), a BCMA-targeted CAR T-cell therapy, in patients with relapsed and refractory multiple myeloma (RRMM): initial KarMMa results. J Clin Oncol. 2020;38(15_suppl): 8503–8503. doi:10.1200/JCO.2020.38.15_suppl.8503.
- Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, Bader P, Verneris MR, Stefanski HE, Myers GD, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. 2018;378(5):439–448. doi:10.1056/NEJMoa1709866.
- Cheng Z, Wei R, Ma Q, Shi L, He F, Shi Z, Jin T, Xie R, Wei B, Chen J, et al. In vivo expansion and antitumor activity of coinfused CD28-and 4-1BB-Engineered CAR-T cells in patients with B cell leukemia. Mol Ther. 2018;26(4):976–985. doi:10.1016/j.ymthe.2018.01.022.
- Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, Smith JP, Walker AJ, Kohler ME, Venkateshwara VR, et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med. 2015;21(6):581–590. doi:10.1038/nm.3838.
- Milone MC, Fish JD, Carpenito C, Carroll RG, Binder GK, Teachey D, Samanta M, Lakhal M, Gloss B, Danet-Desnoyers G, et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther. 2009;17(8):1453–1464. doi:10.1038/mt.2009.83.
- Zhao Z, Condomines M, van der Stegen SC, Perna F, Kloss C, Gunset G, Plotkin J, Sadelain M. Structural design of engineered costimulation determines tumor rejection kinetics and persistence of CAR T cells. Cancer Cell. 2015;28(4):415–428. doi:10.1016/j.ccell.2015.09.004.
- Subklewe M, von Bergwelt-Baildon M, Humpe A. Chimeric antigen receptor T cells: a race to revolutionize cancer therapy. Transfusion Med Hemother. 2019;46(1):15–24. doi:10.1159/000496870.
- Carpenito C, Milone MC, Hassan R, Simonet JC, Lakhal M, Suhoski MM, Varela-Rohena A, Haines KM, Heitjan DF, Albelda SM, et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc Nat Acad Sci. 2009;106(9):3360–3365. doi:10.1073/pnas.0813101106.
- George P, Dasyam N, Giunti G, Mester B, Bauer E, Andrews B, Perera T, Ostapowicz T, Frampton C, Li P, et al. Third-generation anti-CD19 chimeric antigen receptor T-cells incorporating a TLR2 domain for relapsed or refractory B-cell lymphoma: a phase I clinical trial protocol (ENABLE). BMJ Open. 2020;10(2):e034629. doi:10.1136/bmjopen-2019-034629.
- Ramos CA, Rouce R, Robertson CS, Reyna A, Narala N, Vyas G, Mehta B, Zhang H, Dakhova O, Carrum G, et al. In vivo fate and activity of second- versus third-generation CD19-specific CAR-T cells in B cell Non-Hodgkin’s Lymphomas. Mol Ther. 2018;26(12):2727–2737. doi:10.1016/j.ymthe.2018.09.009.
- Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18(4):843–851. doi:10.1038/mt.2010.24.
- Holzinger A, Abken H. CAR T cells: a snapshot on the growing options to design a CAR. HemaSphere. 2019;3(1):e172. doi:10.1097/HS9.0000000000000172.
- Drent E, Poels R, Ruiter R, van de Donk NWCJ, Zweegman S, Yuan H, de Bruijn J, Sadelain M, Lokhorst HM, Groenet al. Combined CD28 and 4-1BB costimulation potentiates affinity-tuned Chimeric Antigen Receptor-engineered T cells. Clin Cancer Res. 2019;25(13):clincanres.2559.2018. doi:10.1158/1078-0432.ccr-18-2559
- DeFrancesco L. CAR-T’s forge ahead, despite Juno deaths. Nat Biotechnol. 2017;35(1):6–7. doi:10.1038/nbt0117-6b.
- Grupp SA, Laetsch TW, Buechner J, Bittencourt H, Maude SL, Verneris MR, Myers GD, Boyer MW, Rives S, De Moerloose B, et al. Analysis of a global registration trial of the efficacy and safety of CTL019 in pediatric and young adults with relapsed/refractory acute lymphoblastic Leukemia (ALL). Blood. 2016;128(22): 221–221. doi:10.1182/blood.V128.22.221.221.
- Locke FL, Neelapu SS, Bartlett NL, Siddiqi T, Chavez JC, Hosing CM, Ghobadi A, Budde LE, Bot A, Rossi JM, et al. Phase 1 results of ZUMA-1: a multicenter study of KTE-C19 anti-CD19 CAR T cell therapy in refractory aggressive Lymphoma. Mol Ther. 2017;25(1):285–295. doi:10.1016/j.ymthe.2016.10.020.
- Ruella M, Barrett DM, Kenderian SS, Shestova O, Hofmann TJ, Perazzelli J, Klichinsky M, Aikawa V, Nazimuddin F, Kozlowski M, et al. Dual CD19 and CD123 targeting prevents antigen-loss relapses after CD19-directed immunotherapies. J Clin Invest. 2016;126(10):3814–3826. doi:10.1172/jci87366.
- Das N, Gupta R, Gupta SK, Bakhshi S, Malhotra A, Rai S, Singh S, Prajapati VK, Sahoo RK, Gogia A, et al. A real-world perspective of CD123 expression in acute leukemia as promising biomarker to predict treatment outcome in B-ALL and AML. Clin Lymphoma Myeloma Leuk. 2020;20(10):e673–e684. doi:10.1016/j.clml.2020.05.004.
- Jordan C, Upchurch D, Szilvassy SJ, Guzman ML, Howard DS, Pettigrew AL, Meyerrose T, Rossi R, Grimes B, Rizzieri DA, et al. The interleukin-3 receptor alpha chain is a unique marker for human acute myelogenous leukemia stem cells. Leukemia. 2000;14(10):1777–1784. doi:10.1038/sj.leu.2401903.
- Taussig DC, Pearce DJ, Simpson C, Rohatiner AZ, Lister TA, Kelly G, Luongo JL, Danet-DesnoyersGH, Bonnet D. Hematopoietic stem cells express multiple myeloid markers: implications for the origin and targeted therapy of acute myeloid leukemia. Blood. 2005;106(13):4086–4092. doi:10.1182/blood-2005-03-1072
- Jin L, Lee EM, Ramshaw HS, Busfield SJ, Peoppl AG, Wilkinson L, Guthridge MA, Thomas D, Barry EF, Boyd A, et al. Monoclonal antibody-mediated targeting of CD123, IL-3 receptor α chain, eliminates human acute myeloid leukemic stem cells. Cell Stem Cell. 2009;5(1):31–42. doi:10.1016/j.stem.2009.04.018.
- Testa U, Pelosi E, Castelli G. CD123 as a therapeutic target in the treatment of hematological malignancies. Cancers (Basel). 2019;11(9):1358. doi:10.3390/cancers11091358.
- Sato N, Caux C, Kitamura T, Watanabe Y, Arai K-i, Banchereau J, Miyajima A. Expression and factor-dependent modulation of the interleukin-3 receptor subunits on human hematopoietic cells. Blood. 1993; Aug 1;82(3):752–61
- Korpelainen EI, Gamble JR, VADAS MA, Lopez AF. IL‐3 receptor expression, regulation and function in cells of the Vasculature. Immunol Cell Biol. 1996;74(1):1–7. doi:10.1038/icb.1996.1.
- Brizzi MF, Garbarino G, Rossi PR, Pagliardi GL, Arduino C, Avanzi GC, Pegoraro L. Interleukin 3 stimulates proliferation and triggers endothelial-leukocyte adhesion molecule 1 gene activation of human endothelial cells. J Clin Invest. 1993;91(6):2887–2892. doi:10.1172/JCI116534.
- Arcangeli S, Rotiroti MC, Bardelli M, Simonelli L, Magnani CF, Biondi A, Biagi E, Tettamanti S, Varani L. Balance of Anti-CD123 chimeric antigen receptor binding affinity and density for the targeting of acute Myeloid Leukemia. Mol Ther. 2017;25(8):1933–1945. doi:10.1016/j.ymthe.2017.04.017.
- Sun Y, Wang S, Zhao L, Zhang B, Chen H. IFN-γ and TNF-α aggravate endothelial damage caused by CD123-targeted CAR T cell. Onco Targets Ther. 2019;12:4907. doi:10.2147/OTT.S205678.
- Cartellieri M, Feldmann A, Koristka S, Arndt C, Loff S, Ehninger A, von Bonin M, Bejestani EP, Ehninger G, Bachmann MP, et al. Switching CAR T cells on and off: a novel modular platform for retargeting of T cells to AML blasts. Blood Cancer J. 2016;6(p):e458. doi:10.1038/bcj.2016.61.
- Bachmann D, Aliperta R, Bergmann R, Feldmann A, Koristka S, Arndt C, Loff S, Welzel P, Albert S, Kegler A, et al. Retargeting of UniCAR T cells with an in vivo synthesized target module directed against CD19 positive tumor cells. Oncotarget. 2018;9(7):7487. doi:10.18632/oncotarget.23556.
- Albert S, Arndt C, Koristka S, Berndt N, Bergmann R, Feldmann A, Schmitz M, Pietzsch J, Steinbach J, Bachmann M, et al. From mono- to bivalent: improving theranostic properties of target modules for redirection of UniCAR T cells against EGFR-expressing tumor cells in vitro and in vivo. Oncotarget. 2018;9(39):25597. doi:10.18632/oncotarget.25390.
- Feldmann A, Arndt C, Bergmann R, Loff S, Cartellieri M, Bachmann D, Aliperta R, Hetzenecker M, Ludwig F, Albert S, et al. Retargeting of T lymphocytes to PSCA- or PSMA positive prostate cancer cells using the novel modular chimeric antigen receptor platform technology “UniCAR”. Oncotarget. 2017;8(19):31368–31385. doi:10.18632/oncotarget.15572.
- Mitwasi N, Feldmann A, Bergmann R, Berndt N, Arndt C, Koristka S, Kegler A, Jureczek J, Hoffmann A, Ehninger A, et al. Development of novel target modules for retargeting of UniCAR T cells to GD2 positive tumor cells. Oncotarget. 2017;8(65):108584. doi:10.18632/oncotarget.21017.
- Pishali Bejestani E, Cartellieri M, Bergmann R, Ehninger A, Loff S, Kramer M, Spehr J, Dietrich A, Feldmann A, Albert S, et al. Characterization of a switchable chimeric antigen receptor platform in a pre-clinical solid tumor model. Oncoimmunology. 2017;6(10):e1342909. doi:10.1080/2162402x.2017.1342909.
- Loff S, Dietrich J, Meyer J-E, Riewaldt J, Spehr J, von Bonin M, Gründer C, Swayampakula M, Franke K, Feldmann A, et al. Rapidly switchable universal CAR-T cells for treatment of CD123-positive Leukemia. Mol Ther Oncolytics. 2020;17:408–420. doi:10.1016/j.omto.2020.04.009.
- Wermke M, Kraus S, Ehninger A, Bargou RC, Goebeler M-E, Middeke JM, Kreissig C, von Bonin M, Koedam J, Pehl M, et al. Proof-of-concept for rapidly switchable universal CAR-T platform with UniCAR-T-CD123 in relapsed/refractory AML. Blood. 2021;137(22):3145–3148. doi:10.1182/blood.2020009759.
- Stirnnagel K, Lüftenegger D, Stange A, Swiersy A, Müllers E, Reh J, Stanke N, Große A, Chiantia S, Keller H. Research analysis of prototype foamy virus particle-host cell interaction with autofluorescent retroviral particles. Retrovirology. 2010; 7(1), 1–17. doi:10.1186/1742-4690-7-45
- Feldmann A, Arndt C, Töpfer K, Stamova S, Krone F, Cartellieri M, Koristka S, Michalk I, Lindemann D, Schmitz M, et al. Novel humanized and highly efficient bispecific antibodies mediate killing of prostate stem cell antigen-expressing tumor cells by CD8+ and CD4+T cells. J Immunol. 2012;189(6):3249–3259. doi:10.4049/jimmunol.1200341.
- Willoughby JE, Kerr JP, Rogel A, Taraban VY, Buchan SL, Johnson PWM, Al-Shamkhani A. Differential impact of CD27 and 4-1BB costimulation on effector and memory CD8 T cell generation following peptide immunization. J Immunol. 2014;193(1):244–251. doi:10.4049/jimmunol.1301217.
- Kontermann RE. Half-life extended biotherapeutics. Expert Opin Biol Ther. 2016;16(7):903–915. doi:10.1517/14712598.2016.1165661.
- Strohl WR. Fusion proteins for half-life extension of biologics as a strategy to make biobetters. BioDrugs. 2015;29(4):215–239. doi:10.1007/s40259-015-0133-6.
- Gill S, Tasian SK, Ruella M, Shestova O, Li Y, Porter DL, Carroll M, Danet-Desnoyers G, Scholler J, Grupp SA, et al. Preclinical targeting of human acute myeloid leukemia and myeloablation using chimeric antigen receptor-modified T cells. Blood. 2014;123(15):2343–2354. doi:10.1182/blood-2013-09-529537.
- Gill SI. How close are we to CAR T-cell therapy for AML? Best Pract Res Clin Haematol. 2019;32(4):101104. doi:10.1016/j.beha.2019.101104.
- Kraus S, Ehninger A, Bargou RC, Goebeler M-E, Kreisig C, Koedam J, Pehl M, Einsele H, Ehninger G, Cartellieri M. Re-activation of UniCAR-T-cells with 2nd cycle of targeting module TM123 in patient with relapsed/refractory AML. EHA-EBMT 3rd European CAR T-Cell Meeting. 2021; Virtual Conference
- Fellermeier S, Beha N, Meyer J-E, Ring S, Bader S, Kontermann RE, Müller D. Advancing targeted co-stimulation with antibody-fusion proteins by introducing TNF superfamily members in a single-chain format. OncoImmunology. 2016;5(11):e1238540. doi:10.1080/2162402X.2016.1238540.
- Caruso HG, Hurton LV, Najjar A, Rushworth D, Ang S, Olivares S, Mi T, Switzer K, Singh H, Huls H, et al. Tuning sensitivity of CAR to EGFR density limits recognition of normal tissue while maintaining potent antitumor activity. Cancer Res. 2015;75(17):3505–3518. doi:10.1158/0008-5472.CAN-15-0139.
- Ghorashian S, Kramer AM, Onuoha S, Wright G, Bartram J, Richardson R, Albon SJ, Casanovas-Company J, Castro F, Popova B, et al. Enhanced CAR T cell expansion and prolonged persistence in pediatric patients with ALL treated with a low-affinity CD19 CAR. Nat Med. 2019;25(9):1408–1414. doi:10.1038/s41591-019-0549-5.
- Park S, Park S-Y, Nishio T, Kamizaki K, Wang Z, Tamada K, Takumi T, Hashimoto R, Otani H, Pazour GJ, et al. Micromolar affinity CAR T cells to ICAM-1 achieves rapid tumor elimination while avoiding systemic toxicity. Sci Rep. 2017;7(1):1–15. doi:10.1038/s41598-017-14749-3.
- Richman SA, Nunez-Cruz S, Moghimi B, Li LZ, Gershenson ZT, Mourelatos Z, Barrett DM, Grupp SA, Milone MC. High-affinity GD2-specific CAR T cells induce fatal encephalitis in a preclinical neuroblastoma model. Cancer Immunol Res. 2018;6(1):36–46. doi:10.1158/2326-6066.CIR-17-0211.
- Chichili GR, Huang L, Li H, Burke S, He L, Tang Q, Jin L, Gorlatov S, Ciccarone V, Chen F, et al. A CD3xCD123 bispecific DART for redirecting host T cells to myelogenous leukemia: preclinical activity and safety in nonhuman primates. Sci Transl Med. 2015;7(289):289ra82–289ra82. doi:10.1126/scitranslmed.aaa5693.
- Hutmacher C, Volta L, Rinaldi F, Murer P, Myburgh R, Manz MG, Neri D. Development of a novel fully-human anti-CD123 antibody to target acute myeloid leukemia. Leuk Res. 2019;84:106178. doi:10.1016/j.leukres.2019.106178.
- Comeau MR, Mitchell D, Gottschalk R, Misher L, Daugherty M, Parr L, Pavlik P, Woodruff B, Fang H, Aguilar M,et al. Abstract 597: Bispecific anti-CD123 x anti-CD3 ADAPTIR™ molecules for redirected T-cell cytotoxicity in hematological malignancies. Cancer Res. 2017;77(13 Supplement):597–597. doi:10.1158/1538-7445.am2017-597
- Sanmamed MF, Pastor F, Rodriguez A, Perez-Gracia JL, Rodriguez-Ruiz ME, Jure-Kunkel M, Melero I. Agonists of co-stimulation in cancer immunotherapy directed against CD137, OX40, GITR, CD27, CD28, and ICOS. Semin Oncol. 2015;42(4):640–655. doi:10.1053/j.seminoncol.2015.05.014
- Hornig N, Kermer V, Frey K, Diebolder P, Kontermann RE, Müller D. Combination of a bispecific antibody and costimulatory antibody-ligand fusion proteins for targeted cancer immunotherapy. J Immunother. 2012;35(5):418–429. doi:10.1097/CJI.0b013e3182594387.
- Arndt C, Feldmann A, von Bonin M, Cartellieri M, Ewen E-M, Koristka S, Michalk I, Stamova S, Berndt N, Gocht A, et al. Costimulation improves the killing capability of T cells redirected to tumor cells expressing low levels of CD33: description of a novel modular targeting system. Leukemia. 2014;28(1):59–69. doi:10.1038/leu.2013.243.
- Dubrot J, Milheiro F, Alfaro C, Palazón A, Martinez-Forero I, Perez-Gracia JL, Morales-Kastresana A, Romero-Trevejo JL, Ochoa MC, Hervás-Stubbs S, et al. Treatment with anti-CD137 mAbs causes intense accumulations of liver T cells without selective antitumor immunotherapeutic effects in this organ. Cancer Immunol Immunother. 2010;59(8):1223–1233. doi:10.1007/s00262-010-0846-9.
- Jetani H, Garcia-Cadenas I, Nerreter T, Thomas S, Rydzek J, Meijide JB, Bonig H, Herr W, Sierra J, Einsele H, et al. CAR T-cells targeting FLT3 have potent activity against FLT3−ITD+ AML and act synergistically with the FLT3-inhibitor crenolanib. Leukemia. 2018;32(5):1168–1179. doi:10.1038/s41375-018-0009-0.
- Dietrich J, Loff S, Spehr J, Riewaldt J, Gründer C, Schreiber M, Bachmann M, Ehninger G, Pehl M, Cartellieri M,et al. Abstract 2209: rapidly switchable universal CAR-T cells with improved safety profile allow for active targeting of PD-L1 expressing solid tumors. Cancer Res. 2020;80(16 Supplement):2209. doi:10.1158/1538-7445.AM2020-2209