
ABSTRACT
Although active immunotherapies are effective strategies to induce activation of CD8+ T cells, advanced stage tumors require further improvements for efficient control. Concerning the burden of cancer-related to Human papillomavirus (HPV), particularly the high incidence and mortality of cervical cancer, our group developed an approach based on a DNA vaccine targeting the HPV-16 E7 oncoprotein (pgDE7h). This immunotherapy is capable of inducing an antitumour CD8+ T cell response but show only partial control of tumors in more advanced growth stages. Here, we combined a chemotherapeutic agent (gemcitabine- Gem) with pgDE7h to overcome immunosuppression and improve antitumour responses in a preclinical mouse tumor model. Our results demonstrated that administration of Gem had synergistic antitumor effects when combined with pgDE7h leading to eradication of both early-stages and established tumors. Overall, the antiproliferative effects of Gem observed in vitro and in vivo provided an optimal window for immunotherapy. In addition, the enhanced antitumour responses induced by the combined therapeutic regimen included enhanced frequencies of antigen-presenting cells (APCs), E7-specific IFN-γ-producing CD8+ T cells, and cytotoxic CD8+ T cells and, concomitantly, less pronounced accumulation of immunosuppressive myeloid-derived suppressor cells (MDSCs) and regulatory T cells (Tregs). These findings demonstrated that the combination of Gem and an active immunotherapy strategy show increased effectiveness, leading to a reduced need for multiple drug doses and, therefore, decreased deleterious side effects avoiding resistance and tumor relapses. Altogether, our results provide evidence for a new and feasible chemoimmunotherapeutic strategy that supports future clinical translation.
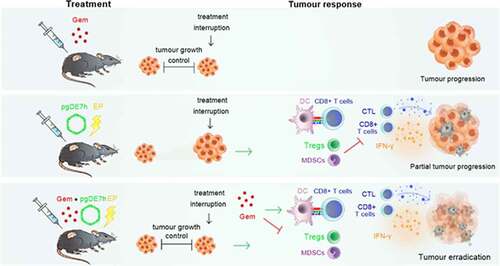
GRAPHICAL ABSTRACT
Introduction
Human papillomavirus (HPV) is the second most common infectious agent associated with cancer in humans, with an estimated worldwide burden of 690,000 cases of cancer per year.Citation1 In particular, HPV-16 and HPV-18 are the most common viral types with oncogenic potential.Citation2 HPV is associated, at different rates, with several types of anogenital and oropharynx cancers and, notably, is the causal factor of almost all cervical cancer cases, the fourth leading cancer type in incidence (570,000/year) and mortality (311,000/year) in women.Citation3 Indeed, the global burden of cervical cancer is expected to increase in the coming years.Citation4 Vaccines to prevent HPV infection have been available for the last ten years, but worldwide coverage is still low, especially in developing countries, where most HPV-associated mortality occurs. People who are unvaccinated or already infected by oncogenic HPV types remain at risk of developing cancer and rely on currently available treatments.
The treatment options for HPV-induced tumors are mainly surgery, chemotherapy, and/or radiotherapy. Considering cervical cancer, the 5-year overall survival rate is approximately 92% for early-stage lesions, but the rate drops to approximately 50% for stage III cancer cases and 17% for more advanced-stage cancer cases.Citation5,Citation6 Although the current treatments provide positive survival outcomes, they are often associated with debilitating adverse effects and resistance to therapy. In addition, tumor relapse is frequently observed, particularly in patients diagnosed with tumors at an advanced stage.Citation7 Thus, the development of alternative therapeutic approaches for HPV-associated tumors with reduced toxicity and enhanced long-term efficacy is urgently needed.
Gemcitabine (Gem) is a nucleoside analogue used in the clinic for a wide range of tumors, such as non-small cell lung cancer, pancreatic cancer, bladder cancer, and breast cancer.Citation8,Citation9 The cytotoxic effects of Gem on tumor cells are associated with the inhibition of DNA replication and DNA repair mechanisms.Citation10 Monotherapy with high doses of Gem is commonly related to hematologic toxicity and resistance following multiple administrations.Citation11,Citation12 The use of lower doses of Gem has been shown to induce milder toxicity, preserving its antitumour effects and improving patient quality of life.Citation13 Alternatively, clinical trials have reported synergistic antitumour effects following combination treatment with Gem and other drugs or immunotherapeutic approaches.Citation14,Citation15 The coadministration of Gem and DC-based immunotherapy has been shown to induce the activation of antigen-specific cytotoxic T lymphocytes (CTLs) against advanced pancreatic cancers.Citation16
Gem can act as an adjuvant to immunotherapy by promoting increased MHC-I expression on tumor cells in vivo and in vitro, favoring tumor cell recognition by cytotoxic T cells.Citation17 Additionally, Gem plays an active role in inhibiting the proliferation of immunosuppressive cells, such as myeloid-derived suppressor cells (MDSCs) and regulatory T cells (Tregs), but does not induce parallel effects on effector T cells.Citation18 Gem was found to inhibit STAT3 phosphorylation in myeloid cells,Citation19 a transcription factor essential for the survival and proliferation of MDSCs and the differentiation of Tregs.Citation20 Therefore, Gem can potentially contribute to modulating some of the immunosuppressive mechanisms present in the tumor microenvironment.
Given the high numbers HPV-attributable tumors and the crucial need to develop alternative therapies with reduced toxicity and enhanced efficacy, we evaluated a new antitumour treatment involving the use of Gem with a DNA vaccine capable of inducing robust HPV-specific T cell responses. The DNA vaccine pgDE7h encodes HPV-16 E7 fused to herpes simplex virus type 1 (HSV-1) glycoprotein D (gD).Citation21,Citation22 Previous results demonstrated that administration of this vaccine therapeutically induced the activation of E7-specific CD8+ T cells and antitumour protection against early-stage tumors in mice.Citation22–24 In the present study, we focused on the combination of Gem and pgDE7h, aiming to improve the treatment efficacy, particularly against tumors in more advanced stages of development. Our results demonstrated the synergistic antitumour effects of the combined therapy with simultaneous control of immunosuppressive responses, induction of tumor-specific cytotoxic CD8+ T cells, and complete eradication of established tumors at experimental conditions.
Materials and methods
TC-1 cells
The TC-1 tumor cell line was kindly provided by Dr. T.C. Wu (Johns Hopkins University, MD, USA) and was generated by transformation of primary C57BL/6 mouse lung epithelial cells with c-Ha-ras and the HPV-16 E6 and E7 oncogenes.Citation25 TC-1 cells were maintained in RPMI 1640 medium (Gibco; MA, USA) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (Vitrocell, SP, BRA), 50 U/ml penicillin G/streptomycin, and 0.4 mg/ml G418 and were maintained at 37°C in a 5% CO2 atmosphere.
Wound healing assay
TC-1 cells (5 × 105) were seeded in 24-well plates and cultured at 37°C in 5% CO2 until 95% confluent, which took approximately 18 to 24 h. Afterward, a wound was made in the monolayer by carefully scratching it with a sterile 200-μL pipette tip. Next, fresh medium containing 50 nM or 500 nM Gem was added to the cells. Untreated cells were used as a control. After 18 h, bright-field images were obtained using an EVOS® FL Cell Imaging System light microscope (Thermo Fisher Scientific, MA, USA).
Spheroids and viability
Multicellular three-dimensional (3D) spheroids of TC-1 cells were prepared according to the following protocol. First, in a 96-well U-bottom plate, 50 µl of a PBS solution with 1% agarose was carefully applied to each well. After 30 min, 7 × 103 TC-1 cells, at a final volume of 200 µL, were carefully added to 50 µl of the agarose matrix previously added to each well. Then, the plate was centrifuged for 5 min at 290 g and room temperature in a swing-out rotor. The cells were cultured at 37°C and 5% CO2 for 4 days until spheroid formation. Next, 100 µl of culture medium was carefully removed from each well, and fresh medium containing Gem was added to the wells such that the final concentration reached 50 or 500 nM. Untreated cells were used as a control group to normalize the data. The spheroids were treated for 72 h. Spheroid images were obtained by bright-field microscopy for spheroid size evaluation. Next, spheroid viability was assessed using ethidium bromide (EB) incorporation in combination with acridine orange (AO) staining (50 µg/mL of each dye). Images were acquired with an EVOS® FL Cell Imaging System (Thermo Fisher Scientific, MA, USA). Green indicates live cells, and red indicates dead cells.
Cell proliferation (WST-1) assay
TC-1 cells were seeded at a concentration of 1 × 104 cells/well in a 96-well flat-bottom plate. Following overnight resting, fresh medium with or without 50 nM or 500 nM Gem was added to the cells. At different incubation time points (4, 6, 8, 12, and 24 h), 10 μL of Cell Proliferation Reagent WST-1 (Roche, BS, SWI, cat. #11644807001) was added to each well, mixed, and incubated for 1 h at 37°C in 5% CO2. Cell viability was estimated by measuring the absorbance at 480 nm. The cell viability percentage was calculated based on the average of triplicate wells using the following calculation: [(Absorbance treated cells/absorbance untreated cells) x 100]; values were normalized by the percentage of the control group (untreated cells).
Cell cycle profiling and DNA damage induction
Cells were treated with 50 or 500 mM Gem and harvested after 24 h. For concomitant cell cycle profiling and DNA damage analysis, cells were detached with trypsin and fixed with formaldehyde (1%) on ice for 15 min. Next, the cells were centrifuged (1500 rpm, 10 min), washed with PBS, centrifuged again, and suspended in 70% ice-cold ethanol. The cells were incubated in ethanol for at least 24 h at −20°C. After this time, the cells were centrifuged (4000 rpm, 5 min), washed with 500 µL of PBS-T-BSA (PBS, 0.2% Triton X-100, and 1% FBS), centrifuged again and permeabilized with PBS-T-BSA for 5 min. Subsequently, the cells were centrifuged, and the pellet was incubated in 50 µL of anti-ɣH2AX antibody (Sigma-Aldrich, MO, USA) overnight at 4°C. Next, the pellet was washed twice with PBS-T-BSA and incubated in anti-mouse-FITC (Sigma-Aldrich, MO, USA) for 1 h in the dark at room temperature. After this period, the cells were washed as previously described. For cell cycle analyses, the same samples were suspended in 200 µL of propidium iodide (200 μg/mL RNase A, 20 μg/mL PI, and 0.1% Triton X-100 in filtered PBS) for 40 min at room temperature. Next, the cells were washed twice with filtered PBS and suspended in 200 µL of filtered PBS for cytometric analysis. Flow cytometry was performed to evaluate the cell cycle profile and DNA damage induction indirectly through the anti-ɣH2AX antibody. The ɣH2AX signal and PI fluorescence in 10,000 cells were analyzed using an Accuri C6 BD cytometer (BD Bioscience, CA, USA) and BD CSampler analysis software.
Mice
Wild-type female C57BL/6 mice aged six to eight weeks were purchased from the Animal Breeding Center at Medical School (University of Sao Paulo). Female C57BL/6 IFN-γ knockout (KO) and C57BL/6 CD8+ T cell KO mice were provided by the facility Animal Breeding Center of the Department of Immunology at the University of São Paulo. All mice were housed at the Microbiology Department of the same university. Experimental procedures were carried out following guidelines established by the National Council for Control of Animal Experimentation (CONCEA), and animal protocols were approved by the Ethics Committee on the Use of Animals (CEUA) on August 18, 2014 (project number 92/2014 and 84/2017).
Tumor cell challenge
Mice were subcutaneously injected with 1 × 105 TC-1 cells per mouse in 100 μl of FBS-free RPMI medium in the right dorsolateral region. Tumor growth was monitored 2 times a week with callipers to determine the smaller (d) and larger (D) diameters of the tumor. Tumor size is expressed as volume, which was calculated according to the formula D x d2/2. Mouse survival was recorded for at least 60 days, mice were euthanized when tumors reached 15 mm of diameter or became necrotic. Tumor-free mice received a second tumor challenge with implantation of 10-fold more TC-1 cells (1 x 106 cells per mouse) 60 days after the first challenge.
Vaccine and immunization
To generate the vaccine pgDE7h, a gene sequence encoding the HPV-16 E7 protein genetically fused to HSV-1 gD was cloned into a pUMVC3 vector (Aldevron, ND, USA) as described previously.Citation26,Citation27 The therapeutic protocol started 5 days after TC-1 cell transplantation (early stage) or when the tumor reached 3 mm in diameter (established tumors). For early-stage immunization protocols, mice were injected with two doses of 50 µg of pgDE7h vaccine with a one-week interval by conventional intramuscular (i.m.) inoculation of the tibialis anterior muscle. In experiments using established tumors, mice were also vaccinated with two doses of the pgDE7h vaccine (5 µg per dose) i.m. associated with in vivo electroporation (EP) using the NEPA21 Super Electroporator (NepaGeneCo., Ltd., Chiba, JPN) as described previously.Citation24 Chemotherapy consisted of intraperitoneal administration of two doses of gemcitabine hydrochloride (80 mg/kg, Gemzar, Lilly, SP, BRA) with a one-week interval.
Immunological assays
For in vivo cytotoxicity assays, splenocyte suspensions from naive mice were stained with carboxyfluorescein diacetate succinimidyl ester (CFSE; Invitrogen) at a final concentration of 0.7 μM (CFSElow) or 7 μM (CFSEhigh). The CFSEhigh population was pulsed with 2.5 μg/mL CD8-specific E7 peptide (49RAHYNIVTF57, GenScript, NJ, USA) for 40 min at 37°C. Equal numbers of pulsed and unpulsed cells were injected intravenously (a total of 2 × 107 cells/mouse) into mice immunized two weeks prior. Splenocytes were harvested 18 h later, and the cell suspension was analyzed for CFSE expression by flow cytometry on an LSRFortessa (BD Biosciences, CA, USA). The percentages of lysed cells were determined by the following formula: [1 – (% CFSEhigh immunized/% CFSElow immunized)/ (% CFSEhigh naive/% CFSElow naive)] x 100. For identification of antigen-specific T cells, a splenocyte suspension was incubated with anti-CD16/CD32 Fc receptor blocking reagent (eBioscience, CA, USA). Splenocytes were then labeled with E7-specific APC-conjugated MHC-class I Dextramer (Immudex, KBH, DNK) according to the manufacturer’s recommendations. Subsequently, the cell surface was stained with anti-CD8a (FITC) and anti-CD3 (PE-Cy5) monoclonal antibodies (mAbs). For memory phenotyping assays and intracellular staining (ICS) for IFN-γ production, splenocytes were cultured at a concentration of 2 × 106 cells/well in the presence of brefeldin A (5 µg/ml – GolgiPlug; BD Biosciences, CA, USA) and recombinant mouse IL-2 (5 ng/ml) with or without a CD8-specific E7 peptide (49RAHYNIVTF57, GeneScript; 1.5 μg/mL) at 37°C and 5% CO2. After 12 h of stimulation, the cell surface was stained with anti-CD3 (PE-Cy5), anti-CD8a (BV605), anti-CD44 (FITC), and anti-CD62L (BV421) mAbs. Next, the surface-stained cells were fixed and permeabilized using Cytofix/Cytoperm (BD Biosciences, CA, USA) and intracellularly stained with an anti-IFN-γ antibody (Alexa Fluor 700). The number of activated cells was determined based on the phenotype CD3+ CD8+ CD44+ IFN-γ+ and effector memory (Tem) cells were detected by the surface phenotype CD3+ CD8+ CD44+ CD62L− T cells. Alternatively, the percentages of CD8+ IFNγ+ cells in the CD8+ CD3+ T cell gate were determined. Results were calculated by subtracting the percentages of unstimulated cells from the percentages of stimulated cells. The phenotypes of myeloid-derived suppressor cells (MDSCs) and dendritic cells (DCs) were determined after surface labeling of cells with anti-CD11b (FITC), anti-Gr1 (APC), anti-CD11 c (PE-Cy7), anti-I-A/IE (MHC-II, BV421), anti-Ly6C (PerCP-Cy5.5), and anti-F4/80 (PE) mAbs. MDSCs were characterized as CD11b+ Gr-1+. For Tregs assays, surface anti-CD3 (APC-Cy7), anti-CD4 (BV605) and anti-CD25 (BB515) mAbs and an anti-FoxP3 (BV421) mAb were used after cell permeabilization with FoxP3 Staining Buffer (eBioscience, CA, USA). All antibodies were purchased from eBioscience, BD, or BioLegend (CA, USA). The samples were analyzed on an LSRFortessa flow cytometer (BD Bioscience, CA, USA), and data were analyzed with FlowJo software. All analyses were performed after the initial exclusion of doublets using the FSC and SSC parameters.
Quantification and statistical analysis
t-distributed stochastic neighbor (t-SNE) analyses were run using concatenated flow cytometry data (5,000 events/sample) under default parameters (iterations: 2,000, perplexity: 50, and u: 0.5) using FlowJo software. t-SNE was applied to expression data for Gr-1, Ly6C, F4/80, MHC-II, and CD11c after downsampling pregating on CD11c+ CD11b+ single cells. The samples were merged to create a single t-SNE map and grouped according to treatment. The Circle shows the clustered population with the MDSCs APC phenotype defined as Gr-1+ Ly6C+ F4/80+ MHC-II+ CD11c+. t-SNE heatmaps show the median fluorescence intensity (MFI) of the markers Gr-1 and MHC-II in all samples overlaid on the t-SNE projection. The scales on the heat maps were individually generated for each surface marker from low to high expression. Additional statistical analyses were performed with GraphPad Prism software, version 8.0. To compare different immunization groups, one- or two-way ANOVA with multiple comparisons and Bonferroni post hoc tests were performed. Survival curves were compared using the log-rank (Mantel-Cox) test. All tests were performed as two-tailed tests, and significance levels were defined as p < .05 (* p < .05; ** p < .01 and *** p < .001) with a confidence interval of 95%. Appropriate methods are indicated in the legends with significant differences marked in all figures. Figures were prepared using GraphPad Prism, Adobe Illustrator 2020 (Adobe Systems, San Jose, CA) and Autodesk SketchBook Pro 2021 software.
Results
Gem limits the proliferation of TC-1 tumor cells in vitro
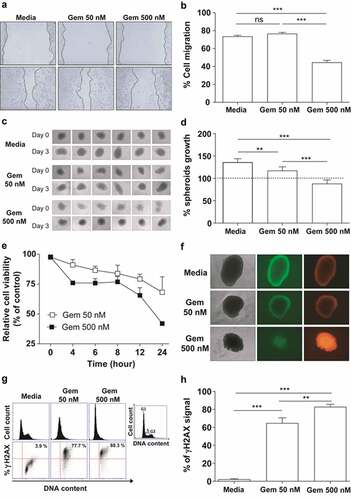
The antitumour effects of Gem were initially evaluated in vitro using TC-1 cells capable of expressing the HPV-16 E6 and E7 oncoproteins, either in monolayer (2D) or spheroid (3D) cultures. The 2D cultures treated with 50 nM Gem did not show any significant inhibitory effects in cell migration assays compared to untreated cultures (). However, exposure to 500 nM Gem significantly reduced TC-1 cell migration (). In contrast, when spheroids were treated with Gem, clear inhibition of cell proliferation was detected with both 50 nM and 500 nM (). Gem also displayed cytotoxic effects on 2D TC-1 cultures at both 50 nM and 500 nM, with viability dropping to 70% and 40%, respectively, after 24 h (). Although 50 nM Gem decreased spheroid growth, this treatment did not induce significant cell death. In contrast, 500 nM Gem halted spheroid growth and caused significant cell death (). Gem also caused dose-dependent cell cycle arrest in TC-1 cells in vitro. In 2D cultures, most of the cells were arrested in G1 phase at both concentrations at 24 h post treatment (). Finally, Gem caused significant DNA damage, as measured by evaluating the phosphorylation of H2AX, resulting in γH2AX formation, a biomarker of genotoxic stress. Notably, both concentrations of Gem increased the formation of γH2AX in treated TC-1 cells ().
Figure 1. In vitro effects of Gem on TC-1 cells. (a) Regenerative growth (cell migration) following disruption of TC-1 cell monolayers incubated in the presence of 50 nM or 500 nM Gem for 15 h. Cells cultured in RPMI medium plus 10% FBS were used as reference controls. (b) Relative TC-1 monolayer regeneration based on the experiment described in (A). Values represent the percentage of TC-1 cells that migrated after Gem treatment. The error bars correspond to the SEM of 8 experiments. (c) Multicellular three-dimensional (3D) spheroids of TC-1 cells cultured for three days with or without Gem at 50 nM or 500 nM. Control cells were cultured in the medium alone. (d) Quantification of TC-1 spheroid growth after treatment with Gem. The dashed line represents the initial area of TC-1 spheroids before treatment with Gem. The values are the mean ± SEM of 6 experiments. (e) Sensitivity of TC-1 cells to Gem (50 nM and 500 nM) evaluated by a WST-1 assay at different time points after exposure. Nontreated cells were used as a control to normalize the values measured for treated cells. (f) Bright-field (left images) and fluorescence microscopy images (central – acridine orange, right – ethidium bromide) of TC-1 3D spheroids to evaluate morphology (density) and viability, respectively, three days after cultivation with or without Gem for three days. (g) TC-1 cell cycle arrest promoted by Gem. Representative histograms of cell cycle profiles (top line) and representative immunostaining for γH2AX (bottom line) to detect genotoxic stress in TC-1 cells by flow cytometry after 24 h of treatment with Gem (50 or 500 nM). (h) Quantification of γH2AX levels in the cells described in (G). All data are presented as the mean ± SEM based on 2 independent experiments performed in triplicate. Statistical significance: (**) p < .01 and (***) p < .001 by ANOVA. (ns) Non-significant
The combination of gem and pgDE7h promotes synergistic antitumour effects on early-stage HPV-associated tumors
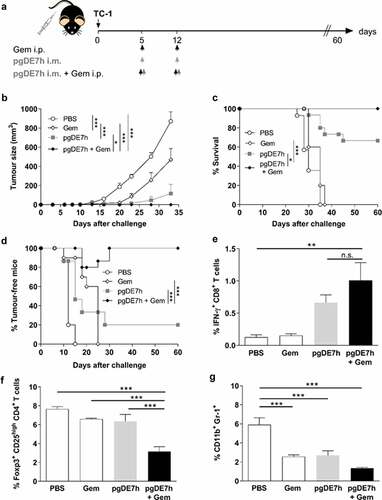
We previously reported that immunizations with pgDE7h conferred partial therapeutic antitumour protection in mice after transplantation of TC-1 cells.Citation22 To evaluate whether Gem can enhance the therapeutic antitumour effects of the therapeutic vaccine, we treated mice with two doses of Gem (80 mg/kg) combined with pgDE7h (50 µg/dose) or not at 5 and 12 days after TC-1 cell transplantation. Gem treatment alone promoted an initial delay in tumor growth, but growth promptly resumed after interruption of treatment, and all animals had to be euthanized before the end of the observation period (). Immunization with pgDE7h showed a slight protective antitumour effect, with only 20% of mice remaining tumor-free (). Nonetheless, the combination of Gem and pgDE7h elicited synergistic antitumour effects and promoted full tumor protection until the end of the observation period (). We also measured CD8+ T cell responses and immunosuppressive cell expansion in mice subjected to the combined treatment. As shown in , mice treated with pgDE7h or with Gem plus pgDE7h showed increased frequencies of E7-specific IFN-γ-producing CD8+ T cells compared with mice treated with only Gem or sham treatment (). In agreement with the literature, mice treated with Gem efficiently block expansion of MDSCs, but did not alter the frequencies of Tregs in the spleen (). In contrast, mice subjected to the combined treatment showed reduced expansion of Tregs and MDSCs in comparison with mice immunized with pgDE7h or treated only with Gem ().
Figure 2. Synergistic antitumour effects observed in mice administered a combination of Gem and pgDE7h to treat early-stage tumors. (a) Schematic representation of the treatment regimen. Mice were transplanted with 1 × 105 TC-1 cells per mouse. On days 5 and 12 post-tumor implantation, the mice received Gem (80 mg/kg) via i.p. injection and/or i.m. immunization with pgDE7h (50 µg/dose). Groups of mice were inoculated with PBS or only Gem as controls. (b) Tumor sizes were measured over time (two-way ANOVA). Each symbol and bar represent the mean tumor volume, and values are expressed as the mean ± SEM. (c) Percentages of surviving and (d) tumor-free mice over time (Mantel-Cox log-rank test). (e) Frequencies of IFN-γ-producing CD8+ T cells on CD3+ CD8+ T cell subsets determined by flow cytometry after overnight stimulation with the peptide for the HPV-16 E7 Kb MHC class I-restricted immunodominant epitope at 2 weeks post-immunization. (f) Frequencies of total Tregs cells were determinate as CD25high FoxP3+ on CD3+ CD4+ subsets, (g) and MDSCs cells were determined as CD11b+ Gr-1+ in spleen cells by flow cytometry. Statistically significant differences were determined with ANOVA and Bonferroni’s test. The results represent data from 2 independent experiments (n= 7–10). Statistical significance: *p< .05, **p< .01, ***p< .001, ns- non-significant
The combination of gem and pgDE7h delivered by electroporation confers full protection against established tumors
We next evaluated whether the combined treatment is capable of controlling tumors at more advanced growth stages. For these experiments, mice bearing tumors with an average diameter of 3 mm were treated with two doses of pgDE7h (5 µg/dose) delivered by electroporation (pgDE7h EP) with or without Gem 10 and 17 days after tumor cell engraftment (). All mice treated with Gem combined with pgDE7h EP showed full eradication of established tumors (). Under the same conditions, mice that received Gem or pgDE7h EP alone displayed significantly reduced tumor growth but showed no or 40% antitumour protection, respectively, until the end of the observation period (). In order to determine whether the combination treatment would prevent tumor relapses, immunized mice were grafted with a second load of TC-1 cell (with 10-fold higher compared with the first tumor cell engraftment) 60 days after the first challenge. The mice treated with pgDE7h EP, with or without combination with Gem, efficiently eliminated TC-1 tumor cells and displayed full protection from tumor recurrence, which demonstrated the long-lasting immune protection induced by the vaccine-based treatment.
Figure 3. Coadministration of Gem and pgDE7h EP controls established tumors. Tumor-bearing mice (3 mm – approximately 10 days after transplantation) received two doses of Gem (80 mg/kg) via i.p. injection and/or i.m. immunization with 2 doses of pgDE7h (5 µg/dose) delivered via electroporation at an interval of 7 days. Groups of mice were inoculated with PBS or only Gem as controls. (a) Schematic representation of the treatment regimen. (b) Tumor sizes in mice subjected to the combined treatment. The tumors were measured over time, and the curves are interrupted when one or more animals developed a tumor at a size that required euthanasia (two-way ANOVA). Statistical significance: ***p< .001 the control group versus all groups on day 25, +++ p< .001 Gem versus pgDE7h EP and pgDE7h EP + Gem on day 33, ### p< .001 pgDE7h versus pgDE7h + Gem on day 50. (c) Percentages of surviving mice over time from different groups. Immunized tumor-free mice were grafted with an additional load of TC-1 tumor cells (10-fold higher) 60 days after the first challenge, and protection was followed for 90 days. Data were (n = 7–14) analyzed by the Mantel-Cox log-rank test. Tumor sizes of (d) CD8 KO mice and (e) IFN-γ KO mice (n = 5). Statistical significance: ***p< .001 versus all groups on day 29. Results represent data from 2 independent experiments. Each symbol and bars represent the mean ± SEM
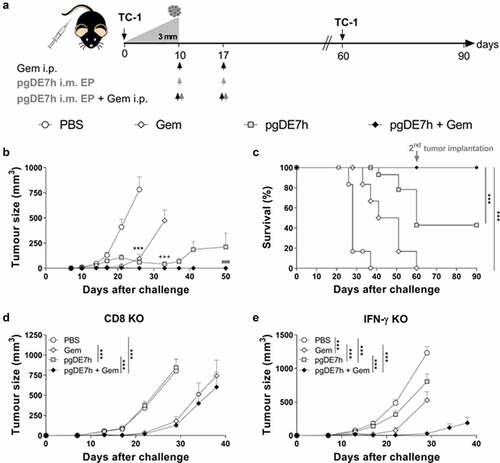
The role of effector CD8+ T cells in the protective effects conferred by the combined treatment was investigated in knockout mice genetically deficient in CD8+ T cells (CD8 KO) or IFN-γ (IFN-γ KO). The antitumour protection elicited in immunocompetent mice was completely abrogated in CD8 KO and IFN-γ KO mice (). Indeed, CD8 KO and IFN-γ KO mice treated with Gem or Gem combined with pgDE7h EP exhibited transient tumor growth and delayed mortality, but none of the treated animals remained tumor-free at the end of the observation period (). Collectively, these results indicate that the antitumour growth effects induced by the combined treatment require activation of CD8+ T cells and IFN-γ responses.
Gem treatment enhances the pgDE7h-induced activation of cytotoxic E7-specific CD8+ T cells
To elucidate the immunological basis of the antitumour effects observed after combination treatment with Gem and pgDE7h EP, CD8+ T cell responses were measured in the spleen of mice subjected to the treatment (). Mice treated with pgDE7h EP showed no significant increase in the relative numbers of E7-specific CD8+ T cells compared with mice treated with only Gem or sham treatment (). However, mice treated with Gem and pgDE7h EP showed a significant increase in the number of E7-Dex+ cells compared to mice treated with Gem or immunized only with pgDE7h EP (). Overall, the combined use of Gem and pgDE7h EP induced enhanced numbers of E7-specific CD8+ T effector memory (Tem) cells (CD8+ CD44high CD62L−) compared to treatment with Gem or pgDE7h EP (). Notably, only mice treated with the combined therapy exhibited high frequencies of activated CD8+ T cells (% CD44+ IFN-γ+ CD8+ T cells) ().
Figure 4. Activation of cytotoxic E7-specific CD8+ T cells in mice subjected to treatment with Gem and pgDE7h. Tumor-bearing mice (3 mm) received two i.p. doses of Gem (80 mg/kg) and/or i.m. immunization with pgDE7h (5 µg/dose) via EP at an interval of 7 days. Groups of mice were inoculated with PBS or only Gem as controls. Two weeks after the second immunization, splenocytes were collected and processed for immunological analyses. (a) Representative flow cytometry plots and (b) percentages of E7-specific CD8+ T cells determined by H2-Db E749–57 (RAHYNIVTF) tetramer staining gated on CD3+ CD8+ T cell subsets. Frequencies of (c) effector memory T cells (CD8+ CD44high CD62L−; Tem) in CD3+ CD8+ T cell subsets and (d) E7-specific CD44high IFN-γ+ in CD3+ CD8+ T cell after stimulation with the HPV-16 E7 Kb MHC class I-restricted immunodominant peptide. (e, f) Two weeks after the second pgDE7h dose, mice received CFSE-labeled splenocytes that were pulsed with the E7-derived peptide or left unpulsed. In vivo cytotoxic activity is expressed as results in a histogram (e) and (f) the percentages of target cell lysis. (n = 10). Values are expressed as the mean ± SEM. Statistical analysis: ANOVA, posttest: Bonferroni. *p < .05, **p < .01, ***p < .001
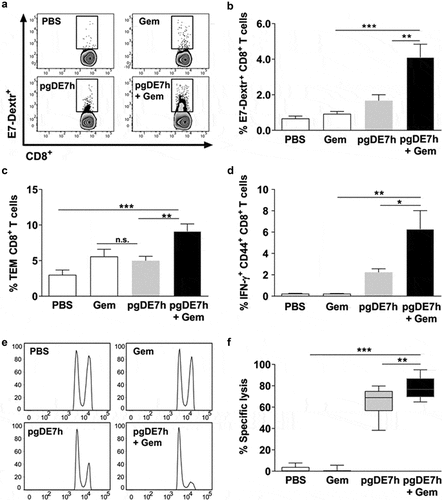
We next determined the in vivo cytotoxic activity of E7-specific CD8+ T cells. Mice treated with pgDE7h EP showed an increased killing capacity directed against target cells loaded with the E7-specific peptide corresponding to the MHC-I-restricted CD8+ T cell epitope (). Moreover, the in vivo E7-specific cytotoxic activity of CD8+ T cells was significantly enhanced in mice treated with Gem and pgDE7h EP compared with those treated with only pgDE7h EP (). Taken together, these results demonstrate that administration of Gem and pgDE7h EP enhances the activation of antigen-specific CD8+ T cells, particularly T cells with the Tem cell phenotype, and endows them with in vivo cytotoxic activities.
We also investigated whether the combination of Gem and pgDE7h EP impacts the modulation of immunosuppressive cells (MDSCs) and promotes the activation of APCs. Mice treated with Gem, with or without combination with pgDE7h EP, showed higher frequencies of splenic DCs (CD11c+ MHC-II+ Ly6C− F4/80−) than mice treated with only pgDE7h EP (). In addition, mice that received any of the treatments showed reduced expansion of MDSCs (CD11b+ Gr-1+) (). The absence of a significant difference in the percentage of MDSCs in this setting between the monotherapies and combination therapy could be due to the mice having tumors of similar size at the time point evaluated.
Figure 5. Mice subjected to combined chemoimmunotherapy showed inhibition of the expansion of immunosuppressive cells and promotion of the expansion of APCs. Tumor-bearing mice (3 mm) received two i.p. doses of Gem (80 mg/kg) and/or i.m. immunization with 2 doses of pgDE7h (50 µg/dose) by electroporation at an interval of 7 days. Groups of mice were inoculated with PBS or only Gem as controls. At two weeks after immunization, splenocytes were collected and processed for immunological analyses. (a) The frequencies of CD11b+ Gr-1+ MDSCs and (b) CD11c+ MHC-II+ (gated out using the markers F4/80 and Ly6C) cells were determined by flow cytometry. (c) t-SNE projection was performed using 2,000 iterations and a perplexity of 50 from splenocyte samples gated on CD11c+ CD11b+. The samples were merged to create a single t-SNE map and grouped according to treatment. A circle marks the clustered population with the MDSCs APC phenotype defined as Gr-1+ Ly6C+ F4/80+ MHC-II+ CD11c+. (d) Heatmaps of the markers Gr-1 and MHC-II in all samples overlaid on the t-SNE projection. (e) The frequencies of MDSCs APC are shown for individual samples defined by the manual gating strategy. Values are expressed as the mean + SEM. Statistical analysis: ANOVA, posttest: Bonferroni. *p < .05, **p < .01, ***p < .001
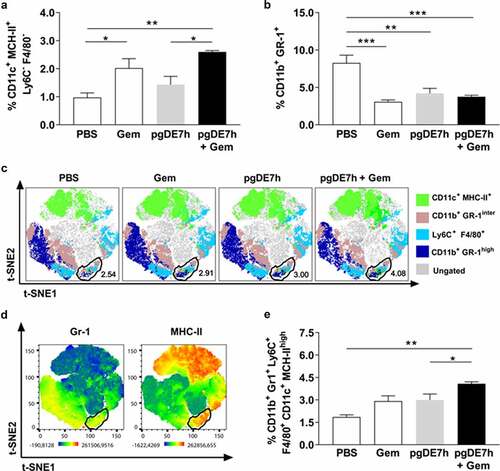
To better demonstrate the specific cell population clusters in mice subjected to the different tested treatments, we applied a t-SNE analysis tool to characterize the heterogeneous profiles of splenic MDSCs and APC populations. Based on this analysis, we identified a GR-1+ cell cluster expressing high MHC-II levels that was also positive for Ly6C and F4/80 (, Supl 1a-b). This result suggested an increase in the MDSCs population with an APC profile. More importantly, the group treated with Gem and pgDE7h had higher frequencies of MHC-IIhigh MDSCs and APCs (CD11b+ Gr-1+ Ly6C+ F4/80+ CD11c+ MHC-IIhigh) than the other tested mouse groups (, Supl 1a-b). Overall, the results indicated that the combination of pgDE7h EP and Gem shifted the phenotype of MDSCs and enhanced the expansion of APCs in treated mice.
Discussion
Herein, we demonstrated the enhanced antitumour therapeutic effects of antigen-specific immunotherapy (pgDE7h) after combination with Gem, leading to eradication of established HPV-associated tumors under experimental conditions. Notably, the tested chemoimmunotherapy combination acted synergistically to increase the anticancer effects induced in treated mice, which were demonstrated by the efficient induction of activated E7-specific cytotoxic CD8+ T cells and reduction in systemic immunosuppressive cellular responses. Administration of two doses of Gem temporarily controlled tumor progression by direct cytotoxic effects on tumor cells without noticeable side effects but was unable to confer long-term protection. In contrast, we demonstrated that the combined Gem and pgDE7h treatment resulted in improved immunomodulatory effects supported by activation of DCs and MDSCs expressing MHC-II, leading to increased antigen presentation and priming of CD8+ T cell responses. More relevantly, all mice with established tumors subjected to the combined treatment exhibited induction of strong immune responses and elimination of tumors and remained tumor-free through the end of the observation period, even after a tumor challenge two months after the initial graft. Altogether, our results provide experimental evidence for the improved efficacy of the combination strategy involving chemotherapy (Gem) and active immunotherapy (pgDE7h) with regard to eradication of established HPV-associated tumors.
Understanding the effects of Gem on tumor cells represents a relevant step toward the establishment of improved anticancer therapies. Several studies have shown the effects of Gem on different tumor cell lines under in vitro and in vivo conditions. We found direct cytotoxic activity of Gem against TC-1 tumor cells in vitro in both 2D and 3D cultures. Such effects included inhibition of cell migration, reduction in cell viability, induction of cell cycle arrest, and genotoxic effects demonstrated by generating γH2AX, suggesting prevention of malignant progression. These effects also translated into a temporary oncostatic effect of the drug in vivo but were restricted to a short time frame close to the administration of Gem. Indeed, Gem monotherapy shows disease stabilization and good tolerability but has minimal antitumour activity in treating recurrent or advanced-stage cervical cancer patients.Citation28,Citation29 Despite its use in the clinic for different types of cancer, Gem is associated with the emergence of drug resistance when used long-term.Citation30 Preclinical studies have demonstrated that long-term use of Gem enhances negative immune checkpoint receptors (PD-L1 and PD-L2) and drug resistance mediated by TGF-β secretion.Citation31 On the other hand, Gem exhibits a positive impact on the exposure of tumor antigens when used at a low dose, which may enhance active immunological responses.Citation32 Such an effect may be directly linked to the enhanced antitumour therapeutic effects achieved when combined with active immunotherapy, favoring presentation and cross-presentation of tumor antigen epitopes by APCs and subsequent induction of antigen-specific cytotoxic T cells.Citation32 The described data suggests that short-term Gem therapy may be an interesting approach when associated with active immunotherapies capable of boosting tumor-specific immune responses, controlling tumor growth and inducing immunological memory to tumor antigens.
Cytotoxic CD8+ T cell responses positively correlate with the control of HPV-16-associated tumors in humans.Citation33,Citation34 Enhanced antitumour effects and generation of E7-specific CD8+ T cells and CTL responses in mice immunized with pgDE7h are observed after EP delivery (Sales et al., 2017). Since DNA vaccines are poorly immunogenic, the use of EP increases cell transfection and the antitumour effects, even at DNA concentrations up to 10-fold lower than the amount required to achieve similar results after conventional i.m. injections. Nonetheless, without any other associated therapy, such effects drop significantly in mice with established tumors following the engraftment of TC-1 cells, a fast-growing tumor cell line.Citation23 In the present study, the combination of pgDE7h EP with Gem resulted in enhanced antitumour activities in mice with established tumors, including activation of tumor-specific CTL responses. In order to demonstrate the synergic adjuvant effects of Gem when combined with immunotherapy, we selected a suboptimal dose of (80 mg/kg) in a two-dose treatment regimen (significantly lower than the dose used in patients – 120 mg/kg), based on previously reported preclinical conditions.Citation35 Our results demonstrate that Gem, when applied concomitantly with immunotherapy at rather low amounts, promotes strong adjuvant effects leading to the synergistic activation of E7-specific CD8+ T cells and control of immunosuppressive responses. Indeed, the generation of CTL responses has been previously identified as a major mechanism underlying the enhanced antitumour effects of Gem when combined with other immunotherapies. Nonetheless, such effects were detected only with repeated administration of Gem at high doses or with concomitant administration of additional adjuvants.Citation36,Citation37 Importantly, previous studies have demonstrated that administration of multiple doses of Gem suppresses CTL responses in mice transplanted with TC-1 cells, with a reduction in antitumour effectiveness.Citation37 In contrast, in our preclinical test, mice treated with two doses of Gem or with Gem + pgDE7h showed no signs of morbidity, weight loss, or a reduction in the number of circulating CD8+ T cells. Moreover, our findings demonstrated that Gem treatment provides a window for immunological intervention that enhances vaccine effectiveness. The enhanced and long-lived therapeutic effects of the combined chemoimmunotherapy approach may thus be attributed to the combined immunomodulatory properties of both Gem and the chimaeric protein encoded by the DNA vaccine vector, as demonstrated by activation of murine and human DCs with a purified gD-E7 protein.Citation38 Of note, the antitumour responses achieved in mice subjected to combined Gem and pgDE7h treatment conferred long-lived immune responses, preventing tumor recurrence even after the subsequent implantation of TC-1 cells. Altogether, the present evidence supports the application of combined therapy for eradication of established HPV-associated tumors.
As highlighted by the present study, combination therapy with Gem and pgDE7h prevented the accumulation of immunosuppressive cells, including MDSCs and Tregs. Circulating and tumor-infiltrating MDSCs are capable of inhibiting the activation of T cells, which leads to immune escape and dismal overall survival with tumor relapse in patients and preclinical models.Citation39–41 Previous evidence has also demonstrated that Gem combined with immunotherapies blocks immunosuppressive mechanisms by reducing the levels of PD-1-expressing cells, MDSCs, tumor-infiltrating Tregs, and tumor-associated macrophages (TAMs).Citation36,Citation42 The inhibitory effects of Gem on these cells may contribute to the tumor protection observed in mice subjected to combination therapy based on pgDE7h. Indeed, our own previous observations clearly demonstrated that the combination of active immunotherapies with blockade of immunosuppressive mechanisms, such as neutralization of IL-10, inhibition of IDO, or depletion of MDSCs,Citation23,Citation24,Citation43,Citation44 represents a trend toward the development of treatments capable of controlling HPV-associated tumors, particularly at advanced growth stages.
Here, we showed that the combination of Gem with pgDE7h increases the frequency of DCs in vivo. Induction of immune responses capable of eradicating established tumors requires the interaction of APCs and CD8+ T cells. Available evidence indicates that Gem promotes the maturation of DCs by upregulating CD80, CD86, and HLA-DR and enhances CTLs under in vitro conditions in cultures with pancreatic cancer cells.Citation45 Similarly, gD target antigens have the potential to induce maturation of mouse and human DC subsets specialized in cross-presentation.Citation38 In addition, our results demonstrated that the decrease in MDSCs in mice subjected to the combined treatment was accompanied by an increase in Gr-1+ cells expressing MHC-IIhigh, Ly6C+, F4/80+, CD11c+, and Ly6C+, suggesting that Gem, in association with pgDE7h, modulates the MDSCs phenotype and maximizes the interactions favoring antigen presentation by APCs. Indeed, modulation of MDSCs to a phenotype favoring the expression of costimulatory molecules was also observed after administration of a combined treatment using carboplatin–paclitaxel-based drugs and a long peptide vaccine in a preclinical TC-1 cell tumor model.Citation46 The present results open interesting perspectives for future studies aiming to elucidate the mechanisms leading to modulation of APC phenotypes by Gem and active immunotherapies.
Treatment of advanced-stage cervical cancer remains a clinical challenge and, so far, mainly involves chemotherapy. The present study provides experimental evidence that concomitant administration of Gem and active immunotherapy (pgDE7h) confers enhanced antitumour responses to treat established HPV-associated tumors using only two doses of the combined treatment. Indeed, the proposed therapy avoids the application of multiple Gem doses and would certainly decrease the incidence of deleterious side effects, a serious concern among patients receiving conventional treatments based on chemotherapy and passive immunotherapies, such as cytokines and monoclonal antibodies. Additionally, the combined treatment led to simultaneous activation of T cell mediated cytotoxic responses with modulation of immunosuppressive cells and induction of immunological memory, which may avoid tumor relapse. Altogether, the present findings support the undertaking of further studies aiming to evaluate this combination therapy under clinical conditions.
Author contributions
Jamile Ramos da Silva, Mariana de Oliveira Diniz, Ana Carolina Ramos Moreno and Luís Carlos de Souza Ferreira conceived the study and the experimental design. Mariana de Oliveira Diniz supervised the experimental work. Jamile Ramos da Silva, Natiely Silva Sales, Mariângela de Oliveira Silva, Luana Raposo de Morais Melo Aps, Natália Cestari Moreno, Karine Bitencourt Rodrigues, Aléxia Adrianne Vesnceslau-Carvalho, Bruna F.M.M. Porchia, Ana Carolina Ramos Moreno, Carlos Frederico M. Menck and Mariana de Oiveira Diniz carried out the experiments, processed the experimental data, and participated in the interpretation of the results. Mariana de Oliveira Diniz, Ana Carolina Ramos Moreno and Luís Carlos de Souza Ferreira discussed the results and contributed to the writing of the manuscript. Jamile Ramos da Silva wrote the manuscript with support from Ana Carolina Ramos Moreno, Mariana de Oliveira Diniz and Luís Carlos de Souza Ferreira.
Supplemental Material
Download ()Acknowledgments
The authors greatly appreciate the helpful technical support of Eduardo G. Martins of the Vaccine Development Laboratory of the University of São Paulo. Additionally, we are grateful for the support for analyses of t-SNE data provided by Luisa Menezes at the Immunology Department of the University of São Paulo.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
Additional information
Funding
References
- De MC, Georges D, Bray F, Ferlay J, Clifford GM. Articles Global burden of cancer attributable to infections in 2018 : a worldwide incidence analysis. Lancet Glob Heal. 2020;8(2):e180–13. doi:10.1016/S2214-109X(19)30488-7.
- Wardak S. Human Papillomavirus (HPV) and cervical cancer. Med Dosw Mikrobiol. 2016;68:73–84.
- Siegel RL, Miller KD. Cancer Statistics, 2020. CA Cancer J Clin. 2020;70(1):7–30. doi:10.3322/caac.21590.
- WHO report on cancer: setting priorities, investing wisely and providing care for all. WHO-CancerReport-2020-Global Profile. Geneva:World Health Organization; 2020.
- Survival rates for cervical cancer. American Cancer Society. 2020. Web page (https://www.cancer.org/cancer/cervical-cancer/detection-diagnosis-staging/survival/en/, accessed January 2021).
- Tseng J, Yen M, Twu N, Lai C, Horng H. Prognostic nomogram for overall survival in stage IIB-IVA cervical cancer patients treated with concurrent chemoradiotherapy. YMOB. 2010;202(2):174.e1–174.e7. doi:10.1016/j.ajog.2009.09.028.
- Gadducci A, Tana R, Cosio S, Cionini L. Treatment options in recurrent cervical cancer (Review). Oncol Lett. 2010:3–11. doi:10.3892/ol.
- Bracci L, Schiavoni G, Sistigu A, Belardelli F. Immune-based mechanisms of cytotoxic chemotherapy: implications for the design of novel and rationale-based combined treatments against cancer. Cell Death Differ. 2014;21(1):15–25. doi:10.1038/cdd.2013.67.
- Carmichael J. The role of gemcitabine in the treatment of other tumours. British Journal of Cancer. 1998;78(S3):21–25. doi:10.1038/bjc.1998.750.
- Mini E, Nobili S, Caciagli B, Landini I, Mazzei T. symposium article Cellular pharmacology of gemcitabine. Gemcitabine Ten Years Clin Exp. Sept 2005;19–21. doi:10.1093/annonc/mdj941. Monast. di Treviso, Italy. 2006;17\(Supplement 5):v7–v12.
- Amrutkar M, Gladhaug IP. Pancreatic cancer chemoresistance to gemcitabine. Cancers (Basel). 2017;9:157. doi:10.3390/cancers9110157.
- De Sousa Cavalcante L, Gemcitabine MG. Metabolism and molecular mechanisms of action, sensitivity and chemoresistance in pancreatic cancer. Eur J Pharmacol. 2014;741:8–16. doi:10.1016/j.ejphar.2014.07.041.
- Sakamoto H, Kitano M, Suetomi Y, Takeyama Y, Ohyanagi H, Nakai T, Yasuda C, Kudo M. Comparison of standard-dose and low-dose gemcitabine regimens in pancreatic adenocarcinoma patients: a prospective randomized trial. J Gastroenterol. 2006;41:70–76. doi:10.1007/s00535-005-1724-7.
- Alexopoulos A, Karamouzis MV, Rigatos G. In vivo synergism between docetaxel and gemcitabine in patients with metastatic breast cancer: general concepts and future perspectives. Semin Oncol. 2004;31(2 SUPPL. 5):25–30. doi:10.1053/j.seminoncol.2004.03.024.
- Hirooka Y, Kawashima H, Ohno E, Ishikawa T, Kamigaki T, Goto S, Takahara M, Goto H. Comprehensive immunotherapy combined with intratumoral injection of zoledronate-pulsed dendritic cells, intravenous adoptive activated T lymphocyte and gemcitabine in unresectable locally advanced pancreatic carcinoma: a phase I/II trial. Oncotarget. 2018;9(2):2838–2847. doi:10.18632/oncotarget.22974.
- Hirooka Y, Itoh A, Kawashima H, Hara K, Nonogaki K, Kasugai T, Ohno E, Ishikawa T, Matsubara H, Ishigami M, et al. A combination therapy of gemcitabine with immunotherapy for patients with inoperable locally advanced pancreatic cancer. Pancreas 2009;38:3. doi:10.1097/MPA.0b013e318197a9e3.
- Liu WM, Fowler DW, Smith P, Dalgleish AG. Pre-treatment with chemotherapy can enhance the antigenicity and immunogenicity of tumours by promoting adaptive immune responses. Br J Cancer. 2010;102:115–123. doi:10.1038/sj.bjc.6605465.
- Suzuki E, Kapoor V, Jassar AS, Kaiser LR, Albelda SM. Gemcitabine selectively eliminates splenic Gr-1+/CD11b+ Myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clin Cancer Res. 2005;11(18):6713–6721. doi:10.1158/1078-0432.CCR-05-0883.
- Eriksson E, Wenthe J, Irenaeus S, Loskog A, Ullenhag G. Gemcitabine reduces MDSCs, tregs and TGFβ-1 while restoring the teff/treg ratio in patients with pancreatic cancer. J Transl Med. 2016;14(1):1–12. doi:10.1186/s12967-016-1037-z.
- Rébé C, Végran F, Berger H, Ghiringhelli F. STAT3 activation. JAK-STAT. 2013;2(1):e23010. doi:10.4161/jkst.23010.
- Diniz MO, Lasaro MO, Ertl HC, Ferreira LCS. Immune responses and therapeutic antitumor effects of an experimental DNA vaccine encoding human papillomavirus type 16 oncoproteins genetically fused to herpesvirus glycoprotein D. Clin Vaccine Immunol. 2010;17(10):1576–1583. doi:10.1128/CVI.00264-10.
- Diniz MO, Cariri FAMO, Aps LRMM, Ferreira LCS. Enhanced therapeutic effects conferred by an experimental DNA vaccine targeting human papillomavirus-induced tumors. Hum Gene Ther. 2013;24(10):861–870. doi:10.1089/hum.2013.102.
- Silva JR, Sales NS, Silva MO, Aps LRMM, Moreno ACR, Rodrigues EG, Ferreira LCS, Diniz MO. Expression of a soluble IL-10 receptor enhances the therapeutic effects of a papillomavirus-associated antitumor vaccine in a murine model. Cancer Immunol Immunother. 2019;68(5):753–763. doi:10.1007/s00262-018-02297-2.
- Sales NS, Silva JR, Aps LRMM, Silva MO, Porchia BFMM, Ferreira LCS, Diniz MO. In vivo electroporation enhances vaccine-mediated therapeutic control of human papilloma virus-associated tumors by the activation of multifunctional and effector memory CD8 + T cells. Vaccine. 2017;(52)7240–7249. doi:10.1016/j.vaccine.2017.11.011.
- Lin KY, Guarnieri FG, Staveley-O’Carroll KF, Levitsky HI, August JT, Pardoll DM, Wu TC. Treatment of established tumors with a novel vaccine that enhances major histocompatibility class II presentation of tumor antigen. Cancer Res. 1996;56:21–26.
- Lasaro MO, Diniz MO, Reyes-Sandoval A, Ertl HC, Ferreira LCS. Anti-tumor DNA vaccines based on the expression of human papillomavirus-16 E6/E7 oncoproteins genetically fused with the glycoprotein D from herpes simplex virus-1. Microbes Infect. 2005;7(15):1541–1550. doi:10.1016/j.micinf.2005.05.024.
- Diniz MO, Ferreira LCS. Enhanced anti-tumor effect of a gene gun-delivered DNA vaccine encoding the human papillomavirus type 16 oncoproteins genetically fused to the herpes simplex virus glycoprotein D. Brazilian J Med Biol Res. 2011;44(5):421–427. = Rev. Bras. Pesqui. médicas e biológicas/Soc. Bras. Biofísica … [et al.].
- Candelaria M, Cetina L, De La Garza J, Dueñas-Gonzálz A. Clinical implications of gemcitabine in the treatment of cervical cancer. Eur J Cancer. 2007;5(1):37–43. doi:10.1016/S1359-6349(07)70014-9.
- Mutch DG, Bloss JD. Gemcitabine in cervical cancer. Gynecol Oncol. 2003;90(2): Pt 2. doi:10.1016/S0090-8258(03)00338-X.
- Jia Y, Xie J. Promising molecular mechanisms responsible for gemcitabine resistance in cancer. Genes Dis. 2015;2(4):299–306. doi:10.1016/j.gendis.2015.07.003.
- Principe DR, Narbutis M, Kumar S, Park A, Viswakarma N, Dorman MJ, Kamath SD, Grippo PJ, Fishel ML, Hwang RF, et al. Long-Term gemcitabine treatment reshapes the pancreatic tumor microenvironment and sensitizes murine carcinoma to combination immunotherapy. Cancer Res. 2020;80(15):3101–3115. doi:10.1158/0008-5472.CAN-19-2959.
- Zhang X, Wang D, Li Z, Jiao D, Jin L, Cong J, Zheng X, Low-Dose Gemcitabine XL. Treatment enhances immunogenicity and natural killer cell-driven tumor immunity in lung cancer. Front Immunol. 2020;11:331. doi:10.3389/fimmu.2020.00331.
- Komdeur FL, Prins TM, Van De Wall S, Plat A, Wisman GBA, Hollema H, Daemen T, Church DN, de Bruyn M, Nijman HW. CD103+ tumor-infiltrating lymphocytes are tumor-reactive intraepithelial CD8+ T cells associated with prognostic benefit and therapy response in cervical cancer. Oncoimmunology 2017;6:9. doi:10.1080/2162402X.2017.1338230.
- Stevanovi´c SS, Draper LM, Langhan MM, Campbell TE, Kwong ML, Wunderlich JR, Dudley ME, Yang JC, Sherry RM, Kammula US, et al. Complete regression of metastatic cervical cancer after treatment with human papillomavirus-targeted tumor-infiltrating T cells. J Clin Oncol. 2015. 10.1200/JCO.2014.58.9093.
- Reiniš M. Genetically modified tumour vaccines producing IL-12 augment chemotherapy of HPV16-associated tumours with gemcitabine. Oncol Rep. 2011. doi:10.3892/or.2011.1221. Mar 18.
- Chang L-S, Yan W-L, Chang Y-W, Yeh Y-C, Chen H-W, Leng C-H, Liu S-J. Gemcitabine enhances antitumor efficacy of recombinant lipoimmunogen-based immunotherapy. OncoImmunology. 2016;5(3):e1095433. doi:10.1080/2162402X.2015.1095433.
- Jang HY, Han BS, Kwon B, Sin JI. Optimized gemcitabine therapy in combination with E7 peptide immunization elicits tumor cure by preventing ag-specific CTL inhibition in animals with large established tumors. DNA Cell Biol. 2018;37(10):850–860. doi:10.1089/dna.2018.4319.
- Porchia BFMM, Moreno ACR, Ramos RN, Diniz MO, Lhtm DA, Rosa DS, Barbuto JAM, Boscardin SB, Ferreira LCS. Herpes simplex virus glycoprotein D targets a specific dendritic cell subset and improves the performance of vaccines to human papillomavirus-associated tumors. Mol Cancer Ther. 2017;16(9):1922–1933. doi:10.1158/1535-7163.MCT-17-0071.
- Wu L, Liu H, Guo H, Wu Q, Yu S, Qin Y, Wang G, Wu Q, Zhang R, Wang L, et al. Circulating and tumor-infiltrating myeloid-derived suppressor cells in cervical carcinoma patients. Oncol Lett. 2018;15(6):9507–9515. doi:10.3892/ol.2018.8532.
- Sevko A, Umansky V. Myeloid-derived suppressor cells interact with tumors in terms of myelopoiesis, tumorigenesis and immunosuppression: Thick as thieves. J. Cancer. 2013;4(1):3–11. doi:10.7150/jca.5047
- Nowak AK, Robinson BWS, Lake RA. Gemcitabine exerts a selective effect on the humoral immune response : implications for combination chemo-immunotherapy. Cancer Res. 2002;62(8):2353–2358.
- Le HK, Graham L, Cha E, Morales JK, Manjili MH, Bear HD. Gemcitabine directly inhibits myeloid derived suppressor cells in BALB/c mice bearing 4T1 mammary carcinoma and augments expansion of T cells from tumor-bearing mice. Int Immunopharmacol. 2009;9(7–8):900–909. doi:10.1016/j.intimp.2009.03.015.
- Diniz MO, Sales NS, Silva JR, Ferreira LCS. Protection against HPV-16-associated tumors requires the activation of CD8+ effector memory t cells and the control of myeloid-derived suppressor cells. Mol Cancer Ther. 2016;15(8):1920–1930. doi:10.1158/1535-7163.MCT-15-0742.
- Moreno ACR, Porchia BFMM, Pagni RL, Souza PDC, Pegoraro R, Rodrigues KB, Barros TB, Aps LRDMM, de Araújo EF, Calich VLG, et al. The combined use of melatonin and an indoleamine 2,3-dioxygenase-1 inhibitor enhances vaccine-induced protective cellular immunity to hpv16-associated tumors. Front Immunol. 2018 August;9:1914. doi:10.3389/fimmu.2018.01914.
- Pei Q, Pan J, Zhu H, Ding X, Liu W, Lv Y, Zou X, Luo H. Gemcitabine-treated pancreatic cancer cell medium induces the specific CTL antitumor activity by stimulating the maturation of dendritic cells. Int Immunopharmacol. 2014;19(1):10–16. doi:10.1016/j.intimp.2013.12.022.
- Welters MJ, Van Der Sluis TC, Van Meir H, Loof NM, Van Ham VJ, Van Duikeren S, Santegoets SJ, Arens R, De Kam ML, Cohen AF, et al. Vaccination during myeloid cell depletion by cancer chemotherapy fosters robust T cell responses. Sci Transl Med. 2016;8:334. doi:10.1126/scitranslmed.aad8307.